
Figure 1: Main cellular types, pathogenic mechanisms and hypotheses in systemic sclerosis.
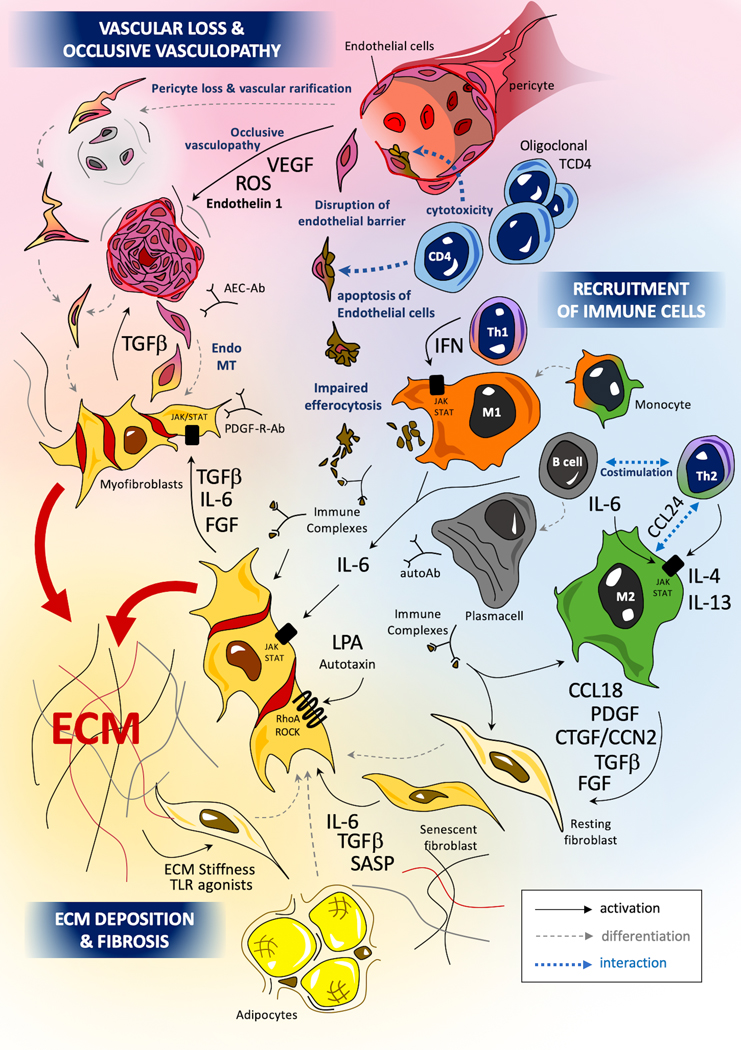
The pathogenesis of SSc involves 3 main mechanisms: occlusive microangiopathy, early inflammatory processes and uncontrolled extra-cellular matrix (ECM) production with resultant fibrosis. Recent studies highlight the role of oligoclonal cytotoxic T-CD4+, driving the apoptosis of endothelial cell (EC) (175,176). Innate immunity, and notably monocytes and macrophages, play a key role in the pathogenesis of SSc (177). Macrophages can adopt various activation profiles depending on their surrounding micro-environment. Interferon (IFN) type II signaling, involving JAK1/TYK2/STAT-1 or TLR-4 agonists induce a classical M1 pro-inflammatory polarization. Th2 cytokines such as IL-4 or IL-13 can induce an alternative profibrotic M2 activation through a STAT3/6 dependent signaling (181): IL-6 also potentiates M2 polarization, notably through the up-regulation of the IL-4 receptor (182). A concomitant excess of CD163highM2 and M1 macrophages has been identified in skin tissues of SSc patients (183) (9). SSc-macrophages show impaired capacities efferocytosis apoptotic cells (phagocytosis of apoptotic cells) with the potential release of internuclear components from these un-eliminated cell debris. The impact of immune complexes composed by autoantibodies and intra-nuclear proteins (topoisomerase, centromere proteins) may also participate to macrophage and fibroblast activation. Myofibroblasts are the major effectors of fibrosis. Myofibroblasts in SSc originate from a variety of tissue-resident mesenchymal progenitor cell types, including fibroblasts, pericytes, microvascular endothelial cells and vascular pre-adipocytes (12). The trans-differentiation of resting fibroblasts and other progenitor cells into pro-fibrotic and inflammatory myofibroblasts is driven by canonical smad-dependent (smad2/3 and 4) and non-canonical smad-independent tumoral growth factor (TGF)-β signaling. Activated myofibroblasts also produce profibrotic mediators such as IL-6 or connective tissue growth factor (CTGF)/CCN2, leading to an autocrine profibrotic pathogenic loop maintaining sustained cellular activation. IL-6 mediates its effects through JAK1/2/TYK2 with subsequent phosphorylation of STAT3 (predominantly) and STAT 1. STAT3 notably promotes the production of key ECM components such as col1a1, col1a2, and profibrotic markers such as CTGF/CCN2 (10). CTGF/CCN2 exerts profibrotic properties notably as a co-factor of TGF-β signaling. CTGF/CCN2 can interact with specific receptors (such as integrins or lipoprotein receptor-related proteins), ECM proteins (such as fibronectin or perlecan) and growth-factors (such as VEGF and TGF-β), with subsequent activation of fibroblast proliferation and myofibroblasts activation (184,185). Uncontrolled production of extra-cellular ECM components such as collagens, tenascin C or fibronectin can in turn activate myofibroblasts either through a direct process involving innate immune sensors such as TLR-4, or through an indirect activation notably depending on mechano-sensing of increased matrix stiffness by integrins (23,24).
IFN= interferon, endoMT=endothelial to mesenchymal transition, Autoab=autoantibodies, PDGF-R-Ab= autoantibodies with agonist effects on PDGF-Receptor, AEC-ab=anti-endothelial cell antibodies, notably including anti endothelin-receptor antibodies with agonists properties, ROS=reactive oxygen species