

SUMMARY
NMDA receptor (NMDAR)-dependent Ca2+ influx underpins multiple forms of synaptic plasticity. Most synaptic NMDAR currents in the adult forebrain are mediated by GluN2A-containing receptors, which are rapidly inserted into synapses during long-term potentiation (LTP); however, the underlying molecular mechanisms remain poorly understood. In this study, we show that GluN2A is phosphorylated at Ser-1459 by Ca2+/calmodulin-dependent kinase IIα (CaMKIIα) in response to glycine stimulation that mimics LTP in primary neurons. Phosphorylation of Ser-1459 promotes GluN2A interaction with the sorting nexin 27 (SNX27)-retromer complex, thereby enhancing the endosomal recycling of NMDARs. Loss of SNX27 or CaMKIIα function blocks the glycine-induced increase in GluN2A-NMDARs on the neuronal membrane. Interestingly, mutations of Ser-1459, including the rare S1459G human epilepsy variant, prolong the decay times of NMDAR-mediated synaptic currents in heterosynapses by increasing the duration of channel opening. These findings not only identify a critical role of Ser-1459 phosphorylation in regulating the function of NMDARs, but they also explain how the S1459G variant dysregulates NMDAR function.
Graphical Abstract
In brief
Yong et al. identify that activity-dependent phosphorylation of Ser-1459 in the GluN2A C-terminal domain by CaMKIIα promotes its interaction with the SNX27-retromer complex, thereby enhancing the surface expression of NMDARs during synaptic potentiation. Mutations of Ser-1459 prolong the decay times of NMDAR-mediated synaptic currents by increasing the duration of channel opening.
INTRODUCTION
NMDA receptors (NMDARs) are ionotropic glutamate receptors that act as “coincidence detectors” of presynaptic glutamate release and postsynaptic membrane depolarization. NMDAR-mediated excitatory postsynaptic currents (EPSCs) mediate the flux of calcium (Ca2+) into the postsynaptic compartment, triggering downstream Ca2+-dependent signaling cascades that are crucial for neuronal development, synaptic and structural plasticity, learning, and memory (Bosch and Hayashi, 2012; Morris, 2013; Nicoll and Roche, 2013; Paoletti et al., 2013). Pharmacological and genetic manipulations that disrupt the expression and function of NMDARs often cause impairments in synaptic plasticity and cognitive deficits in animal models. Importantly, NMDAR dysfunction has also been implicated in many human neurological disorders, including stroke, epilepsy, Alzheimer’s disease, neuropathic pain, and schizophrenia (Zhou and Sheng, 2013). Moreover, genes that encode NMDAR subunits are remarkably intolerant to mutations, which have been associated with various human neurodevelopmental and neuropsychiatric disorders such as epilepsy, autism spectrum disorders, intellectual disability, and schizophrenia (Myers et al., 2019; XiangWei et al., 2018).
Most NMDARs in the forebrain are heterotetramers composed of two obligatory GluN1 subunits and two identical (diheteromeric) or different (triheteromeric) GluN2 subunits (Paoletti et al., 2013; Sanz-Clemente et al., 2013; Stroebel et al., 2018; Vieira et al., 2020). Among the four different glutamate-binding GluN2 subunits, GluN2A and GluN2B, each of which confers NMDARs with distinct ion channel properties and intracellular trafficking pathways (Sanz-Clemente et al., 2013; Vieira et al., 2020; Wyllie et al., 2013), are highly expressed in the hippocampus and cortex (Gray et al., 2011). The expression of synaptic NMDARs is regulated during development as they undergo a switch in their subunit composition from GluN2B- to GluN2A-containing receptors (Monyer et al., 1994; Sheng et al., 1994). In the developing visual cortex, the switch in NMDAR subunit composition during the critical period can be rapidly driven by sensory experience (Quinlan et al., 1999). The same phenomenon has also been observed following the induction of long-term potentiation (LTP) in acute hippocampal slices from young mice (Bellone and Nicoll, 2007), organotypic hippocampal slices (Barria and Malinow, 2002; Grosshans et al., 2002), and primary neuronal cultures (Swanger et al., 2013; Zhang et al., 2015). Given that GluN2A-containing NMDARs have a higher channel open probability and a faster deactivation time than do those containing the GluN2B subunit, such an activity-dependent switch in NMDAR subunit composition at synapses will have major implications for dendritic integration, circuit refinement, and synaptic plasticity (Barria and Malinow, 2005; Kirkwood et al., 1996; Shipton and Paulsen, 2013; Yashiro and Philpot, 2008). Despite this, the molecular mechanisms underlying the activity-dependent trafficking of GluN2A-containing NMDARs during synaptic plasticity remain poorly understood.
The precise subcellular localization, membrane trafficking, and synaptic targeting of GluN2-containing NMDARs are largely determined by protein-protein interactions and post-translational modifications in the cytoplasmic C-terminal tails (Lussier et al., 2015; Vieira et al., 2020). Sorting nexin 27 (SNX27) is a highly conserved regulator of cargo retrieval from endosomes to the plasma membrane that directly interacts with various GluN2 subunits of NMDARs through its N-terminal PDZ (postsynaptic density 95/disc-large/zona occluden-1) domain (Cai et al., 2011; Clairfeuille et al., 2016; Mota Vieira et al., 2020). SNX27 forms a complex with retromer (a heterotrimer of VPS26, VPS29, and VSP35) via its direct interaction with VPS26, and it acts as a cargo adaptor for retromer-mediated transport from intracellular endosomes to the cell surface (Cullen and Korswagen, 2011; Gallon et al., 2014). Genetic deletion of SNX27 causes a profound loss of total and surface NMDAR expression due to a defect in the endosomal trafficking pathway, underscoring its critical role in regulating NMDAR recycling in the brain (Wang et al., 2013). The high-affinity binding of the SNX27 PDZ domain to its cargo molecules generally involves the formation of an “electrostatic clamp,” which is formed constitutively by acidic residues at the (−3) and (−5) positions upstream of the PDZ binding motif, or, alternatively, by phosphorylation of serine or threonine residues in these positions (Clairfeuille et al., 2016). The corresponding amino acid at the (−5) position within the GluN2A C-terminal tail is a serine residue (Ser-1459, see Figure 1A), which has recently been shown to be a substrate of Ca2+/calmodulin-dependent kinase IIα (CaMKIIα) (Mota Vieira et al., 2020). Importantly, a rare genetic variant that involves the S1459G substitution has been reported in a patient with epilepsy (Bowling et al., 2017). Although the phosphorylation state of Ser-1459 can regulate the interaction between GluN2A and SNX27, as well as receptor trafficking under basal conditions (Mota Vieira et al., 2020), the role of Ser-1459 phosphorylation in controlling the activity-dependent endosomal recycling of NMDARs during synaptic potentiation is unclear.
Figure 1. GluN2A Ser-1459 phosphorylation state regulates NMDAR recycling.

(A) Schematic domain structure of a single GluN2A subunit depicting the CaMKIIα phosphorylation site, Ser-1459, which is located adjacent to the C-terminal PDZ binding motif (PDZbm).
(B) Phosphorylation of Ser-1459 does not regulate GluN2A surface expression under basal conditions. Primary hippocampal neurons were transfected with plasmids encoding SEP-GluN2A, either wild-type (WT), or the phospho-deficient S1459A (SA) or phospho-mimetic S1459D (SD) mutant at DIV12. Representative images of surface and total SEP-GluN2A in a neuron from each group, together with enlarged images of the boxed regions, are shown. Scale bars, 50 μm and 10 μm (enlarged images).
(C) Quantification of the surface/total GluN2A ratio normalized to the value of control neurons expressing SEP-GluN2A WT. Data are presented as mean ± SEM (WT, n = 30 neurons; S1459A, n = 29; and S1459D, n = 30; from three independent cultures).
(D) Phosphorylation of Ser-1459 does not affect GluN2A internalization under basal conditions. Transfected neurons were incubated with rabbit anti-GFP antibodies at 25°C for 10 min and were allowed to internalize in the medium at 37°C for 30 min.
(E) Quantification of the SEP-GluN2A internalization index normalized to the value of control neurons expressing SEP-GluN2A WT. Data are presented as mean ± SEM (WT, n = 30 neurons; S1459A, n = 27; and S1459D n = 27; from three independent cultures).
(F) The phospho-deficient S1459A mutant reduces the constitutive recycling of GluN2A. Transfected neurons were incubated with rabbitanti-GFP antibodies and receptors were allowed to internalize for 30 min as above. Non-internalized SEP-GluN2A was blocked with anti-rabbit Fab fragments. Neurons were transferred back to 37°C for 30 min to allow for internalized receptors to recycle back to the plasma membrane.
(G) Quantification of the SEP-GluN2A recycling index normalized to the value of control neurons expressing SEP-GluN2A WT that were fixed prior to receptor recycling. Data are presented as mean ± SEM (WT, n = 28 neurons; S1459A, n = 26; and S1459D n = 20; from three independent cultures). *p < 0.05, **p < 0.01 using one-way ANOVA with a Tukey’s multiple comparison test.
In this study, we tested the hypothesis that glycine stimulation, a validated method of chemically inducing LTP in primary neuronal cultures (Lu et al., 2001), rapidly induces CaMKIIα-mediated phosphorylation of GluN2A at Ser-1459 and consequently enhances GluN2A interaction with SNX27, which is required for the upregulation of GluN2A-containing NMDARs on the neuronal plasma membrane during synaptic potentiation. We also investigated the effects of Ser-1459 phosphomutants, as well as the rare S1459G human epilepsy variant, on NMDAR-mediated EPSCs by using an engineered “artificial” synapse preparation that allows us to examine the biophysical properties of NMDARs with a defined subunit composition (Biederer and Scheiffele, 2007). Finally, we also performed single-channel recordings from excised HEK293 membrane patches containing diheteromeric GluN2A-NMDARs to investigate the effects of Ser-1459 mutations on the gating mechanism of NMDA channels.
