
Figure 3:
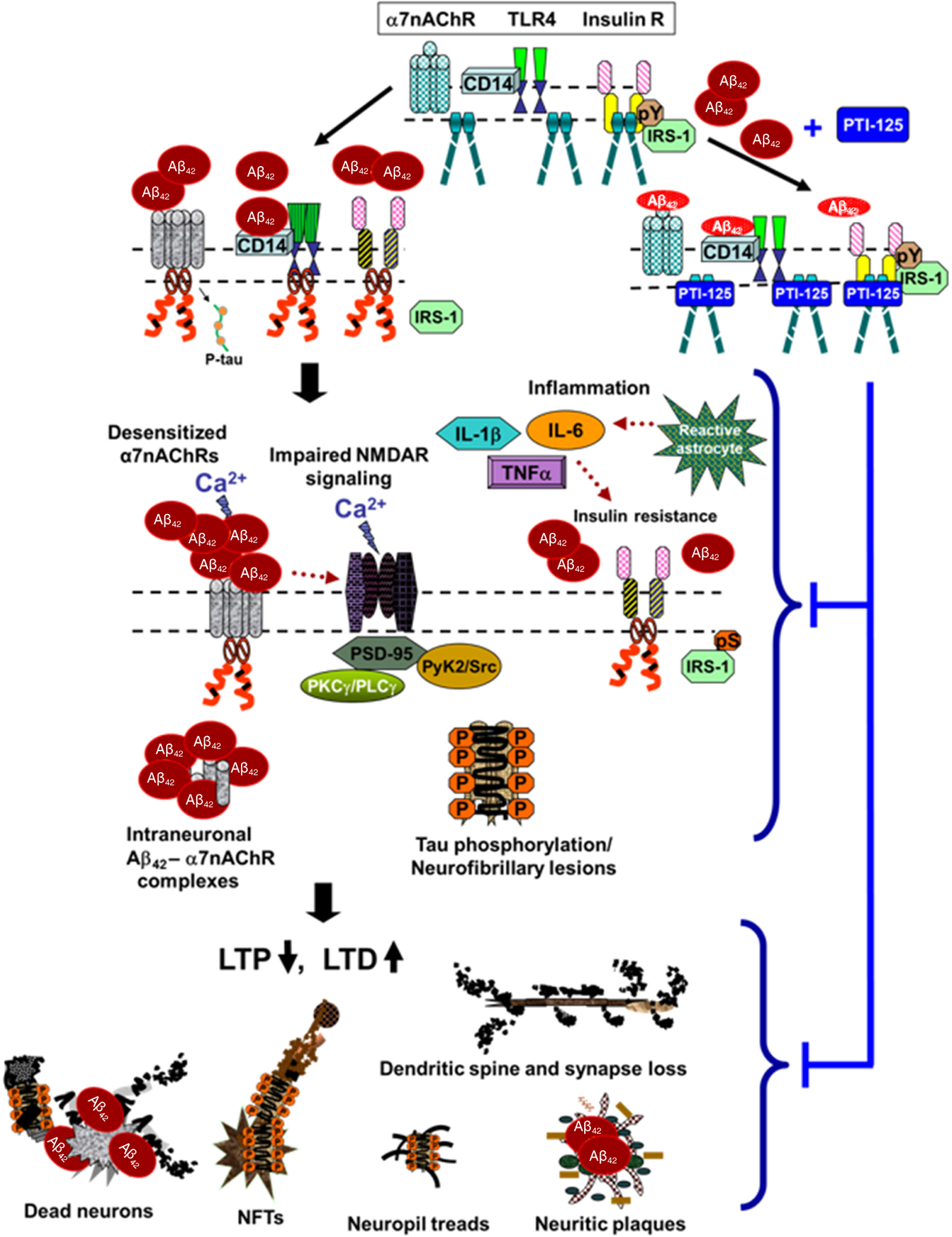
Proposed model of pathological consequences of altered FLNA-enabled Aβ42 signaling via α7nAChR and TLR4. Soluble Aβ42 monomers or small oligomers bind α7nAChR or CD14, complexed with TLR4, inducing recruitment of FLNA to these receptors. Dimers of native FLNA, coupled to insulin receptors but not to α7nAChR or TLR4, are depicted as straight rods; red curly FLNA depicts the altered form, which is recruited to α7nAChR and TLR4 (and possibly also insulin receptors). Enabled by altered FLNA’s new linkages, Aβ42 activates α7nAChR to hyperphosphorylate tau and persistently activates TLR4 to induce inflammatory cytokine release (TNFα, IL-1β and IL-6) by reactive astrocytes. This neuroinflammation likely contributes to insulin receptor desensitization[57]. Although the insulin receptor is constitutively associated with native FLNA, it is possible that altered FLNA also contributes to the insulin receptor dysfunction in AD. Aβ42’s aberrant signaling through α7nAChR impairs function of α7nAChR and of NMDARs, restricting calcium influx through both receptors. Increasing Aβ42 piling onto α7nAChR leads to intraneuronal Aβ42-α7nAChR complexes. The hyperphosphorylated tau dissociates from microtubules, disrupting microtubule stability, axonal transport and neuronal function. Along with dysfunctional tau, impaired NMDARs reduce LTP and heighten LTD. Dendritic spines and synapses are lost. Neuritic plaques, neuropil treads and neurofibrillary tangles are formed, and neurons degenerate. FLNA: filamin A; Aβ: amyloid beta; α7nAChR: α7 nicotinic acetylcholine receptor; TLR4: toll-like-receptor 4; TNFα: tumor necrosis factor-α; IL: interleukin; AD: Alzheimer’s disease; NMDAR: N-methyl-D-aspartate receptor; LTP: long-term potentiation; LTD: long-term depression