

Abstract
Although large genome-wide association studies (GWAS) of major depressive disorder (MDD) have identified many significant loci, the SNP-based heritability remains notably low, which might be due to etiological heterogeneity in existing samples. Here, we test the utility of targeting the severe end of the MDD spectrum through genome-wide SNP genotyping of 2725 cases who received electroconvulsive therapy (ECT) for a major depressive episode (MDE) and 4035 controls. A subset of cases (n = 1796) met a narrow case definition (MDE occurring in the context of MDD). Standard GWAS quality control procedures and imputation were conducted. SNP heritability and genetic correlations with other traits were estimated using linkage disequilibrium score regression. Results were compared with MDD cases of mild-moderate severity receiving internet-based cognitive behavioral therapy (iCBT) and summary results from the Psychiatric Genomics Consortium (PGC). The SNP-based heritability was estimated at 29–34% (SE: 6%) for the narrow case definition, considerably higher than the 6.5–8.0% estimate in the most recent PGC MDD study. Our severe MDE cases had smaller genetic correlations with neurodevelopmental disorders and neuroticism than PGC MDD cases but higher genetic risk scores for bipolar disorder than iCBT MDD cases. One genome-wide significant locus was identified (rs114583506, P = 5e−8) in an intron of HLA-B in the major histocompatibility locus on chr6. These results indicate that individuals receiving ECT for an MDE have higher burden of common variant risk loci than individuals with mild-moderate MDD. Furthermore, severe MDE shows stronger relations with other severe adult-onset psychiatric disorders but weaker relations with personality and stress-related traits than mild-moderate MDD. These findings suggest a different genetic architecture at the severest end of the spectrum, and support further study of the severest MDD cases as an extreme phenotype approach to understand the etiology of MDD.
Subject terms: Depression, Genetics
Introduction
Major depressive disorder (MDD) has a lifetime prevalence of 15–20% [1]. Given its high prevalence and the tendency for chronicity, MDD is associated with considerable morbidity, mortality, as well as personal and societal costs [2]. Identifying causal genetic variants associated with MDD could improve understanding of underlying biological networks and lead to identification or repurposing of pharmacotherapies to treat patients suffering from this debilitating disorder.
For many years, genome-wide association studies (GWAS) yielded no significant findings for MDD [3], likely related to its relatively low twin-heritability of 31–42% [4]. Concerted efforts to massively increase sample size [5–7] resulted in a meta-analysis of 130,664 MDD cases and 330,470 controls that identified 44 significant loci [7], followed by 102 significant variants in a discovery cohort of 246,363 cases and a replication sample of 414,055 cases [5]. These studies began to illuminate mechanisms underlying MDD, such as altered gene expression in the prefrontal and anterior cingulate cortex and connection to antidepressant mechanisms of action [7].
The most recent SNP-based heritability estimate for MDD is still low (8.9%) [5], which might reflect etiological heterogeneity wherein MDD cases are a mix of mild, moderate, and severe forms of MDD with differing etiological process and genetic architectures across the severity spectrum. Increasing sample size to enhance GWAS power required inclusion of less severe MDD cases, resulting in greater phenotypic—and hence possibly etiological—heterogeneity [5, 6, 8]. For instance, the MDD case definition for large sample sizes from 23andMe was based on self-report of having received an MDD diagnosis by a medical professional and not by direct clinical evaluation or health records [9]. Although the genetic correlation between self-reported and clinically ascertained samples with MDD is high (rg = 0.86) [10], it is similarly high for neuroticism (rg = 0.84). Genetic findings in MDD cohorts of lesser severity may confer risk that is not specific for MDD [11].
An alternative strategy to uncover insights about the genetic underpinnings of MDD is to focus on patients with greater severity [12, 13]. An obvious choice is patients treated with electroconvulsive therapy (ECT). ECT is an effective treatment for severe major depressive episodes (MDE) [14, 15], and is not prescribed for mild cases [16–18]. As described at length elsewhere [19], the rationale for focusing on individuals receiving ECT for MDE is threefold. First, clinical applicability is higher given that patients with mood disorders receiving ECT are among the most severely ill people seen in clinical psychiatry. Second, studying extreme phenotypes may increase statistical power. For example, a GWAS of individuals with very high IQ (mean 170, or top 0.03%) yielded three genome-wide significant variants with only 1238 cases [20], while a population-based sample (mean IQ: ~100) yielded no genome-wide significant variants despite almost 18,000 cases [21]. Third, studying extreme forms of MDD may reduce etiological heterogeneity. For example, CONVERGE identified two genome-wide significant associations in clinically ascertained Han Chinese women with recurrent MDD [22].
For these reasons, we conducted a GWAS of individuals who had received ECT for an MDE. Our aims were to estimate SNP-based heritability and genetic correlation structure of severe MDE compared to milder forms of MDD, and to conduct a case–control GWAS for severe MDE.
Methods
Cases
Participants were selected from the Predictors For ECT (PREFECT) study. PREFECT enrolled people from the Swedish National Quality Register for ECT between 2013 and 2017 (Q-ECT) [23, 24]. All Swedish hospitals administering ECT report to Q-ECT, which collects clinical and demographic data including the indication for the current ECT series and the self-rated Montgomery–Åsberg depression rating scale scores (MADRS-S) [25]. The validity of diagnoses recorded in the Q-ECT register has recently been investigated. The accuracy ranged from 95 to 99% when tested against the ICD-10 diagnostic codes indicating ECT in the medical records [26]. All participants provided informed consent, and the study was approved by the ethical review board in Stockholm (Approval Nos. 2012/1969–31/1 and 2020-10151). PREFECT participants were included in this study if the indication for ECT was an MDE. The “broad case definition” (n = 2904, or 2725 after genotyping quality control (QC)) included MDE in the context of (a) MDD (ICD-10 codes F320–F323, F328–F333, F338, F339, F412, F530), (b) bipolar disorder (F313–F315, F318, F319), (c) another mood disorder with a pretreatment MADRS-S score ≥20 (mixed anxiety and depressive disorder F412, schizoaffective disorder F259, or mood disorder NOS F349), or (d) an MDE as indicated by free text or a pretreatment MADRS-S score ≥20 but for which the specific indication was missing in Q-ECT. The “narrow case definition” was a subset of the broad group consisting of those receiving ECT for an MDE in the context of MDD (n = 1922, or 1796 after QC, ICD-10 codes given above). Most participants (47.7%) had received ECT more than once (mean 2.2 courses). If there was more than one course of ECT and if diagnostic indications differed, the first indication was selected after ranking indications by severity, with MDD indications ranked first. Patients who had both broad and narrow indications were categorized in the broad, not narrow, group to be conservative. For example, a patient who had received ECT for F323 (MDD, single episode, severe with psychotic features) and once for F259 (schizoaffective disorder) was classified into the broad group.