RESULTS
The phosphorylation state of Ser-1459 regulates the basal recycling of GluN2A-containing NMDARs
Phosphorylation of NMDAR subunits in the C-terminal domain is a common mechanism controlling receptor trafficking and function (Lussier et al., 2015). A recent study has shown that GluN2A is phosphorylated on its C-terminal domain at Ser-1459 by CaMKIIα both in vitro and in cells (Mota Vieira et al., 2020), a finding which we confirmed here (Figure S1). Co-expression of truncated CaMKIIα (tCaMKIIα) lacking the regulatory domain, which is constitutively active, and GST (glutathione S-transferase)-tagged GluN2A C-tails (residues 1364–1464) in heterologous HEK293T cells led to a robust increase in GluN2A phosphorylation on Ser-1459 as detected by a specific antibody against GluN2A phosphorylated (phospho-)S1459 (Figures S1A and S1B). Pharmacological activation of protein kinase A (PKA) or protein kinase C (PKC) with forskolin or phorbol ester (phorbol myristate acetate [PMA]) did not affect the levels of GluN2A phosphorylation at Ser-1459 (Figures S1C and S1D). Immunoreactivity was completely abolished in cells expressing the GST-GluN2A C-tail S1459A mutant, confirming the specificity of the phospho-antibody.
To determine the effect of Ser-1459 phosphorylation on the steady-state expression of surface GluN2A, we transfected rat primary hippocampal neurons with super-ecliptic pH-sensitive GFP (SEP)-tagged GluN2A, either wild-type or the S1459A phospho-deficient or S1459D phospho-mimetic mutant, and performed a surface staining assay with anti-GFP antibodies that recognize the extracellular SEP. No significant differences in the levels of surface GluN2A were observed across genotypes (Figures 1B and 1C). Next, we carried out an antibody-feeding assay with GFP antibodies to measure the degree of SEP-GluN2A internalization and recycling in live transfected hippocampal neurons over a period of 1 h (30 min of internalization plus 30 min of recycling). Although we did not observe any significant differences in the amount of endocytosed SEP-GluN2A among the three transfected groups (Figures 1D and 1E), the GluN2A-S1459A phospho-deficient mutant caused a marked reduction in the level of NMDAR recycling back to the plasma membrane compared to wild-type or the GluN2A-S1459D phospho-mimetic mutant (Figures 1F and 1G). These results indicate that the phosphorylation of Ser-1459 promotes the endosomal recycling of GluN2A-containing NMDARs in primary hippocampal neurons under basal conditions.
CaMKIIα-dependent phosphorylation of GluN2A at Ser-1459 enhances SNX27-retromer binding
SNX27 is known to interact with GluN2 subunits of NMDARs and play a role in receptor trafficking through the intracellular endosomal system (Cai et al., 2011; Clairfeuille et al., 2016; Mota Vieira et al., 2020; Wang et al., 2013). Given that a mechanism underlying SNX27 cargo selection involves the phosphorylation of serine or threonine residues upstream of the PDZ binding motif, we predicted that phosphorylation of GluN2A at Ser-1459 would increase the binding affinity toward SNX27. To test this, we first performed an isothermal titration calorimetry (ITC) assay and measured the binding affinity of the purified recombinant SNX27 PDZ domain with GluN2A peptides (Figure 2A; Table S1). Our results showed that the substitution of serine to glutamic acid at residue 1459 enhanced SNX27 binding (dissociation constant [Kd] from 46 to 25 μM), whereas serine-to-alanine substitution did not have any significant effect (Kd = 49 μM). As expected, a serine-to-glutamate mutation at Ser-1462 (−2 position) completely abolished the interaction between the GluN2A peptide and SNX27, confirming that complex formation is dependent on the PDZ domain. We next performed a pull-down assay using total lysates of HEK293T cells co-expressing myc-SNX27 and GST-GluN2A C-tails (residues 1213–1464) with phosphorylation-deficient (S1459A) or phospho-mimetic (S1459E) mutations. Compared with wild-type GluN2A, the phospho-mimetic mutant significantly increased SNX27 binding (Figures 2B and 2C). Similar results were obtained when we performed the same pull-down assay using lysates of cells co-expressing GST-SNX27 and full-length SEP-GluN2A (Figures S2A and S2B). In addition, GST-GluN2A C-tails were able to pull down components of the endogenous retromer complex, VPS26 and VPS35, the interaction of which was also dramatically enhanced by the GluN2A S1459E mutant (Figures 2B, 2D, and 2E). The V1464E PDZ binding motif-defective mutant or GST alone failed to pull down myc-SNX27 and the endogenous retromer complex, confirming the specificity of the assay (Figures 2B-2E). Furthermore, mutation of a single conserved histidine residue in the SNX27 PDZ binding motif interacting pocket (H112A) abolished SNX27 binding to both wild-type GluN2A and the phospho-mimetic mutant (Figure 2F). Taken together, these data demonstrate a role for the Ser-1459 phospho-mimetic mutant in promoting the PDZ-dependent interaction between SNX27 and GluN2A.
Figure 2. GluN2A Ser-1459 phosphorylation enhances SNX27-retromer binding.

(A) ITC experiments comparing the binding of GluN2A C-terminal peptides, either WT (blue), or the S1459A phospho-deficient (orange), S1459E phospho-mimetic (green), or S1462E PDZ binding-deficient (purple, negative control) mutant, to the purified SNX27 PDZ domain protein. The thermodynamic parameters of binding are given in Table S1.
(B) GST pull-down experiments from HEK293T cells co-transfected with plasmids encoding myc-SNX27 and GST alone or GST-GluN2A C-tails including WT, S1459A (SA), S1459E (SE), and the V1464E PDZbm mutant (PDZd). Total cell lysates (input) and bound proteins (pull-down) were resolved by SDS-PAGE and analyzed by western blotting with specific antibodies against myc, VPS35, VPS26, and GST.
(C–E) Quantification of the levels of myc-SNX27 (C), VPS35 (D), and VPS26 (E) binding to GST-GluN2A C-tails. Data represent mean ± SEM of band intensities normalized to WT values (n = 3–4; from three independent experiments). *p < 0.05, ***p < 0.001, ****p < 0.0001 using one-way ANOVA with a Tukey’s multiple comparison test.
(F) GST pull-down experiments revealed that the H112A mutation in myc-SNX27, which disrupts binding to PDZbm, failed to bind both WT and the S1459E phospho-mimetic GST-GluN2A C-tails in HEK293T cells.
(G) HEK293T cells were co-transfected with plasmids encoding myc-SNX27, GST-GluN2A C-tails (WT or S1459A), together with pEGFP or pEGFP-tCaMKIIa (constitutively active truncated CaMKIIα). Cells were lysed and pulled down with GSH-Sepharose. Bound proteins and total lysates were analyzed by immunoblotting with specific antibodies against myc, GST, GluN2A pS1459, and GFP.
(H) Quantification of the level of myc-SNX27 binding to GST-GluN2AC-tails. Data represent mean ± SEM of band intensities normalized to WT values(n = 3; from three independent experiments). *p < 0.05 using two-way ANOVA with a Sidak’s multiple comparison test.
To directly examine whether phosphorylation of Ser-1459 increases the binding affinity between SNX27 and GluN2A, we next performed an ITC assay and found that a phosphorylated version of the GluN2A C-terminal peptide bound to the PDZ domain of SNX27 with an even stronger affinity than the S1459E phospho-mimetic peptide (Kd = 14 μM, Table S1). We then performed the same pull-down assay using lysates of HEK293T cells expressing myc-SNX27, GST-GluN2A C-tails (either wild-type or S1459A phospho-deficient mutant) in the presence of GFP or GFP-tCaMKIIα. As expected, overexpression of GFP-tCaMKIIα significantly enhanced myc-SNX27 binding to the GST-GluN2A C-tail, an effect that was abolished by the GluN2A-S1459A phospho-deficient mutant (Figures 2G and 2H). Moreover, the same results were obtained when full-length SEP-GluN2A was used in the pull-down assay (Figures S2C and S2D). Collectively, these data provide strong evidence supporting the role of CaMKIIα-mediated phosphorylation of Ser-1459 in promoting the interaction between GluN2A and theSNX27-retromer complex.
SNX27 is required for an activity-dependent increase in surface GluN2A receptors
To assess whether SNX27 regulates the expression of surface GluN2A-containing NMDARs, we transduced primary cortical neurons with lentiviral particles expressing GFP alone, specific short hairpin RNA (shRNA) against SNX27 with GFP (Hussain et al., 2014), or myc-SNX27 cDNA. Surface biotinylation assays revealed that neither the gain or loss of SNX27 function affected the steady-state expression of total and surface GluN2A under basal conditions (Figure S3). Next, we examined the role of SNX27 in mediating the trafficking of pseudo-phosphorylated GluN2A (S1459D). We co-transfected primary hippocampal neurons with SEP-GluN2A (wild-type, S1459A, or S1459D) and myc-SNX27 constructs, either wild-type or mutants that disrupt the interaction with the retromer complex (L65A) or the PDZ ligands (H112A) (Gallon et al., 2014; Lauffer et al., 2010). Quantification of the surface-to-total SEP-GluN2A ratio in the dendrites revealed a robust increase in the level of the SEP-GluN2A S1459D phospho-mimetic on the plasma membrane of neurons overexpressing myc-SNX27 wild-type (Figures 3A and 3B). In contrast, expression of myc-SNX27 L65A or H112A mutants failed to promote surface expression of pseudo-phosphorylated SEP-GluN2A (Figures 3A and 3B). Moreover, overexpression of myc-SNX27 (wild-type or mutants) did not result in significant changes in the levels of SEP-GluN2A wild-type or the S1459A mutant on the cell surface. Taken together, these results support the notion that SNX27 promotes the activity-induced endosomal recycling of phosphorylated GluN2A at Ser-1459 in a PDZ- and retromer-dependent manner.
