
FIGURE 1.
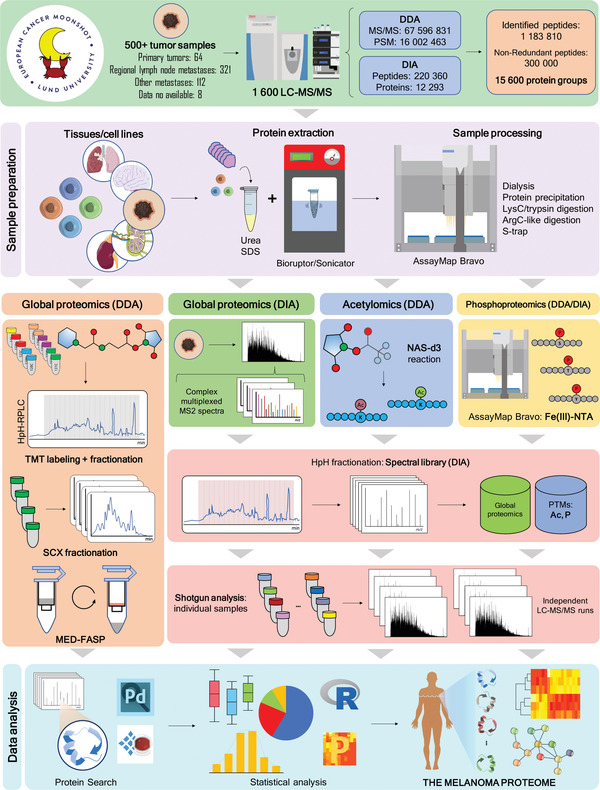
Comprehensive view of proteomic workflows used in the MM500 study. (Upper panel) 505 melanoma tissue samples and four cultured cell lines were analyzed. 1549 LC‐MS/MS experiments produced a proteomic signature of melanoma based on the quantification of 15 973 protein groups representing more than 360 000 nonredundant peptides. (Sample preparation) Several protocols were used which included protein extraction in the presence of urea or SDS with the aid of a Sonifier or a Bioruptor, followed by manual or automatic enzymatic digestion. (Global proteomics) This was performed using both DDA and DIA. DDA data was generated by TMT 11‐plex technology combined with high pH RP‐HPLC fractionation; by SCX stepwise separation of peptide mixtures, by the analysis of fractions derived from the MED‐FASP method, and also by shotgun proteomics. (Acetylomics) DIA‐MS was used to determine naturally occurring protein acetylation sites. This was achieved by modifying protein‐free lysine e‐amino groups with deuterium‐labeled acetyl groups, which upon MS peptide identification and quantitation allowed distinguishing chemically labeled acetylation from endogenous acetylation. 80 (Phosphoproteomics) Enrichment of phosphopeptides was performed in the Bravo AssayMap robot 110 and isolated phosphopeptides were directly analyzed by DDA or DIA. (Spectral Libraries of DIA‐MS) MS/MS spectral libraries for DIA‐MS global proteomics acetylomics and phosphoproteomics were built out of DDA‐LC‐MS/MS data. This included shotgun analysis of the very same samples submitted to DIA‐MS, of other samples from melanoma tissues and cultured cells used in this meta‐study, as well as the analysis of a mixture of these samples previously fractionated by high pH RP‐HPLC. (Shotgun analysis) Individual samples were submitted to LC‐MS/MS analysis either in DDA or DIA modes. (Data analysis) The programs Proteome Discoverer and Spectronaut were used throughout all the experiments for protein identification and quantitation