Demographic and health information (e.g., education level, occupation, number of suicide attempts) were collected from the Patient Register [27] and Longitudinal integration database for health insurance and labor market studies 1990–2013 [28]. We calculated the number of antidepressant or antipsychotic medication prescriptions using data from the Prescription Drug Register (July 2005 to May 2018). Each of the ATC codes below was considered as one type: antidepressants including N06AA (tricyclics), N06AB (SSRI), and N06AF, N06AG, N06AX (other antidepressants); mood stabilizers including N03AG (valproate), N05AN (lithium), and N03AF, N03AX (other mood stabilizers); antipsychotics including N05AH (clozapine), and N05AA, N05AB, N05AC, N05AD, N05AE, N05AF, N05AH, N05AL, N05AX (other antipsychotics).
PREFECT blood sampling occurred retrospectively or prospectively. In the retrospective collection, an information letter was sent to all persons registered in the Q-ECT. A brief telephone interview was completed for those who volunteered to participate, and a blood sampling kit was sent out. Participants donated blood at their nearest lab. In the prospective collection, patients scheduled for an ECT series with at least six planned sessions were eligible for inclusion. Patients were recruited from seven psychiatric hospitals in Sweden (Danderyd, Huddinge, Hudiksvall, Sahlgrenska, Umeå, Uppsala, and Örebro). Prior to the study, participants were informed about the study and provided informed consent. Blood samples were drawn prior to the first ECT. All blood samples were sent to Karolinska Institutet Biobank where DNA was extracted and stored.
Controls
Controls were obtained from the Swedish arm of the Anorexia Nervosa Genetics Initiative (ANGI) [29, 30]. ANGI controls were population based (identified by Statistics Sweden, n = 1035) or archived controls from LifeGene (n = 3000), a population-based study of genes, environment, lifestyle, and health [31]. Controls were excluded from the original ANGI study if they reported a history of an eating disorder. For purposes of the current study, we also excluded individuals with a self-reported lifetime history of MDD, bipolar disorder, schizophrenia, or schizoaffective disorder. As expected for an eating disorder cohort, ANGI controls were 98% female. Blood samples were provided at special test centers (LifeGene controls) or the nearest lab (population-based controls) and transported to Karolinska Institutet Biobank where DNA was extracted and stored.
MDD comparison cohorts
Two MDD cohorts were selected for comparison with the more severely ill PREFECT cohort. The iCBT cohort consists of n = 964 MDD cases who were treated for MDD with internet-based cognitive behavior therapy (iCBT) at the Internet Psychiatry Clinic in Stockholm [32]. The iCBT study excluded individuals with severe depression and moderate to high risk of suicide, and included only individuals who sought psychotherapy and showed no communication difficulties that would impact treatment. Consequently, this sample comprises patients with higher average educational attainment than PREFECT patients (any university/college: PREFECT 39.5%, iCBT 67.7% [32]). The Psychiatric Genomic Consortium (PGC) MDD cohort comprises n = 170,756 MDD cases (PGC MDD and UK Biobank, excluding the 23andMe cohort) with a range of severity including individuals with a self-reported history of help-seeking for depression [5]. The iCBT cohort was used to compare genetic risk scores (GRS) and the PGC MDD cohort was used to compare GWAS summary statistics.
Genotyping
DNA was extracted from peripheral blood for all samples and genotyped on Illumina GSA-MD SNP arrays (v1). Genotyping was conducted at high-volume core facilities, either Life and Brain GmbH (Bonn, Germany) for PREFECT and iCBT cases or the Broad Institute (Cambridge, MA, USA) for ANGI and LifeGene controls.
Genotype quality control and imputation
Raw SNP data from cases and controls were each harmonized to have the same markers and allele coding, then merged and treated as a single data set. The following inclusion criteria were applied to case and control genotypes separately: marker names identical in cases and controls, call rate > 0, and minor allele frequency > 0.01. All SNP alleles were recoded to the “+” strand using Illumina annotation files. Pseudo-autosomal region markers and markers with poor genotype clustering in cases or controls after manual inspection were removed. The same type of arrays were used for all samples and careful comparisons revealed no important biases by genotyping site. QC was applied with the PGC Ricopili pipeline [33]. For QC, all subjects were included but the final association analysis was performed on PREFECT subsets meeting broad and narrow case definitions. Samples were excluded for genotype missingness > 0.02 (after first removing SNPs with call rate < 0.95), genotypic sex ambiguity or not matching phenotypic data, or autosomal heterozygosity |F| > 0.2. SNPs were excluded for call rates < 0.99, difference in missingness between cases and controls > 0.005, MAF < 0.01, or deviation from Hardy–Weinberg equilibrium in cases or controls (P < 10−6). Ancestry outliers were identified by projecting study samples on principal components (PCs) generated in 1000 Genomes reference data (phase 3, version 5). Individuals > 3 standard deviations from the European reference population mean in PC 1 or 2 were excluded. After outlier exclusion, PCs were generated in the study sample, and one from each pair of identified related individuals flagged for exclusion (estimated IBD sharing > 0.2). Samples passing QC were imputed to the HRC r1.1 reference panel using the Sanger Imputation Service using Eagle2 and PBWT for phasing and imputation [34]. The genome build was hg19.