Figure 3. SNX27 is required for activity-dependent membrane delivery of the GluN2A-containing NMDARs.

(A) At DIV12, primary hippocampal neurons were co-transfected with plasmids encoding SEP-GluN2A, either WT, or the phospho-deficient S1459A (SA), or phospho-mimetic S1459D (SD) mutant, with various pRK5-myc-SNX27 constructs, including WT, the retromer-associated VPS26 binding-deficient mutant (L65A), and the PDZ-dead mutant (H112A). Representative images of surface and total SEP-GluN2A in a neuron from each group, together with enlarged images of the boxed regions, are shown. Scale bars, 50 μm and 10 μm (enlarged images).
(B) Quantification of the surface/total GluN2A ratio normalized to the value of control neurons co-expressing SEP-GluN2A WT and myc-SNX27 WT. Data are presented as mean ± SEM (WT-WT, n = 39 neurons; WT-L65A, n = 20; WT-H112A, n = 38; S1459A-WT, n = 40; S1459A-L65A, n = 21; S1459A-H112A, n = 39; S1459D-WT, n = 37; S1459D-L65A, n = 25; and S1459D-H112A, n = 36; from three independent cultures). ****p < 0.0001 using one-way ANOVA with a Tukey’s multiple comparison test.
(C) Primary cortical neurons were transduced with lentiviral particles expressing GFP alone (control) or SNX27 shRNA at DIV9. At DIV15, surface biotinylation assays were performed in SNX27 knockdown and control neurons following 5 min of glycine stimulation (cLTP). The relative amounts of surface and total proteins were assessed by western blotting using specific antibodies against GluN2A, SNX27, and β-actin.
(D) Quantification of the surface/total ratio of GluN2A in SNX27 knockdown and control neurons following glycine stimulation. Data represent mean ± SEM of band intensities relative to their respective control values (dashed line; n = 7; from three independent experiments). *p < 0.05 using a Mann-Whitney’s test.
(E) Hippocampal neurons expressing either GFP-SNX27 or GFP alone were stimulated with glycine and subjected to a PLA assay. Representative images showing the PLA signals (top) and GFP fluorescence (bottom) in a 50-μm segment of a primary dendrite of a neuron in each group. Scale bar, 10 μm.
(F and G) Quantification of the number of PLA puncta in the primary dendrite of neurons expressing GFP-SNX27 (F) orGFP alone (G) following glycine stimulation. Data are presented as mean ± SEM (GFP-SNX27, control, n = 31 neurons, cLTP, n = 30; GFP, control, n = 16, cLTP, n = 14; from four independent cultures). ***p < 0.001 using a Mann-Whitney test.
GluN2A-containing NMDARs are rapidly inserted into neuronal membrane during LTP (Barria and Malinow, 2002; Bellone and Nicoll, 2007; Grosshans et al., 2002; Swanger et al., 2013; Zhang et al., 2015), which can be chemically mimicked (cLTP) by bath applying glycine in low Mg2+ artificial cerebrospinal fluid (ACSF) in primary neuronal cultures. To examine the role of SNX27 in regulating the surface targeting of GluN2A during cLTP, we performed a surface biotinylation assay and western blot on primary cortical neurons that were transduced with lentiviral particles expressing either GFP alone or SXN27 shRNA with GFP. As previously reported (Swanger et al., 2013; Zhang et al., 2015), glycine stimulation resulted in a rapid and significant increase in surface GluN2A (Figures 3C and 3D). However, glycine-induced enhancement of surface GluN2A was inhibited in SNX27-depleted neurons (Figures 3C and 3D). To investigate whether glycine stimulation increases the interaction between GluN2A and SNX27, we performed a proximity ligation assay (PLA) in primary hippocampal neurons that expressed either GFP-SNX27 or GFP alone, using antibodies that recognize endogenous GluN2A and GFP. We found that glycine stimulation significantly increased the number of PLA puncta in the primary dendrite of neurons expressing GFP-SNX27 (Figures 3E and 3F). In contrast, no significant changes in the PLA signals between GFP and endogenous GluN2A were observed in control neurons overexpressing soluble GFP alone (Figures 3E and 3G). Collectively, these data underscore the essential role of SNX27 in controlling activity-dependent, but not basal trafficking of, GluN2A-containing NMDARs in primary neurons.
CaMKIIα-dependent Ser-1459 phosphorylation is required for the glycine-induced increase in surface GluN2A receptors
Although the phosphorylation of GluN2A on Ser-1459 is dynamically regulated by neuronal activity in vivo (Mota Vieira et al., 2020), it is not known whether its phosphorylation by CaMKIIα can also be modulated by cLTP stimulation in primary neurons. To address this, we stimulated primary cortical neurons with glycine for 5 min and immunoprecipitated neuronal lysates with anti-GluN2A-pS1459 antibodies. Quantitative western blot analyses revealed a robust increase in the level of Ser-1459 phosphorylation following cLTP, an effect that was blocked by a pharmacological inhibitor of CaMKIIα, KN-93 (Figures 4A and 4B). To corroborate these findings, we performed the same experiments in neurons that were transduced with lentiviral particles expressing Cas9-GFP alone or Cas9-GFP with a specific single guide RNA (sgRNA) against rat CaMKIIα. In agreement with the pharmacological inhibition, deleting CaMKIIα blocked glycine-induced phosphorylation of the GluN2A subunit on Ser-1459 (Figures 4C and 4D). Furthermore, the application of KN-93 prevented glycine-induced upregulation of surface GluN2A (Figures 4E-4G), suggesting that CaMKIIα-dependent phosphorylation of Ser-1459 promotes the endosomal recycling of GluN2A during synaptic potentiation. Finally, we performed a surface staining assay with anti-GFP antibodies on cultured hippocampal neurons that were transfected with SEP-GluN2A, either wild-type or the S1459A phospho-deficient mutant, following cLTP stimulation. Consistent with our surface biotinylation data, glycine treatment led to a robust increase in surface SEP-GluN2A wild-type (Figures 4H and 4I). However, this effect was abolished by the S1459A mutation. In summary, these findings demonstrate a critical role of CaMKIIα-dependent GluN2A phosphorylation on Ser-1459 in promoting SNX27-retromer binding and activity-induced recycling of NMDARs during synaptic potentiation.
Figure 4. CaMKIIα-mediated phosphorylation of GluN2A at Ser-1459 is required for glycine-induced membrane delivery of NMDARs.

(A) Primary cortical neurons were treated with KN-93 or water (control) and stimulated with glycine for 5 min. Endogenous GluN2A phospho-S1459 was enriched from total lysates by immunoprecipitation using anti-pS1459 antibodies and analyzed by western blotting.
(B) Quantification of the levels of Ser-1459 phosphorylation normalized to the vehicle-treated neurons (control, n = 8; KN-93, n = 5; from four independent experiments). Data represent mean ± SEM. **p < 0.01 using one-way ANOVA with a Sidak’s multiple comparison test.
(C) Cortical neurons expressing Cas9 alone (control) or Cas9 with a specific CaMKIIα sgRNA were stimulated with glycine. The levels of GluN2A phosphorylation at Ser-1459 were determined by western blotting.
(D) Quantification of the levels of Ser-1459 phosphorylation normalized to the vehicle-treated neurons (control, n = 4–7; CaMKIIα sgRNA, n = 5–6; from three independent experiments). Data represent mean ± SEM. *p < 0.05 using one-way ANOVA with a Sidak’s multiple comparison test.
(E) Primary cortical neurons were treated with KN-93 or water, stimulated with glycine, and subjected to surface biotinylation assays. The relative amounts of surface and total proteins were assessed by western blotting.
(F and G) Quantification of the surface/total ratio of GluN2A (F) and the total GluN2A/β-actin ratio (G) in each group. Data represent mean ± SEM of band intensities relative to the control values of unstimulated vehicle-treated neurons (n = 8–10 per group). *p < 0.05 using one-way ANOVA with a Sidak’s multiple comparison test.
(H) Hippocampal neurons expressing SEP-GluN2A, either WT or the phospho-deficient S1459A (SA) mutant, were stimulated with glycine and subjected to an antibody-feeding assay. Representative images of surface and total SEP-GluN2A in a neuron from each group are shown. Scale bars, 50 μm and 10 μm (enlarged images).
(I) Quantification of the surface/total GluN2A ratio normalized to the value of unstimulated neurons expressing SEP-GluN2A WT. Data represent mean ± SEM (WT-control, n = 34 neurons; WT-cLTP, n = 34; S1459A-control, n = 33; and S1459A-cLTP, n = 36; from three independent cultures). ****p < 0.0001 using one-way ANOVA with a Tukey’s multiple comparison test.
GluN2A Ser-1459 is a critical residue that regulates the synaptic current and gating of diheteromeric GluN1/2A NMDARs
In addition to its role in regulating glutamate receptor trafficking, phosphorylation of the intracellular C-terminal tail can also modulate the biophysical properties and kinetics of glutamategated ion channels (Traynelis et al., 2010). We next evaluated the impact of Ser-1459 phosphorylation on NMDAR-mediated synaptic currents by measuring spontaneous EPSCs in HEK293 cells that form heterosynapses with mature primary cortical neurons (Figure 5A). This experimental setup allowed us to measure EPSCs that are mediated by NMDARs with defined subunit compositions, namely diheteromeric wild-type GluN1/2A (GluN1/2AWT), GluN1/2AS1459A, or GluN1/2AS1459D receptors. Our results revealed that neither the S1459A phospho-deficient nor the S1459D phospho-mimetic mutant affected the peak synaptic current amplitudes (Figures 5A and 5B). However, mutations of this serine residue significantly altered the NMDAR kinetics (Figures 5A, 5C, and 5D). The measurement of mean normalized currents demonstrated that these mutations slowed the 10%–90% rise times (wild-type, 4.3 ± 0.4 ms; S1459A, 11.3 ± 1.1 ms; S1459D, 14.6 ± 1.5 ms; Figure 5C), as well as the decay phase of the EPSCs (wild-type, 39.1 ± 4.4 ms; S1459A, 72.7 ± 8.6 ms; S1459D, 84.5 ± 8.5 ms; Figure 5D). These data clearly demonstrate that Ser-1459 is a critical determinant that regulates the kinetics of GluN2A-mediated EPSCs, regardless of its phosphorylation status.
Figure 5. Mutations of GluN2A Ser-1459 result in prolonged excitatory postsynaptic currents (EPSCs) in heterosynapses.

(A) Examples of EPSCs together with normalized, averaged current overlays (top, right) mediated by GluN1/2AWT (blue), GluN1/2AS1459A (orange), GluNI/2AS1459D (green), and GluN1/2AS1459G (red).
(B–D) Summary box-and-whisker plots of mean synaptic current peak amplitudes (B), mean 10%–90% activation times (C), and mean decay time constants (D) for the indicated receptors. Boxes indicate the median, 25th, and 75th percentiles, and the whiskers mark the minimum and maximum values (WT, n = 19 cells; S1459A, n = 12; S1459D, n = 18; and S1459G, n = 30). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 using one-way ANOVA with a Dunnett’s multiple comparison test.
Genetic de novo mutations and rare variants occurring in GRIN2A, which encodes the GluN2A subunit, are associated with various neurological and neurodevelopmental disorders, in particular epilepsy/seizures (Myers et al., 2019; XiangWei et al., 2018). A de novo mutation in GRIN2A c.4375A>G (ClinVar, 224108), which results in the missense variant on the GluN2A Ser-1459 phosphorylation site (S1459G), has been reported in an affected individual with focal epilepsy and speech disorder (Bowling et al., 2017). Having demonstrated that the GluN2A S1459G variant did not affect the steady-state expression of surface GluN2A-NMDARs in primary hippocampal neurons (Figure S4), we next sought to determine the properties of EPSCs mediated by diheteromeric GluN1/2AS1459G at heterosynapses. As expected, the S1459G mutant exhibited a slower activation time and deactivation kinetics (Figures 5A, 5C, and 5D). Surprisingly, the peak EPSCs mediated by GluN1/2AS1459G were markedly larger than those mediated by GluN1/2AWT (wild-type, 112.6 ± 16.8 pA; S1459G, 360.3 ± 58.5 pA; Figures 5A and 5B). Taken together, these alterations to NMDAR-EPSCs would result in an increase in the charge transfer per synaptic event and represent a clear gain of function conferred by all three tested mutations.
To further dissect the underlying mechanisms, we examined microscopic NMDAR currents by exposing excised HEK293 membrane patches containing diheteromeric NMDARs to 1 mM glutamate and 100 μM glycine in the absence of extracellular Mg2+. Individual GluN1/2AWT, GluN1/2AS1459G, and GluN1/2AS1459D receptors were activated in well-defined periods of variable duration (Figure 6A). No differences in the single-receptor current amplitude were observed at −70 mV for the three receptors (wild-type, 2.60 ± 0.03 pA; S1459G, 2.68 ± 0.05 pA; S1458A, 2.69 ± 0.04 pA). Unitary current-voltage (I–V) experiments were carried out for wild-type receptors, which exhibited a mild inward rectification and a current reversal potential at −3.2 mV (Figure S5). Assuming that mutant receptors have the same reversal potential, we calculated the unitary conductance for all receptors and found no significant differences among groups (Table S2). These relatively large and uniform unitary currents allowed us to measure the real-time opening and closing rates of NMDARs. Although we found no significant differences in the mean open channel probability (PO ~0.65 for all groups, Figure 6B), the open channel duration of individual active receptors was significantly enhanced in both GluN1/2AS1459G and GluN1/2AS1459D receptors (wild-type, 0.63 ± 0.08 s; S1459G, 1.58 ± 0.48 s; S1459D, 1.73 ± 0.23 s; Figure 6C). Because the mean active period duration is a determinant of the ensemble current deactivation rate (Dixon et al., 2014; Scott et al., 2015; Wyllie et al., 1998), the increase in the open channel duration of mutant receptors was consistent with the observed increase in the synaptic current deactivation kinetics at heterosynapses (Figure 5D). These results therefore suggest a pathomechanism whereby the enhanced excitatory drive caused by the increased charge transfer per synaptic event would promote seizure activity in individuals carrying the GRIN2A c.4375A>G variant.
Figure 6. Ser-1459 mutation regulates the gating of GluN1/2A diheteromeric NMDARs.

(A) Continuous sweep of single-receptor currents mediated by GluN1/2AWT (top, blue), GluN1/2AS1459G (middle, red), and GluN1/2AS1459D (bottom, green) receptors recorded in the presence of 1 mM glutamate, 0 mM extracellular Mg2+, and 100 μM glycine at −70 mV. Note the similar current amplitudes but longer active durations in the mutant receptors.
(B and C) Summary box-and-whisker plots of intra-activation PO (B) and active duration (C) of individual receptors. Boxes indicate the median, 25th, and 75th percentiles, and the whiskers mark the minimum and maximum values (WT, n = 119 single receptor activations from 8 membrane patches; S1459G, n = 101 activations from 5 patches; S1459D, n = 149 activations from 16 patches). *p < 0.05, ***p < 0.001 using Kruskal-Wallis one-way ANOVA with a Dunn’s multiple comparison test.
DISCUSSION
It is well established that NMDARs undergo a rapid switch in subunit composition from GluN2B- to GluN2A-containing receptors during LTP (Bellone and Nicoll, 2007). Despite this, our understanding of how GluN2A-NMDARs are dynamically trafficked into and out of the neuronal plasma membrane in response to neuronal activity is very limited. The present study establishes a mechanism by which activity-dependent phosphorylation of the GluN2A subunit at Ser-1459 by CaMKIIα promotes the association between NMDARs and the SNX27-retromer complex, subsequently enhancing their endosomal trafficking to the neuronal surface during cLTP (Figure 7). In addition, we also found that molecular modifications to Ser-1459 can dramatically influence the gating of NMDARs. Given that a phosphorylationmimicking mutation (S1459D) slowed the synaptic current decay rate, we infer that the phosphorylation of Ser-1459 would do likewise, thereby inducing an increase in net charge transfer and Ca2+ influx during synaptic potentiation.
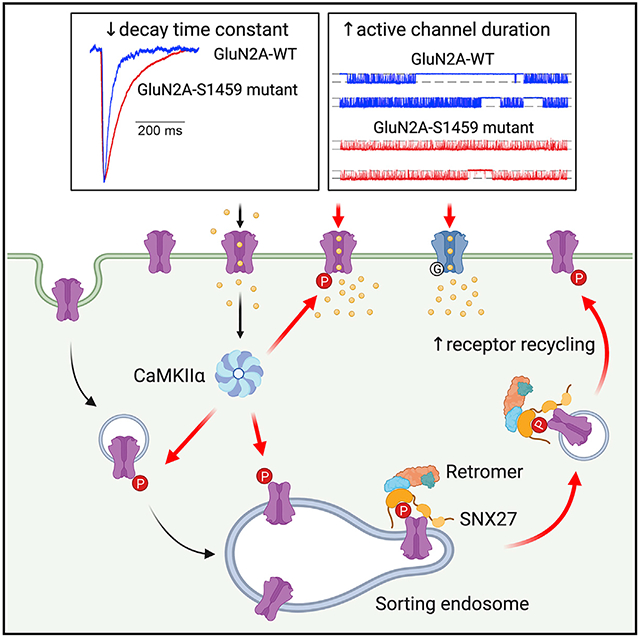
Figure 7. Proposed model for the role of Ser-1459 phosphorylation in regulating NMDAR functions.

During LTP, NMDAR-dependent influx of Ca2+ into the postsynaptic compartment activates the protein kinase CaMKIIα. Phosphorylation of GluN2A at Ser-1459 by CaMKIIα in the endocytic vesicles and/or sorting endosomes promotes its interaction with the SNX27-retromer complex, thereby enhancing the rate of GluN2A recycling back to the plasma membrane. CaMKIIα can also phosphorylate existing GluN2A-containing NMDARs on the surface and affect the gating of these ion channels by prolonging the duration of single channel opening, which in turn leads to enhanced NMDAR current density and Ca2+ influx. Importantly, the GluN2A S1459G variant, which is associated with epilepsy, also displays augmentation in synaptic NMDAR currents due to a decrease in the receptor deactivation kinetics and an increase in active channel duration. Image was created with BioRender.com.