Statistical analysis
Genome-wide association on imputed data was performed using logistic regression in the Ricopili pipeline, adjusting for the first four ancestry PCs. To explore potential bias introduced by the sex imbalance between cases and controls, we performed sensitivity analyses with sex as an additional covariate. SNP-based heritability and genetic correlations with other traits were estimated from the GWAS results using LD score regression [35]. A sensitivity analysis with genome-wide complex trait analysis (GCTA-GREML, gcta version 1.93.2beta [36]) was conducted to confirm heritability findings. Analyses were performed on directly genotyped markers, and further repeated using only common markers (MAF ≥ 5%). LD score regressions were also repeated using common markers. Conversion from observed to liability scale SNP-based heritability requires an estimate of lifetime prevalence (K) of MDE treated with ECT. Using the Q-ECT register, we identified the unique individuals who had received ECT in Sweden from 2014 to 2018. These estimates were extrapolated to age 85, adjusted upward by 5% to account for missing treatments, and adjusted downward by 15% to remove ECT indications that we excluded (e.g., for manic episodes); this yielded an estimate of K = 1.49%. We report SNP-based heritability on the liability scale for K in the range of 0.8–1.6%. Genetic correlations were compared between PREFECT and the less severe PGC MDD cohort for psychiatric, neurological, and other lifestyle and disease traits.
Genetic risk scores (GRS)
We generated GRS to compare severely ill PREFECT cases with mild-moderately ill cases receiving iCBT. As training data, we used GWAS summary statistics for MDD [5], bipolar disorder [37], schizophrenia [38], educational attainment [39], and cognition [40]. None of the PREFECT cases nor controls were part of any of the training samples. We excluded indels and strand-ambiguous SNPs, and kept one SNP from the extended MHC region (chr6: 25–34 Mb). We applied LD-clumping to remove extensive correlations among SNPs, discarding variants within 500 kb of and in LD r2 > 0.1 with the most associated SNP in a region. We computed GRS for each individual as the sum of the number of risk alleles weighted by ß. For interpretability, these were scaled to represent increased risk and standardized to mean 0 and SD 1. We generated GRS for thresholds of PT ≤ 0.05, 0.1, 0.5, and 1. As a primary analysis, we compared the mean differences in various GRS with PT ≤ 0.05 controlling for the first five ancestry PCs. Educational attainment and intelligence GRS were used as positive controls given the large demographic difference between PREFECT and iCBT cohorts in educational attainment.
Results
Demographics and depression severity
Demographic and clinical characteristics of PREFECT cases receiving ECT for an MDE are shown in Table 1. As expected for an MDE sample, approximately two-thirds (63%) were female [41]. A majority (85%) had completed secondary or university education, and 48.6% were born in an urban area. ECT cases had had a mean of 2.2 previous courses of ECT (SD = 2.2, range: 1–26), 4.0 previous psychiatric hospital admissions (SD = 5.1, range: 1–71), and 5.5 different antidepressant or antipsychotic trials (SD = 2.2, range: 1–14). Nearly one-third (30.9%) had attempted suicide at least once, and 65.2% were diagnosed with a severe depressive episode.
Table 1.
Participant demographic and clinical characteristics at time of ECT.
| Variable | Cases receiving ECT for MDE | Controls | |
|---|---|---|---|
| Age, mean (SD) | 51.6 (16.4) | 33.1 (9.96) | |
| Male, n (%) | 1015 (37.2%) | 65 (2.0%) | |
| Education level |
15.0% compulsory education or less (≤9 years) 45.5% any upper secondary (≤12 years) 39.5% any college/university |
n/a | |
| Born in urban areaa | 48.6% | n/a | |
| Occupation |
8.1% unskilled labor 55.2% craftsmen and skilled labor 17.0% technicians 17.2% professionals 2.4% management work 0.1% military |
n/a | |
| Indication for ECT | Broad case group (n = 2725) | Narrow case group (n = 1796) | |
|---|---|---|---|
| MDD, mild (F320/F330) | 5 (0.2%) | 5 (0.3%) | |
| MDD, moderate (F321/F331) | 363 (13.3%) | 327 (18.2%) | |
| MDD, severe (F322/332) | 1202 (44.1%) | 1038 (57.8%) | |
| MDD, severe with psychotic features (F323/333) | 295 (10.8%) | 268 (14.9%) | |
| MDD, severity unspecified (F329/F339) | 230 (8.4%) | 214 (11.9%) | |
| MDD, postpartum (F530) | 15 (0.6%) | 15 (0.8%) | |
| Other depressive episodes (F328/F338) | 5 (0.2%) | 4 (0.2%) | |
| Other MDEb | 27 (1.0%) | 5 (0.3%) | |
| Bipolar disorder, current MDE, mild/moderate (F313) | 132 (4.8%) | 0 | |
| Bipolar disorder, current MDE, severe (F314) | 229 (8.4%) | 0 | |
| Bipolar disorder, current MDE, severe with psychotic features (F315) | 34 (1.2%) | 0 | |
| Bipolar disorder, severity unspecified (F319) | 26 (1.0%) | 0 | |
| Other bipolar affective disorders (F318) | 6 (0.2%) | 0 | |
| Mixed anxiety and depressive disorder (F412)c | 62 (2.3%) | 20 (1.1%) | |
| Persistent mood disorder, unspecified (F349)c | 1 (<0.1%) | 0 | |
| Schizoaffective disorder (F259) | 22 (0.8%) | 0 | |
| Missing and MADRS-S > 19d | 171 (6.3%) | 0 |
n/a not available.
aCounties containing Stockholm, Gothenburg, Malmö, and Uppsala.
bICD-10 code missing; licensed psychiatrist reviewed treating clinician’s free text description of indication for ECT and classified into narrow or broad group.
cPatients only included if their pre-ECT MADRS-S score was >19.
dICD-10 code missing; subject included if MADRS-S score > 19, indicating severe depression.
Although cases and controls were of similar genetic ancestries and were genotyped on the same SNP arrays, sex was imbalanced due to ascertainment of cases and controls. In our experience, differences in case–control genotyping platform or ancestry is more likely to introduce bias in GWAS results than sex differences (indeed, we observed massive test statistic inflation when we first attempted analyses with a different control sample better matched on sex but genotyped on a different SNP array). In contrast, the present case–control imbalance in sex did not lead to notable bias as there was no evidence of test statistic inflation for the broad case definition (λGC = 1.036, λ1000 = 1.012, and LD score regression intercept 0.98, SE: 0.0078) or for the narrow case definition (λGC = 1.031, λ1000 = 1.013, and LD score regression intercept of 0.99, SE: 0.0074). Furthermore, sensitivity analyses adding sex as a covariate yielded very similar test statistics and no evidence of inflation (broad definition: λGC = 1.028, λ1000 = 1.009, and LD score regression intercept of 0.99, SE: 0.0074; narrow definition: λGC = 1.026, λ1000 = 1.011, an LD score regression intercept of 0.99, SE: 0.0067). Genetic correlations between analyses adjusted for ancestry PCs and sensitivity analyses adjusted for sex were 1.02 (SE: 0.014) for the broad case definition and 1.05 (SE: 0.029) for the narrow case definition. Sensitivity analysis results using GCTA gave somewhat lower point estimates for SNP-based heritability, but were not inconsistent with the LD score results when comparing 95% confidence intervals (see Supplementary Table S1). The same held true for LD score regressions using only common markers (Supplementary Table S2).
Genome-wide association analysis
We conducted GWAS with 2725 cases meeting a broad case definition and a subset of 1796 meeting a narrow case definition (MDE in the context of MDD) compared to 3290 controls. The broad case definition identified three SNPs reaching genome-wide significance (P < 5e–8) (Fig. 1A). The SNP rs114583506 (MAF = 0.04, P = 3.6e−8) was located in an intron of HLA-B in the major histocompatibility region on chromosome 6 and nearby markers had similar P values. Two other significant SNPs had minor allele frequencies near 0.01 but with few or no neighboring markers in LD supporting the association, suggesting false positive findings. In analyses adjusted for sex, the top three SNPs remained the same, and there was minimal change in the effect size for the genome-wide significant SNP in the MHC region: in the primary analysis OR = 0.56 (SE: 0.093) and sex-adjusted analysis OR = 0.60 (SE: 0.103). For the narrow case definition (Fig. 1B), one SNP reached genome-wide significance but had MAF close to 0.01 and no supporting markers in LD consistent with a false positive association. Regional association plots are shown in Figs. S1 and S2 and association results are in Tables S3 and S4.
Fig. 1. Genome-wide association results.

Manhattan plot of P values for single nucleotide polymorphism (SNPs) for the genome-wide association analysis of ECT-treated major depressive episodes (MDE) in the A broad case definition and B narrow case definition of MDE occurring in the context of major depressive disorder. The red line (P < 5e−8) depicts the threshold above which results are considered significant. The MHC SNP alone showed neighboring markers in linkage disequilibrium (LD) supporting the association.
SNP-based heritability and genetic correlations
SNP-based heritability (liability scale) for the broad case definition was 0.35 (SE: 0.05) and 0.31 (SE: 0.06) for the narrow phenotype assuming a lifetime prevalence (K) of 0.01 for ECT-treated MDE. In contrast, the estimated population prevalence for MDD of any severity is 0.08–0.16, and the PGC MDD cohort (which includes MDD cases of varying severity) reported SNP-based heritability estimates of 0.06–0.08. Figure 2 shows SNP-based heritability estimates for different values of K for severe MDE in PREFECT broad and narrow phenotypes as well as for MDD of varying severity from PGC MDD cases (these include a wide range of MDD definitions, from self-report in community samples to those treated by psychiatrists in outpatient and inpatient settings but with small numbers receiving ECT). Across a range of values of K, the SNP-based heritability in cases receiving ECT for an MDE is considerably greater than in PGC MDD cases (SNP-based heritability for broad case definition 0.33–0.39, and narrow case definition 0.29–0.34).
Fig. 2. Comparison of SNP-based heritability (h2) estimates and impact of population prevalence.
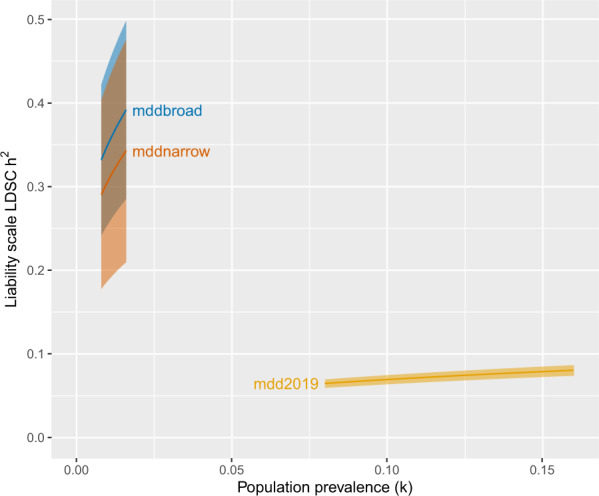
SNP-h2 was computed with linkage disequilibrium score regression (LDSC) on the liability scale for a range of population prevalence (K) values for severe major depressive episode (mddnarrow and mddbroad PREFECT case definitions) and for a large sample of more moderate cases (mdd2019, Psychiatric Genomics Consortium—Major Depressive Disorder Working Group sample excluding 23andMe). Pointwise 95% confidence intervals shaded.