Unlike GluN2B, GluN2A-containing NMDARs are preferentially sorted into late endosomes for degradation (Lavezzari et al., 2004). The SNX27-retromer complex is one of the major endosomal retrieval complexes that prevent lysosomal degradation of many transmembrane proteins, including AMPA receptors (AMPARs) (Hussain et al., 2014; Loo et al., 2014; Temkin et al., 2017; Wang et al., 2013), by recycling them back to the plasma membrane (Steinberg et al., 2013). Our results show that SNX27 itself is not required to maintain the steady-state expression of surface and total GluN2A in primary neurons. Overexpression of SNX27 or the GluN2A S1459D phospho-mimetic mutant does not result in increased levels of surface GluN2A unless they are co-expressed in the same neuron. These data suggest that SNX27 and pseudo-phosphorylated GluN2A are required but not sufficient on their own to drive the endosomal recycling of NMDARs. This is in contrast with the results of a recent study, which found that the GluN2A S1459A phospho-deficient mutant causes a slight but significant reduction in the surface expression of NMDARs (Mota Vieira et al., 2020). The reasons for this discrepancy include differences in the age and density of the neurons, the experimental protocols, and the method of analysis. Notwithstanding these technical variations, both studies concur that phosphorylation of Ser-1459 regulates the basal recycling of GluN2A-NMDARs in neurons.
It appears that SNX27 plays a more prominent role in maintaining the steady-state expression of AMPARs in neurons (Hussain et al., 2014; McMillan et al., 2020). Although all AMPAR subunits possess C-terminal PDZ binding motifs, they do not interact directly with SNX27 (Clairfeuille et al., 2016; McMillan et al., 2020). Instead, their interaction is mediated by the synaptic adhesion molecule leucine-rich repeat and fibronectin type-III domain-containing protein 2 (LFRN2), which interacts with SNX27 through its PDZ binding motif (McMillan et al., 2020). LFRN2 binds to the PDZ domain of SNX27 with a low micromolar affinity due to the presence of an acidic residue at the (−3) position (Asp-786) that forms electrostatic and hydrogen bonds with Arg-58 of SNX27 (Clairfeuille et al., 2016; McMillan et al., 2020). This almost 30-fold difference in LRFN2 (Kd = 1.6 μM) and GluN2A (Kd = 46 μM) binding affinity toward SNX27 may explain the differential requirement of SNX27 in regulating the trafficking of AMPARs versus NMDARs under basal conditions.
One of the major findings of this study is that CaMKIIα-mediated phosphorylation at Ser-1459, the level of which is elevated during synaptic potentiation, enhances the interaction between GluN2A and the SNX27-retromer complex. This provides a mechanism that essentially switches the fate of internalized NMDARs from degradation to the recycling pathway in an activity-dependent manner. These results are consistent with the essential roles of SNX27 and the retromer complex in NMDAR-dependent LTP (Temkin et al., 2017; Wang et al., 2013). Our results therefore indicate that the SNX27-retromer complex is required to deliver not only AMPARs (Hussain et al., 2014; Loo et al., 2014; Temkin et al., 2017; Wang et al., 2013), but also GluN2A-containing NMDARs to the plasma membrane during LTP.
CaMKIIα, one of the most abundant proteins in the brain and an essential regulator of NMDAR-mediated EPSCs and LTP (Bayer and Schulman, 2019; Incontro et al., 2018; Silva et al., 1992), is known to interact with the distal C-terminal domain of GluN2A (Gardoni et al., 2001; Gardoni et al., 1999). More recently, CaMKIIα has been shown to phosphorylate GluN2A at Ser-1459 in the C-tail (Mota Vieira et al., 2020), a finding that we confirmed in the current study. In addition to modulating the binding of GluN2A and SNX27, the phosphorylation state of Ser-1459 also affects PSD-95 binding and the density of dendritic spines in neurons (Mota Vieira et al., 2020). However, the impact of Ser-1459 phosphorylation on NMDAR-mediated EPSCs was not investigated. Using a HEK293-primary neuron co-culture system, we found that both the S1459A and S1459D phosphomutants cause a deceleration in the synaptic current rise time and a slowing in the deactivation kinetics without affecting the amplitude of the peak currents. Mechanistically, the slower rate of receptor deactivation is due to a prolonged duration of single-channel openings. These results suggest that mutations increase the net charge transfer, establishing a critical role for Ser-1459 as part of the gating mechanism for GluN2A-containing receptors. It is conceivable that post-translational modification of Ser-1459 through CaMKIIα-mediated Ser-1459 phosphorylation has the ability to enhance excitatory input and Ca2+ signaling of target neurons, including the maintenance of CaMKIIα activity and AMPAR-mediated potentiation during LTP. Overall, these findings further emphasize the importance of protein phosphorylation of the intracellular C-terminal domain of glutamate receptors as a general regulatory mechanism for receptor gating, which is best exemplified by the effects of GluA1 phosphorylation by PKA and CaMKIIα on AMPAR open probability and channel conductance (Banke et al., 2000; Derkach et al., 1999; Kristensen et al., 2011).
Many genetic GRIN2A variants have been found in patients with epilepsy. Most of these variants are located in the extracellular and transmembrane domains where they typically result in an overall increase in NMDAR-mediated currents (Myers et al., 2019; XiangWei et al., 2018). However, a small number of mutations in the intrinsically disordered intracellular C-terminal domain have also been reported, including the missense variant on the GluN2A Ser-1459 phosphorylation site (S1459G) in an individual with focal epilepsy and speech disorder (Bowling et al., 2017). The GluN2A-S1459G variant exhibits defects in SNX27 and PSD-95 binding, and consequently causes deficits in receptor recycling, reduced dendritic spine density, and a decrease in the frequency of AMPAR-mediated mini-EPSCs when overexpressed in cultured hippocampal neurons (Mota Vieira et al., 2020). In the present study, we directly measured the effects of the S1459G mutation on NMDAR-mediated currents at heterosynapses. As expected, the S1459G variant altered the kinetics of receptor activation and deactivation, accompanied by an aberrant enhancement in the peak current amplitude. Single-channel recordings from excised patches confirmed the prolonged duration of single-channel opening as the underlying mechanism. Our findings therefore identify functional consequences of the S1459 variant that lead to both gain and loss of NMDAR function, highlighting the complex nature of a GluN2A C-terminal variant in disease etiology. Indeed, rare variants that cause missense mutations in the M2 channel pore-forming loop of various NMDAR subunits have previously been reported to cause a gain of channel function accompanied by reduced surface expression of the receptors (Li et al., 2019). This highlights the need to study the complex effects of NMDAR rare variants on neuronal function throughout development, which could potentially be of clinical importance in the move toward personalized medicine.
In conclusion, our findings describe dual mechanisms by which CaMKIIα-mediated phosphorylation of GluN2A at Ser-1459 regulates the trafficking and gating of NMDARs during synaptic potentiation (Figure 7). We propose that, during LTP, NMDAR-dependent influx of Ca2+ into the postsynaptic compartment activates the protein kinase CaMKIIα. Phosphorylation of GluN2A at Ser-1459 by CaMKIIα in the endocytic vesicles and/or sorting endosomes promotes its interaction with SNX27. The assembly of the SNX27-retromer complex further enhances the binding affinity toward phosphorylated GluN2A, and subsequently increases the rate of GluN2A recycling back to the plasma membrane. However, in the present work, we cannot exclude a possible role of SNX27 in promoting the forward trafficking of GluN2A-containing NMDARs within the intracellular pool when they become phosphorylated during synaptic potentiation. CaMKIIα can also phosphorylate existing GluN2A-containing NMDARs on the surface and prolong the duration of single-channel opening, thereby augmenting the NMDAR current density and Ca2+ influx. On a broader scale, these mechanisms may be important in the expression and maintenance of bidirectional plasticity, dendritic integrity, learning, and memory.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to, and will be fulfilled by the Lead contact, Victor Anggono (v.anggono@uq.edu.au).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact.
Data and code availability
This study did not generate datasets/code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Rats
Adult female Sprague-Dawley rats and their embryos (males and females, embryonic day 18) were used for the preparation of primary hippocampal neurons. All research procedures involving the use of animals were conducted in accordance with the Australian code of practice for the care and use of animals for scientific purposes and were approved by the University of Queensland Animal Ethics Committee.
HEK293T cells
HEK293T cells were obtained from ATCC (CRL-3216). Cells were grown in DMEM with 4.5g/L glucose (GIBCO) supplemented with 10% FBS (Invitrogen) and 50 U/ml penicillin, 50 μg/ml streptomycin (GIBCO) in a humidified 5% CO2 incubator at 37°C.
METHOD DETAILS
DNA constructs
The plasmid encoding SEP-GluN2A was created by subcloning rat GluN2A cDNA into the pRK5 vector and subsequently inserting SEP cDNA after the signal peptide (residues 1-34). GST-tagged GluN2A C-terminal tails were generated by amplifying GluN2AcDNA encoding residues 1213-1464 and 1364-1464 with the following primers: residues 1213-1464 FP, 5′-ATCGTCGACCCATTGCA GAAGCTGCCTTTCGAAT-3′; residues 1364-1464 FP, 5′-ATCGTCGACCGATAATCCTTTCCTCCACACGTAT-3′; common RP, 5′-TCGAGCGGCCGCTCTTAAACATCAGATTC-3′. The PCR products were subsequently cloned into the unique SalI and NotI sites in the pCIS-GST vector. Full-length SNX27 cDNA was subcloned from pRK5-myc-SNX27 into the same pCIS-GST or FUW-myc lentiviral vector using the unique SalI and NotI sites. GluN2A (S1459A, S1459D, S1459E, S1459G and V1464E) and SNX27 (H112A and L65A) point mutants were generated with an overlapping PCR protocol. The shRNA targeting the rat SNX27 sequence (5′-AACCAGGTAATAGCGTTTGAA-3′) was cloned into the FG12 lentiviral vector, which has been validated previously (Hussain et al., 2014). The sgRNA targeting the rat CaMKIIα sequence (5′-CTCTTCCGTGAATCGGGTGC-3′) was cloned into the pLentiCRISPR-GFP vector (Incontro et al., 2018). Other plasmids encoding untagged-GluN1 (Chen et al., 2020), HA-neuroligin-1B (Chih et al., 2006), GFP-tCaMKIIα (Opazo et al., 2010), myc-SNX27 (Hussain et al., 2014) and GST-SNX27 PDZ domain (Gallon et al., 2014) have been reported previously. The pEGFP-C1 vector was obtained from Clontech.