PREFECT cases showed significantly smaller genetic correlations with neurodevelopmental disorders (autism spectrum disorder rg = 0.02 and ADHD rg = 0.26), neuroticism (rg = 0.42), and smoking initiation (rg = 0.18) compared to more diverse PGC MDD cases (Fig. 3 and Table S5). PREFECT cases showed no association with type 2 diabetes, coronary artery disease, or alcohol consumption, which all correlated positively with the less severe PGC MDD cohort. Finally, PREFECT cases showed a significantly stronger negative correlation with intelligence (rg = −0.20), and a negative correlation with BMI (rg = −0.08), while the PGC MDD cases showed a positive significant BMI correlation.
Fig. 3. Genetic correlations comparing a sample with severe major depressive episodes (green, mddnarrow, and blue, mddbroad PREFECT case definitions) to a large sample of more moderate cases (orange, mdd2019, Psychiatric Genomics Consortium—Major Depressive Disorder Working Group sample excluding 23andMe) on psychiatric, neurological, and central nervous system disorders along with other traits.

Results are presented with 95% confidence intervals and computed with linkage disequilibrium (LD) score regression. Asterisks and daggers denote significant differences in correlations of mddbroad and mddnarrow, respectively, with more moderate mdd2019. Comparisons were analyzed using a two-sample z-test with P < 0.05. For data sources see Table S5.
Genetic risk score (GRS) comparisons to mild and moderate MDD samples
To generate GRS, we merged mild-moderate MDD cases receiving iCBT with PREFECT ECT cases, and repeated QC and imputation to avoid bias in subsequent analyses due to SNPs with varying QC qualities in the two case samples. There was no evidence of test statistic inflation (λ1000 = 1.01). After applying the same sample exclusion criteria as for PREFECT case–control QC, 905 iCBT cases, 2757 broad case definition, and 1819 PREFECT narrow cases definition were available for calculation of GRS. When comparing the severely ill PREFECT cases with the mild-moderately ill iCBT cases, PREFECT cases (narrow group) had slightly higher mean GRS for MDD (P = 0.02). The two case sets differed more strongly in GRS for bipolar disorder and cognitive traits, where PREFECT cases carried a significantly higher GRS burden for bipolar disorder and lower GRSs for educational attainment and IQ compared to iCBT cases (P < 1e−4, Fig. 4A, B), for both narrow and broad case definitions.
Fig. 4. Genetic risk scores (GRS) in severe ECT-treated major depressive disorder (MDD) cases (PREFECT MDD) compared with more mild/moderate internet-based cognitive behavioral therapy (iCBT) MDD cases.

The case–case comparison was conducted with the A broad case definition and B narrow case definition with GRS associations with MDD, bipolar disorder (BIP), schizophrenia (SCZ), educational attainment (Edu), and intelligence quotient (IQ).
Discussion
We conducted a GWA analysis in 2725 patients who received ECT for an MDE in the Swedish PREFECT study. Given that ECT is used to treat severe depressive episodes, this group potentially represents a more homogenous subset of patients with MDD than has been studied in most previous GWAS. Taken together, our findings show that the polygenic prediction of severe MDE is not only stronger (as apparent from the high SNP-based heritability), but also different (as revealed by genetic correlations and GRS analyses) than studies of less severe cases. This suggests that larger studies focusing on the severest end of the clinical spectrum have the potential to offer more power and uncover new insights about the genetic basis of MDD.
A previous study of another severe MDD subgroup, recurrent MDD, reported two significant loci but no SNP-heritability estimate [22]. Hence, we provide one of the first SNP-heritability estimates for severe MDE. Our SNP-based heritability estimate of 0.29–0.34 appears to be markedly higher than those from studies of milder MDD and community ascertained samples such as the PGC MDD cohort, where the latest report estimated SNP-based heritability to 0.065–0.080 [5]. Consistent with this observation, twin/pedigree-heritability estimates are also higher in selected MDD subtypes including recurrent, early onset, and/or postpartum MDD than general MDD samples [3, 4, 42]. Furthermore, more stringent definitions of MDD in UK Biobank participants yield higher estimates of SNP-based heritability (0.11–0.13 for self-reported help-seeking, and 0.26–0.32 for self-reported meeting DSM-5 MDD symptom and duration criteria) [11]. SNP-based heritability calculations for discrete traits depend on the estimated population prevalence of the phenotype. In Sweden, ~15% of individuals diagnosed with an MDE receive ECT at some point [23]. We estimate the prevalence of ECT in Sweden to range from 0.008 to 0.016 to yield SNP-based heritability of 0.29–0.34. That SNP-based heritability is higher for severe MDD supports the extreme phenotype strategy, which has been successful for other traits [20], to elucidate the genetics of MDD. Indeed, an international consortium was recently formed to study the genomics of ECT-treated MDD [19].
We also compared the PREFECT and PGC MDD cohorts with respect to genetic correlations with other disorders and traits. Most genetic correlations observed in our PREFECT sample mirrored the correlations in the PCG-MDD cohort with less severe cases. However, we found significantly lower correlations with neuroticism, ADHD, autism, intelligence, BMI, smoking, alcohol use, type 2 diabetes, and coronary heart disease in the severely ill PREFECT cohort than in the PGD-MDD cohort. This different correlation pattern could reflect important underlying biological and neurocognitive differences between severe and mild-moderate MDE. For example, neuroticism has been tightly linked to MDD due to its relationship to rumination and worry, two cognitive processes that maintain depressive symptoms [43]. The smaller correlation with neuroticism in severe MDE possibly indicates that these classic cognitive processes are less important for maintaining symptoms among the severely ill.
Furthermore, we conducted GRS analyses comparing our cohort with less severe cases who underwent internet-based cognitive behavioral therapy (iCBT) to treat MDD. Our severe ECT-treated cases carried a significantly higher GRS burden for bipolar disorder, and, as expected due to sample ascertainment by treatment type, lower GRS burdens for educational attainment and IQ, than cases in the iCBT cohort. This was true also for the narrow phenotype definition (i.e., MDE in the context of MDD). Taken together with the genetic correlations, these findings suggest that severe MDD is less likely to share genetic risk with neuroticism, neurodevelopmental disorders (autism, ADHD), and traits and diseases associated with environment and/or stress (coronary artery disease, smoking, alcohol use, type 2 diabetes, and BMI), but more likely to share genetic risk with adult-onset affective disorders than less severe MDD cases. These results fit well in the genetics of the mood disorder spectrum, where more severe subtypes of MDD were found to be more genetically similar to bipolar disorder and schizophrenia [44]. It is possible that the PREFECT cohort reflects not only a severe subtype but also other clinical variables associated with reasons for ECT such as the progression of illness or prognosis. Larger studies are required to address these potential subtypes within MDD and to reveal whether the etiology of severe/ECT-treated MDD is distinct from mild-moderate MDD as has been suggested [11]. Indeed, in a comprehensive analysis of the UK Biobank, we found that SNP-heritability estimates are considerably greater across multiple more severe subtypes of MDD (manuscript in preparation, Yi et al.). Others point to evidence for a common factor of psychopathology (p) that describes a general liability to psychiatric conditions, citing comorbidities between disorders, a single latent factor emerging from analyses of multiple different symptom questionnaires, and genetic correlations between disorders [45–47]. Our pattern of genetic correlations and GRS results may point to a common factor of severe affective psychopathology potentially distinct from milder forms of psychopathology.
In a GWAS comparing the PREFECT cohort with controls, we found a genome-wide significant association with rs114583506, which lies in an intron of HLA-B in the major histocompatibility locus on chromosome 6. This region contains the greatest number of GWAS associations in the genome, and has been previously associated with multiple psychiatric disorders including schizophrenia, MDD, and PTSD [7, 38, 48].
To our knowledge, this is the largest GWAS of severe MDE to date. Despite this strength, the sample size is small by contemporary standards, and we have formed a global consortium to obtain more cases [19]. Moreover, although controls and cases were genotyped with the same Illumina array, cases and controls were not well-matched on sex and age; however, any potential bias introduced by these differences is likely to be conservative as many young, female controls will develop MDD later in life. Furthermore, with few exceptions (e.g., waist–hip ratio), multiple large-scale studies have reported no sex differences in heritability across hundreds of complex traits, and no sex differences in allele frequencies of common variants [49]. The exclusion criteria of self-reported major psychiatric disorders are comparable to most PGC control cohorts [5]. Most importantly, our sensitivity analyses did not change GWAS results meaningfully, and showed a genetic correlation indistinguishable from one with the original results. Finally, this study relied on diagnoses recorded in the Q-ECT register. Although these have been found to be valid when tested against medical records [26], they were nevertheless made in a real-world clinical setting and psychiatric diagnoses ascertained by clinicians might differ from those ascertained with structured research interviews.
Conclusion
Severe MDE has a far higher SNP-based heritability than mild-moderate MDD. Cases with severe MDE may have a distinctive genetic architecture from milder MDD. Future studies with larger samples of ECT-treated MDE will elucidate these questions.
Supplementary information
Acknowledgements
We first thank the study participants. We also thank the staff at participating ECT-units in Danderyd, Huddinge, Hudiksvall, Sahlgrenska, Umeå, Uppsala, and Örebro for recruiting patients; the Swedish National Quality Register for ECT (Q-ECT) for providing data; and the BBMRI.se and KI Biobank at Karolinska Institutet for professional biobank service. We thank PREFECT data collectors Marie Lundin, Birgitta Ohlander, Milka Krestelica, Radja Dawoud, Martina Wennberg, and PREFECT data manager Bozenna Illadou. The PREFECT study was funded by the Swedish foundation for Strategic Research (KF10-0039), and grants from the Swedish Research Council (2018-02653). PFS was supported by the Swedish Research Council (Vetenskapsrådet, award D0886501), the Horizon 2020 Program of the European Union (COSYN, RIA Grant Agreement No. 610307), and US NIMH (U01 MH109528 and R01 MH077139). CMB was supported by Swedish Research Council (Vetenskapsrådet, award: 538-2013-8864) and US NIMH (R01MH120170, R01MH119084, and U01 MH109528). CR was supported by the Swedish Research Council (2018-02487) and the Swedish Research Council for Health, Working Life and Welfare (2018-00221). CCC was supported by a US Fulbright grant. YL is in part supported by a 2018 NARSAD Young Investigator Grant from the Brain & Behaviour Research Foundation and US NIMH (R01 MH123724).
Data availability
Summary statistics are available for download on the Psychiatric Genomic Consortium website.
Compliance with ethical standards
Conflict of interest
ML declares that, over the past 36 months, he has received lecture honoraria from Lundbeck pharmaceutical. PFS reports the following potentially competing financial interests. Current: Lundbeck (advisory committee, grant recipient), RBNC Therapeutics (advisory committee, stock ownership). CMB reports: Shire (grant recipient, Scientific Advisory Board member); Idorsia (consultant); Pearson (author, royalty recipient). AJ is currently employed at the Swedish Medical Products Agency, SE-75103 Uppsala, Sweden. The views expressed in this paper are the personal views of the authors and not necessarily the views of the government agency. AJ’s contribution to this work was done before he started his employment at the MPA.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Patrick F. Sullivan, Mikael Landén
Contributor Information
Patrick F. Sullivan, Email: pfsulliv@med.unc.edu
Mikael Landén, Email: mikael.landen@gu.se.
Supplementary information
The online version of this article (10.1038/s41380-020-00984-0) contains supplementary material, which is available to authorized users.