Antibodies
The rabbit polyclonal antibody against GluN2A phospho-S1459 was custom-made and has been validated previously (Mota Vieira et al., 2020). The following antibodies were purchased from commercial sources: mouse anti-β-actin (Cat# sc-47778, Santa Cruz Biotechnology), rabbit anti-CaMKII (Cat# 4436, Cell Signaling Technology), chicken anti-GFP (Cat# GFP-1020, Aves Labs); rabbit anti-GFP (Cat# 504302-AP, Proteintech), rabbit anti-GluN2A (Cat# 04-901, Millipore), mouse anti-GFP (Cat# ab1218, Abcam), mouse anti-GST (Cat# 66001-2-Ig, Proteintech), mouse anti-myc (Cat# sc-40, Santa Cruz Biotechnology), rabbit anti-SNX27 (Cat# 16329-1-AP, Proteintech), rabbit anti-VPS26 (Cat# ab181352, Abcam), rabbit anti-VPS35 (Cat# 10236-1-AP, Proteintech) and chicken anti-MAP2 (cat# ab92434, Abcam). The polyclonal rabbit antibody against GFP (JH4030) was a gift from Dr. Richard Huganir (Anggono et al., 2011). Alexa-conjugated and HRP-conjugated secondary antibodies were purchased from Thermo Scientific and GE Healthcare, respectively.
Isothermal titration calorimetry
Recombinant rat SNX27 PDZ domain (residues 38-135) was expressed in E. coli BL21(DE3) cells and purified on glutathione-Sepharose columns, as described previously (Clairfeuille et al., 2016). ITC experiments were carried out on a Microcal iTC200 instrument in buffer consisting of 50 mM HEPES (pH 7.5), 2 mM DTT and 100 mM NaCl. Peptides (in the range of 2-50 mM) were titrated into 50-100 μM SNX27 PDZ domain at 25°C. Data were processed with ORIGIN (OriginLab) to extract the thermodynamic parameters ΔH, Kd (1/Ka) and the stoichiometry, n. ΔG and ΔS were derived from the relationship: ΔG = −RTlnKa and ΔG = ΔH - TΔS. The thermodynamic binding parameters are reported in Table S1.
HEK293T cells and GST pull-down assays
HEK293T cells were grown in DMEM with 4.5g/l glucose supplemented with 10% FBS, 50 U/ml penicillin and 50 μg/ml streptomycin in a humidified 5% CO2 incubator at 37°C. Cells expressing functional GluN1/2A NMDARs were maintained in medium supplemented with 40 mM MgCl2 to avoid excessive Ca2+ influx and cell toxicity. GST pulldown assays were carried out as previously described (Yong et al., 2020). Briefly, HEK293T cells were co-transfected with either pCIS empty (GST), pCIS-GluN2A-C-tails (GST-GluN2A C-tails) or pCIS-SNX27 (GST-SNX27) in combination with other plasmids by the calcium-phosphate precipitation method. To activate the PKA and PKC signaling pathways, cells were treated with forskolin (Sigma, 10 μM) or PMA (Sigma, 1 μM), respectively, for 10 min prior to cell lysis. Cells were lyzed 48 h post-transfection with ice-cold cell lysis buffer (1% Triton X-100, 1mM EDTA, 1mM EGTA, 50mM NaF, 5mM Na-pyrophosphate in PBS) supplemented with EDTA-free protease inhibitor cocktail. Lysates were centrifuged at 20,627g for 20 min at 4°C and incubated with glutathione agarose beads (Thermo Scientific) overnight at 4°C. Beads were washed three times with ice-cold cell lysis buffer and bound proteins were eluted with 2X SDS sample buffer at 50°C for 30 min and analyzed by western blotting. Blots were analyzed using the enhanced chemiluminescence method. Images were acquired on the Odyssey Fc imaging system (LI-COR) and band intensities were quantified using Image Studio Lite software (LI-COR).
Primary neuronal cultures and transfection
Sprague-Dawley rat embryos (males and females, embryonic day 18) were used for the preparation of primary hippocampal and cortical neurons (Widagdo et al., 2015). All research procedures involving the use of animals were conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes and were approved by the University of Queensland Animal Ethics Committee (QBI/047/18). Briefly, hippocampi and cortices were isolated and dissociated separately with 30U of papain suspension (Worthington) for 20 min in a 37°C water bath. A single-cell suspension was obtained by triturating tissues with fire-polished glass Pasteur pipettes and then plated at a density of 8 × 104 cells (for hippocampal neurons, percoverslip), 2 × 105 cells (for cortical neurons, per well) or 1 × 106 cells (for cortical neurons, per dish) on poly-L-lysine-coated 12-well plates or 6 cm dishes in Neurobasal growth medium supplemented with 2 mM Glutamax, 1% penicillin/streptomycin, and 2% B-27. Neurons were kept in a humidified 5% CO2 tissue culture incubator at 37°C. Hippocampal and cortical neurons were maintained in Neurobasal medium containing 0% or 1% FBS, respectively, and fed twice a week. For cortical neurons, uridine (Sigma, 5 μM) and 5′-fluoro-2′-deoxyuridine (Sigma, 5 μM) were added to the culture medium at days in vitro (DIV) 5 to stop glial proliferation. Transfection of hippocampal neurons was carried out at DIV 12-14 with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Cells were processed at DIV 15-17.
Lentivirus preparation and transduction
HEK293T cells were transfected by the calcium-phosphate co-precipitation method using 10 μg of the plasmid of interest and 5 μg each of pMD2.G envelope plasmid, pRSV-Rev encoding plasmid and pMDLg/pRRE packaging construct (Incontro et al., 2018; Tan et al., 2017). Forty-eight hours after transfection, culture supernatants were collected and passed through a 0.45 μm cellulose acetate low binding protein membrane filter. Lentiviral particles were harvested either by ultracentrifugation at 106,559 g for 2 h at 4°C using a Beckman SW 32 Ti rotor or using the PEG-it Virus Precipitation Solution (System Biosciences) according to the manufacturer’s instructions. Concentrated virus particles were resuspended in Neurobasal medium, flash frozen with liquid nitrogen and stored at −80°C. Neurons were transduced with lentiviral particles between DIV 6-9 for 6 h or overnight, after which they were further incubated for 6-9 days prior to harvesting.
Glycine-induced cLTP
cLTP was performed on primary rat hippocampal and cortical neurons at DIV 15-17 as previously described, with some modifications (Tan et al., 2017). Briefly, neurons were washed and incubated in pre-warmed low Mg2+ artificial cerebrospinal fluid (ACSF; 120 mM NaCl, 2 mM CaCl2, 5 mM KCl, 0.4 mM MgCl2, 30 mM glucose, 25mM HEPES, pH 7.4) containing 0.5 μM tetrodotoxin (Abcam), 20 mM bicuculline (Abcam) and 5 μM strychnine (Sigma) at 37°C for 45 min. cLTP was induced by incubating neurons with low Mg2+ ACSF supplemented with 200 μM glycine, 20 μM bicuculline and 5 μM strychnine at room temperature for 5 min. To inhibit the activity of CaMKIIα, neurons were treated with KN-93 (Abcam, 10 μM) for 15 min prior to and during cLTP induction.
Surface staining and antibody-feeding assays
To study the trafficking of NMDARs in primary hippocampal cultures, we used an antibody-feeding approach as described previously (Anggono et al., 2013; Chiu et al., 2019). Surface GluN2A was labeled by incubating live primary hippocampal neurons transfected with various SEP-GluN2A constructs with rabbit anti-GFP antibody (JH4030, 1:250) for 30 min at 4°C prior to 10 min fixation in ice-cold parafix solution (4% paraformaldehyde, 4% sucrose in PBS). Following cell permeabilization (0.25% Triton X-100 in PBS, 10 min) and blocking (10% normal goat serum, 1 h) at room temperature, total SEP-GluN2A was labeled with chicken anti-GFP antibody (Abcam, 1:5,000) at 4°C overnight. The surface and total SEP-GluN2A were subsequently visualized by Alexa-568-conjugated anti-rabbit and Alexa 488-conjugated anti-chicken secondary antibodies, respectively. To determine the amount of receptor internalization, surface SEP-GluN2A was first labeled with rabbit anti-GFP antibody in live neurons at room temperature for 10 min. The neurons were washed with warm ACSF (25 mM HEPES, 120 mM NaCl, 5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 30 mM D-glucose, pH 7.4) and incubated at 37°C for 30 min to allow receptor internalization to occur. Surface-bound and non-internalized antibodies were blocked with an excess amount of unconjugated donkey anti-rabbit Fab fragments (Jackson ImmunoResearch, 1:50) at room temperature for 20 min. After washing, the neurons were fixed, blocked, and incubated with Alexa-647-conjugated anti-rabbit secondary antibody at room temperature for 30 min. Neurons were subsequently washed, permeabilized, blocked, and incubated with chicken anti-GFP antibody at 4°C overnight. The total and internalized SEP-GluN2A were visualized by Alexa 488-conjugated anti-chicken and Alexa-568-conjugated anti-rabbit secondary antibodies, respectively. To measure the level of receptor recycling, neurons expressing SEP-GluN2A were fed with rabbit anti-GFP antibody and allowed to be endocytosed for 30 min. After the neurons were incubated with an excess amount of unconjugated donkey anti-rabbit Fab fragments, they were transferred back into a humidified 37°C tissue culture incubator for 30 min, which allowed internalized receptors to recycle back to the plasma membrane. The neurons were then fixed, blocked, and incubated with Alexa-647-conjugated anti-rabbit secondary antibody which labels recycled receptors on the plasma membrane. They were subsequently washed, permeabilized, blocked, and incubated with chicken anti-GFP antibody at 4°C overnight. The total and intracellular SEP-GluN2A were visualized by Alexa 488-conjugated anti-chicken and Alexa-568-conjugated anti-rabbit secondary antibodies, respectively. Images were collected with a 63X oil-immersion objective on a Zeiss LSM510 confocal microscope. Fluorescence intensities were quantified using ImageJ software (National Institutes of Health) for surface, internalized, and total GluN2A. Data were expressed as the surface/total GluN2A ratio, internalized/(surface+internalized) GluN2A (internalization index) or recycled/ (surface+internalized) GluN2A (recycling index).