References
- 1.Hasin DS, Sarvet AL, Meyers JL, Saha TD, Ruan WJ, Stohl M, et al. Epidemiology of adult DSM-5 major depressive disorder and its specifiers in the United States. JAMA Psychiatry. 2018;75:336–46. doi: 10.1001/jamapsychiatry.2017.4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ekman M, Granström O, Omerov S, Jacob J, Landen M. The societal cost of depression: evidence from 10,000 Swedish patients in psychiatric care. J Affect Disord. 2013;150:790–7. doi: 10.1016/j.jad.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 3.Levinson DF, Mostafavi S, Milaneschi Y, Rivera M, Ripke S, Wray NR, et al. Genetic studies of major depressive disorder: why are there no genome-wide association study findings and what can we do about it? Biol Psychiatry. 2014;76:510–2. doi: 10.1016/j.biopsych.2014.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry. 2000;157:1552–62. doi: 10.1176/appi.ajp.157.10.1552. [DOI] [PubMed] [Google Scholar]
- 5.Howard DM, Adams MJ, Clarke T-K, Hafferty JD, Gibson J, Shirali M, et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci. 2019;22:343–52. doi: 10.1038/s41593-018-0326-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McIntosh AM, Sullivan PF, Lewis CM. Uncovering the genetic architecture of major depression. Neuron. 2019;102:91–103. doi: 10.1016/j.neuron.2019.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet. 2018;50:668. doi: 10.1038/s41588-018-0090-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sullivan PF, Geschwind DH. Defining the genetic, genomic, cellular, and diagnostic architectures of psychiatric disorders. Cell. 2019;177:162–83. doi: 10.1016/j.cell.2019.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hyde CL, Nagle MW, Tian C, Chen X, Paciga SA, Wendland JR, et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat Genet. 2016;48:1031–6. doi: 10.1038/ng.3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Howard DM, Adams MJ, Shirali M, Clarke T-K, Marioni RE, Davies G, et al. Genome-wide association study of depression phenotypes in UK Biobank identifies variants in excitatory synaptic pathways. Nat Commun. 2018;16:1470. doi: 10.1038/s41467-018-03819-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai N, Revez JA, Adams MJ, Andlauer TFM, Breen G, Byrne EM, et al. Minimal phenotyping yields genome-wide association signals of low specificity for major depression. Nat Genet. 2020;52:437–47. doi: 10.1038/s41588-020-0594-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guey LT, Kravic J, Melander O, Burtt NP, Laramie JM, Lyssenko V, et al. Power in the phenotypic extremes: a simulation study of power in discovery and replication of rare variants. Genet Epidemiol. 2011;35:236–46. doi: 10.1002/gepi.20572. [DOI] [PubMed] [Google Scholar]
- 13.Li D, Lewinger JP, Gauderman WJ, Murcray CE, Conti D. Using extreme phenotype sampling to identify the rare causal variants of quantitative traits in association studies. Genet Epidemiol. 2011;35:790–9. doi: 10.1002/gepi.20628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brus O, Cao Y, Gustafsson E, Hultén M, Landen M, Lundberg J, et al. Self-assessed remission rates after electroconvulsive therapy of depressive disorders. Eur Psychiatry. 2017;45:154–60. doi: 10.1016/j.eurpsy.2017.06.015. [DOI] [PubMed] [Google Scholar]
- 15.Nordenskjöld A, Knorring L, von, Engström I. Predictors of the short-term responder rate of electroconvulsive therapy in depressive disorders—a population based study. BMC Psychiatry. 2012;12:115. doi: 10.1186/1471-244X-12-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carney S, Cowen P, Geddes J, Goodwin G, Rogers R, Dearness K, et al. Efficacy and safety of electroconvulsive therapy in depressive disorders: a systematic review and meta-analysis. Lancet. 2003;361:799–808. doi: 10.1016/S0140-6736(03)12705-5. [DOI] [PubMed] [Google Scholar]
- 17.Lisanby SH. Electroconvulsive therapy for depression. N Engl J Med. 2007;357:1939–45. doi: 10.1056/NEJMct075234. [DOI] [PubMed] [Google Scholar]
- 18.Pagnin D, de Queiroz V, Pini S, Cassano GB. Efficacy of ECT in depression: a meta-analytic review. J ECT. 2004;20:13. doi: 10.1097/00124509-200403000-00004. [DOI] [PubMed] [Google Scholar]
- 19.Soda T, McLoughlin DM, Clark SR, Oltedal L, Kessler U, Haavik J, et al. International Consortium on the Genetics of Electroconvulsive Therapy and Severe Depressive Disorders (Gen-ECT-ic). Eur Arch Psychiatry Clin Neurosci. 2019. 10.1007/s00406-019-01087-w. [DOI] [PMC free article] [PubMed]
- 20.Zabaneh D, Krapohl E, Gaspar HA, Curtis C, Lee SH, Patel H, et al. A genome-wide association study for extremely high intelligence. Mol Psychiatry. 2018;23:1226–32. doi: 10.1038/mp.2017.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benyamin B, Pourcain BS, Davis OS, Davies G, Hansell NK, Brion MJA, et al. Childhood intelligence is heritable, highly polygenic and associated with FNBP1L. Mol Psychiatry. 2014;19:253–8. doi: 10.1038/mp.2012.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.CONVERGE Consortium. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature. 2015;523:588–91. doi: 10.1038/nature14659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nordenskjöld A. Kvalitetsregister ECT—Årsrapport 2014. Region Örebro län; Örebro, Sweden. 2014.