Proximity ligation assay (PLA)
Following glycine-induced cLTP, the association between GFP-SNX27 and endogenous GluN2A was detected using the Duolink In Situ PLA kit (Sigma) according to the manufacturer’s recommendation. Neurons were fixed, permeabilized, blocked, and incubated with rabbit anti-GluN2A (1:200) and mouse anti-GFP (1:1000) antibodies overnight at room temperature. After washing with buffer A, primary antibodies were probed using Duolink PLA probes mouse PLUS and rabbit MINUS diluted in Duolink antibody diluent at 37°C for 1 h. The ligation reaction containing the oligonucleotide and ligase was incubated for 30 min at 37°C, followed by two washes. Rolling circle amplification and probe binding labeled with a fluorophore was carried out at 37°C for 100 min followed by washes with buffer B. Post-labeling with chicken anti-MAP2 was subsequently carried out to visualize neuronal structure. Samples were post-fixed briefly for 2 min, washed, and blocked prior to incubating with chicken anti-MAP2 (1:2000) for 1 h followed by five washes with 1X PBS and visualization by Alexa-647-conjugated anti-chicken secondary antibodies. The number of PLA puncta per 50 μm segment of a primary dendrite from a neuron were counted and normalized against non-glycine treated control cells. Data were expressed as the number of PLA puncta per 50 μm of primary dendritic segment.
Surface biotinylation assay
The surface biotinylation assay was carried out at DIV 15-17 to measure the amount of endogenous GluN2A protein on the plasma membrane (Tan et al., 2017). Briefly, live neurons were washed once in ice-cold ACSF (pH 8.2), followed by a 30 min incubation with 1 mg/ml of sulfo-NHS-SS-biotin (CovaChem) at 4°C. Free biotin was quenched by washing the cells three times with ice-cold Tris-buffered saline (TBS, 50 mM Tris-HCl, 150 mM NaCl, pH 7.4). Neurons were lyzed in RIPA buffer (1% Triton X-100, 0.5% Na-deoxycholate, 0.1% SDS, 2 mM EDTA, 2 mM EGTA, 50 mM NaF, 10 mM Na-pyrophosphate in TBS, pH 7.4) supplemented with EDTA-free protease inhibitor cocktail (Sigma) at 4°C. Lysates were centrifuged at 20,627g for 20 min at 4°C and incubated with Neutravidin agarose beads (Thermo Scientific) overnight at 4°C. Beads were washed three times with ice-cold RIPA buffer and bound proteins were eluted with 2X SDS sample buffer at 50°C for 30 min and analyzed by western blotting. The absence of β-actin on the surface fraction confirmed the specificity of the assay.
Immunoprecipitation assays
To determine the level of GluN2A-S1459 phosphorylation in primary cortical neurons, we performed immunoprecipitation assays using rabbit anti-GluN2A-pS1459 antibodies as previously described (Mota Vieira et al., 2020). Briefly, neurons were lyzed in ice-cold RIPA buffer containing protease and phosphatase inhibitor cocktails. Lysates were centrifuged at 14,000 rpm for 20 min at 4°C and cleared with protein A-Sepharose beads (Thermo Scientific). Pre-cleared lysates were then incubated with anti-GluN2A-pS1459 coupled to protein A-Sepharose overnight at 4°C. After four washes with ice-cold lysis buffer, bound proteins were eluted in 2X SDS sample buffer at 50°C for 30 min. Both total and eluted proteins were analyzed by western blotting with specific antibodies against GluN2A, CaMKII and β-actin.
Artificial synapse recordings
Methods for preparing neurons and HEK293 cells for artificial synapse recordings have previously been described (Chen et al., 2020). Briefly, HEK293 cells were transfected with cDNAs encoding human GluN1 and SEP-tagged rat GluN2A subunits, plus empty pEGFP and mouse neuroligin-1B (Addgene #15261, in the pCAGGS expression vector) in a ratio of 1:1:1:0.5:1, using a calcium-phosphate co-precipitation protocol. Cortical neurons from embryonic day 18 rat embryos (ethics approval number: QBI/142/16/NHMRC/ARC) were plated on poly-D-lysine-coated coverslips at a density of 1 × 105 cells per coverslip in DMEM supplemented with 10% FBS. The medium was replaced after 24 h with Neurobasal medium containing 2% B-27 and 1% Glutamax. After one week, half of this medium was replaced with fresh medium. Neurons were allowed to grow for 3-4 weeks before freshly transfected HEK293 cells were plated onto the neurons. Artificial synaptic connections typically formed spontaneously within 24 h and EPSCs in HEK293 cells were recorded 2-5 d later. Artificial synapse recordings were done in the whole-cell patch-clamp recording configuration at a holding potential of −70 mV unless otherwise stated, at 22 ± 2°C. Patch pipettes were fabricated from borosilicate hematocrit tubing (Harvard Apparatus) and had tip resistances of 2-5 MΩ when filled with the intracellular solution which contained (in mM): 145 CsCl, 2 CaCl2, 2 MgCl2, 10 HEPES, and 10 EGTA, adjusted to pH 7.4 with CsOH. Cells were perfused with extracellular solution, which contained (in mM): 140 NaCl, 5 KCl, 2 CaCl2, 10 HEPES and 10 D-glucose, adjusted to pH 7.4 with NaOH. EPSCs were filtered (−3dB, 4-pole Bessel) at 4 kHz, and then sampled at 10 kHz and recorded using a Multiclamp 700B amplifier and pClamp 10 software (Molecular Devices). Recordings with series resistances > 20 MΩ were discarded and series resistance compensation was not applied to the recorded cell. Solutions containing defined concentrations of glutamate, glycine and Mg2+ were applied to cells by gravityinduced perfusion microtubules.
Single-channel recordings
Single-receptor recordings were performed at room temperature (22 ± 1°C) in the outside-out patch configuration. The intracellular solution contained (in mM): 145 CsCl, 2 CaCl2, 2 MgCl2,10 HEPES, and 10 EGTA, adjusted to pH 7.4 with CsOH. The extracellular solution comprised (in mM): 140 NaCl, 5 KCl, 2 CaCl2, 0.020 Na2EDTA, 10 HEPES, and 10 D-glucose. NaOH was used to adjust both solutions to a pH of 7.4. The concentration of EDTA was chosen to chelate Mg2+ and other contaminant divalent ions without affecting Ca2+. The single-channel conductance was determined by constructing current-voltage (I-V) plots from data obtained at clamped voltages of (in mV) ± 70, ± 35, ± 15 and 0. The conductance was calculated using
| eqn.1 |
Where Vp is the holding potential (—70 mV), Vljp is the liquid junction potential and Vrev is the reversal potential. Vljp was calculated using JPCalWin and Vrev was estimated directly from the I-V plots. Single channel currents were recorded using an EPC 10 USB Heka Patch Clamp Amplifier, filtered (−3dB, 4-pole Bessel) at 2.9 kHz and sampled at 50 kHz. QuB software was used to determine the mean duration of single receptor activations and the intra-activation open probability (PO). Single channel currents were idealized using a resolution dead-time of 70 μs and the critical shut period that separated one discrete activation from the next was 200 ms. Activations with fewer than 20 events were excluded from analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
The sample size (n) reported in figure legends represents the number of individual neurons or wells generated from at least three independent experiments, unless otherwise stated. Statistical analysis was performed using Graph Pad Prism 9.0. One-way analysis of variance (ANOVA) with a Tukey’s, Sidak’s or Dunnet’s post hoc multiple comparisons test was used to compare the parameters for more than two groups. For the analyses of active duration and PO of single-channel recordings, a Kruskal-Wallis one-way ANOVA with the Dunn’s post hoc multiple comparisons test was used. For comparison between two groups, a two-tailed Mann-Whitney’s test was employed. All data are reported as mean ± standard error of the mean (SEM), or median with minimum and maximum values.