- 24.Nordanskog P, Hultén M, Landén M, Lundberg J, von Knorring L, Nordenskjöld A. Electroconvulsive therapy in Sweden 2013: data From the National Quality Register for ECT. J ECT. 2015;31:263–7. [DOI] [PMC free article] [PubMed]
- 25.Svanborg P, Åsberg M. A comparison between the Beck Depression Inventory (BDI) and the self-rating version of the Montgomery Åsberg Depression Rating Scale (MADRS) J Affect Disord. 2001;64:203–16. doi: 10.1016/S0165-0327(00)00242-1. [DOI] [PubMed] [Google Scholar]
- 26.Ahmad I. Validity of diagnoses and treatment dates in the Swedish National Quality Register for Electroconvulsive Therapy. Örebro: Örebro University; 2020. [DOI] [PubMed]
- 27.Ludvigsson JF, Andersson E, Ekbom A, Feychting M, Kim J-L, Reuterwall C, et al. External review and validation of the Swedish national inpatient register. BMC Public Health. 2011;11:450. doi: 10.1186/1471-2458-11-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ludvigsson JF, Svedberg P, Olén O, Bruze G, Neovius M. The longitudinal integrated database for health insurance and labour market studies (LISA) and its use in medical research. Eur J Epidemiol. 2019;34:423–37. doi: 10.1007/s10654-019-00511-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thornton LM, Munn-Chernoff MA, Baker JH, Juréus A, Parker R, Henders AK, et al. The Anorexia Nervosa Genetics Initiative (ANGI): overview and methods. Contemp Clin Trials. 2018;74:61–9. doi: 10.1016/j.cct.2018.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watson HJ, Yilmaz Z, Thornton LM, Hübel C, Coleman JRI, Gaspar HA, et al. Genome-wide association study identifies eight risk loci and implicates metabo-psychiatric origins for anorexia nervosa. Nat Genet. 2019;51:1207–14. doi: 10.1038/s41588-019-0439-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Almqvist C, Adami H-O, Franks PW, Groop L, Ingelsson E, Kere J, et al. LifeGene—a large prospective population-based study of global relevance. Eur J Epidemiol. 2011;26:67–77. doi: 10.1007/s10654-010-9521-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andersson E, Crowley JJ, Lindefors N, Ljótsson B, Hedman-Lagerlöf E, Boberg J, et al. Genetics of response to cognitive behavior therapy in adults with major depression: a preliminary report. Mol Psychiatry. 2019;24:484–90. doi: 10.1038/s41380-018-0289-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lam M, Awasthi S, Watson HJ, Goldstein J, Panagiotaropoulou G, Trubetskoy V, et al. RICOPILI: rapid imputation for COnsortias PIpeLIne. Bioinformatics. 2020;36:930–3. doi: 10.1093/bioinformatics/btz633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48:1279–83. doi: 10.1038/ng.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bulik-Sullivan BK, Loh P-R, Finucane HK, Ripke S, Yang J, Patterson N, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47:291–5. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stahl EA, Breen G, Forstner AJ, McQuillin A, Ripke S, Trubetskoy V, et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat Genet. 2019;51:793–803. doi: 10.1038/s41588-019-0397-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pardiñas AF, Holmans P, Pocklington AJ, Escott-Price V, Ripke S, Carrera N, et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat Genet. 2018;50:381–9. doi: 10.1038/s41588-018-0059-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee JJ, Wedow R, Okbay A, Kong E, Maghzian O, Zacher M, et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat Genet. 2018;50:1112. doi: 10.1038/s41588-018-0147-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Savage JE, Jansen PR, Stringer S, Watanabe K, Bryois J, de Leeuw CA, et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat Genet. 2018;50:912–9. doi: 10.1038/s41588-018-0152-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salk RH, Hyde JS, Abramson LY. Gender differences in depression in representative national samples: meta-analyses of diagnoses and symptoms. Psychol Bull. 2017;143:783–822. doi: 10.1037/bul0000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Viktorin A, Meltzer-Brody S, Kuja-Halkola R, Sullivan PF, Landén M, Lichtenstein P, et al. Heritability of perinatal depression and genetic overlap with nonperinatal depression. Am J Psychiatry. 2016;173:158–65. doi: 10.1176/appi.ajp.2015.15010085. [DOI] [PubMed] [Google Scholar]
- 43.Segerstrom SC, Tsao JCI, Alden LE, Craske MG. Worry and rumination: repetitive thought as a concomitant and predictor of negative mood. Cogn Ther Res. 2000;24:671–88. doi: 10.1023/A:1005587311498. [DOI] [Google Scholar]
- 44.Coleman JRI, Gaspar HA, Bryois J, Bipolar Disorder Working Group of the Psychiatric Genomics Consortium, Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium, Breen G. The genetics of the mood disorder spectrum: genome-wide association analyses of more than 185,000 cases and 439,000 controls. Biol Psychiatry. 2020;88:169–84. [DOI] [PMC free article] [PubMed]
- 45.Caspi A, Moffitt TE. All for one and one for all: mental disorders in one dimension. Am J Psychiatry. 2018;175:831–44. doi: 10.1176/appi.ajp.2018.17121383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brainstorm Consortium, Anttila V, Bulik-Sullivan B, Finucane HK, Walters RK, Bras J, et al. Analysis of shared heritability in common disorders of the brain. Science. 2018;360:eaap8757. [DOI] [PMC free article] [PubMed]
- 47.Selzam S, Coleman JRI, Caspi A, Moffitt TE, Plomin R. A polygenic p factor for major psychiatric disorders. Transl Psychiatry. 2018;8:1–9. doi: 10.1038/s41398-018-0217-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nievergelt CM, Maihofer AX, Klengel T, Atkinson EG, Chen C-Y, Choi KW, et al. International meta-analysis of PTSD genome-wide association studies identifies sex- and ancestry-specific genetic risk loci. Nat Commun. 2019;10:4558. doi: 10.1038/s41467-019-12576-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khramtsova EA, Davis LK, Stranger BE. The role of sex in the genomics of human complex traits. Nat Rev Genet. 2019;20:173–90. doi: 10.1038/s41576-018-0083-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Summary statistics are available for download on the Psychiatric Genomic Consortium website.