Supplementary Material
KEY RESOURCES TABLES
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa 488-conjugated goat anti-chicken IgY antibody | Thermo Scientific | Cat# A32931TRRRID: AB_2866499 |
| Alexa 568-conjugated goat anti-rabbit IgG antibody | Thermo Scientific | Cat# A-11036RRID: AB_10563566 |
| Alexa 647-conjugated goat anti-rabbit IgG antibody | Thermo Scientific | Cat# A-21245RRID: AB_2535813 |
| β-actin (mouse monoclonal, clone C4, used 1:5000 in WB) | Santa Cruz Biotechnology | Cat# sc-47778; RRID:AB_2714189 |
| CaMKII (pan) (rabbit monoclonal, clone D11A10, used 1:1000 in WB) | Cell Signaling Technology | Cat# 4436RRID:AB_10545451 |
| Fab fragment donkey anti-rabbit IgG | Jackson ImmunoResearch | Cat# 711-007-003RRID: AB_2340587 |
| GFP (rabbit polyclonal, used 1:5000 in WB) | Proteintech | Cat# 50430-2-APRRID:AB_11042881 |
| GFP (rabbit polyclonal, JH4030, used 1:250 in ICC) | A gift from R. Huganir (Johns Hopkins University) (Anggono et al., 2011) | N/A |
| GFP (chicken polyclonal, used 1:5000 in WB & ICC) | Aves labs | Cat# GFP-1020RRID:AB_10000240 |
| GFP (mouse monoclonal, used 1:1000 in ICC) | Abcam | Cat# ab1218RRID: AB_298911 |
| GluN2A (rabbit monoclonal, clone A12W, used 1:3000 in WB, 1:200 in ICC) | Millipore | Cat# 04-901RRID:AB_1163481 |
| GluN2A phospho-S1459 (1:1000 used in WB, 2ul used in co-IP) | Mota Vieira et al., 2020 | N/A |
| GST (mouse monoclonal, clone 3G12B10, used 1:5,000 in WB) | Proteintech | Cat# 66001-2-lg |
| MAP2 (chicken polyclonal, used 1:2000 in ICC) | Abcam | Cat# ab92434RRID:AB_2138147 |
| Myc (mouse monoclonal, clone 9E10, used 1:5,000 in WB, 1:100 in ICC) | Santa Cruz Biotechnology | Cat# sc-40; RRID:AB_627268 |
| Peroxidase-conjugated donkey anti-rabbit IgG antibody | GE Healthcare | Cat# NA934; RRID:AB_772206 |
| Peroxidase-conjugated sheep anti-mouse IgG antibody | GE Healthcare | Cat# NA931; RRID:AB_772210 |
| Peroxidase-conjugated goat anti-chicken IgY antibody | Santa Cruz Biotechnology | Cat# sc-2428; RRID:AB_650514 |
| SNX27 (rabbit polyclonal, used 1:1000 in WB) | Proteintech | Cat# 16329-1-APRRID:AB_10888628 |
| VPS26 (rabbit monoclonal, clone EPR13456, used 1:1000 in WB) | Abcam | Cat# ab181352; RRID:AB_2665924 |
| VPS35 (rabbit polyclonal, used 1:500 in WB) | Proteintech | Cat# 10236-1-AP; RRID:AB_2215216 |
| Chemicals, peptides, and recombinant proteins | ||
| Bicuculline methiodide | Abcam | Cat# ab120108 |
| Forskolin | Sigma | Cat# F3917 |
| Complete, EDTA-free protease inhibitor cocktail | Sigma-Aldrich | Cat# 11836170001 |
| Duolink in situ PLA probe anti-mouse PLUS | Sigma-Aldrich | Cat#DUO92001 |
| Duolink in situ detection reagents red | Sigma-Aldrich | Cat#DUO92008 |
| Glutathione agarose | Thermo Scientific | Cat# 16100 |
| KN-93 | Abcam | Cat# ab120980 |
| Lipofectamine 2000 | Thermo Scientific | Cat# 11668019 |
| Neutravidin agarose | Thermo Scientific | Cat# 29204 |
| Papain suspension | Worthington | Cat# LS003126 |
| Phorbol 12-myristate 13-acetate (PMA) | Sigma | Cat# P8139 |
| Phosphatase inhibitor cocktail | AG Scientific | Cat# P-1517 |
| Strychnine hydrochloride | Sigma | Cat# S8753 |
| Sulfo-NHS-SS-biotin | CovaChem | Cat# 14207-100 |
| Tetrodotoxin citrate | Abcam | Cat# ab120055 |
| Experimental models: Cell lines | ||
| Human: HEK293T | ATCC | Cat# CRL-3216; RRID:CVCL_0063 |
| Experimental models: Organisms/strains | ||
| Rat: Sprague-Dawley | University of Queensland Biological Resources |
RRID:RGD_70508 |
| Recombinant DNA | ||
| FG12 | Qin et al., 2003 | Addgene plasmid #14884 |
| FG12-SNX27-shRNA | This paper | N/A |
| FUW-SNX27 WT | This paper | N/A |
| pCAG-HA-neuroligin-1B | Chih et al., 2006 | Addgene plasmid #15261 |
| pCIS | A gift from R. Huganir (Johns Hopkins University) | N/A |
| pCIS-GluN2A-Ctail (1213-1464) WT | This paper | N/A |
| pCIS-GluN2A-Ctail (1213-1464) S1459A | This paper | N/A |
| pCIS-GluN2A-Ctail (1213-1464) S1459E | This paper | N/A |
| pCIS-GluN2A-Ctail (1213-1464) V1464E | This paper | N/A |
| pCIS-GluN2A-Ctail (1364-1464) WT | This paper | N/A |
| pCIS-GluN2A-Ctail (1364-1464) S1459A | This paper | N/A |
| pCIS-GluN2A-Ctail (1364-1464) S1459D | This paper | N/A |
| pCIS-GluN2A-Ctail (1364-1464) V1464E | This paper | N/A |
| pCIS-SNX27 WT | This paper | N/A |
| pEGFP-C1 | Clontech | N/A |
| pEGFP-SNX27 WT | Clairfeuille et al., 2016 | N/A |
| pEGFP-truncated CaMKIIα | A gift from P. Opazo (University of Queensland) (Opazo et al., 2010) | N/A |
| pGEX4T-2-SNX27 PDZ domain (38-135) | Clairfeuille et al., 2016 | N/A |
| pLentiCRISPR-GFP | A gift from S. Incontro (University of California, San Francisco) (Incontro et al., 2018) | N/A |
| pLentiCRISPR-CaMKIIα-sgRNA (rat specific) | This paper | N/A |
| pMD2.G | A gift from D. Trono (Swiss Federal Institute of Technology Lausanne) | Addgene plasmid #12259 |
| pMDLg/pRRE | A gift from D. Trono (Swiss Federal Institute of Technology Lausanne) | Addgene plasmid #12251 |
| pRK5-GluN1 | Chen et al., 2020 | N/A |
| pRK5-SEP-GluN2A WT | This paper | N/A |
| pRK5-SEP-GluN2A S1459A | This paper | N/A |
| pRK5-SEP-GluN2A S1459D | This paper | N/A |
| pRK5-SEP-GluN2A S1459G | This paper | N/A |
| pRK5-myc-SNX27 WT | Hussain et al., 2014 | N/A |
| pRK5-myc-SNX27 L65A | This paper | N/A |
| pRK5-myc-SNX27 H112A | This paper | N/A |
| pRSV-Rev | A gift from D. Trono (Swiss Federal Institute of Technology Lausanne) | Addgene plasmid #12253 |
| Software and algorithms | ||
| Fiji | National Institutes of Health | RRID:SCR_002285; https://fiji.sc |
| Image Studio Lite | LI-COR | RRID:SCR_015795; https://www.licor.com/bio/image-studio-lite/ |
| GraphPad Prism 9 | GraphPad | RRID:SCR_002798; https://www.graphpad.com/ |
| pClamp10 | Molecular Devices | RRID:SCR_011323; https://www.moleculardevices.com/products/software/pclamp.html |
| QuB software | Nicolai and Sachs, 2013 | RRID:SCR_018076; https://qub.mandelics.com/ |
Highlights.
CaMKIIα phosphorylates GluN2A at Ser-1459 in response to glycine stimulation
CaMKIIα and SNX27 are required for the glycine-induced increase in surface GluN2A
GluN2A Ser-1459 is a critical residue that controls the gating of NMDA receptors
The epilepsy-associated GluN2A S1459G variant prolongs open channel duration
ACKNOWLEDGMENTS
We would like to thank Richard Huganir, David Baltimore, Didier Trono, Peter Scheiffele, Salvatore Incontro, and Patricio Opazo for providing DNA plasmids and antibodies, as well as Rowan Tweedale for critical reading of this manuscript. Imaging was performed at the Queensland Brain Institute’s Advanced Microscopy Facility. This work was supported by grants from the Australian Research Council (ARC; DP190101390 to V.A., B.M.C., and A.K.), the National Health and Medical Research Council (NHMRC; GNT1099114 to B.M.C. and V.A. and GNT1156673 to A.K.), the National Institute of Neurological Disorders and Stroke Intramural Research Program (to K.W.R.), the Portuguese Foundation for Science and Technology (FCT [Fundação para a Ciência e a Tecnologia] grant SFRH/BI/106010/2015 to M.M.V.), and the Clem Jones Centre for Ageing Dementia Research to V.A. B.M.C. is supported by an NHMRC Senior Research Fellowship (APP1136021). J.W. is currently supported by a University of Queensland Amplify Fellowship and previously held an ARC Discovery Early Career Researcher Award (DE170100112). X.L.H.Y. is supported by a Research Training Program Scholarship from the Australian Government and the University of Queensland, as well as the Ian Lindenmayer PhD Topup Scholarship.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109338.
REFERENCES
- Anggono V, Clem RL, and Huganir RL (2011). PICK1 loss of function occludes homeostatic synaptic scaling. J. Neurosci 31, 2188–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anggono V, Koç-Schmitz Y, Widagdo J, Kormann J, Quan A, Chen CM, Robinson PJ, Choi SY, Linden DJ, Plomann M, and Huganir RL (2013). PICK1 interacts with PACSIN to regulate AMPA receptor internalization and cerebellar long-term depression. Proc. Natl. Acad. Sci. USA 110, 13976–13981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, and Traynelis SF (2000). Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J. Neurosci 20, 89–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A, and Malinow R (2002). Subunit-specific NMDA receptor trafficking to synapses. Neuron 35, 345–353. [DOI] [PubMed] [Google Scholar]
- Barria A, and Malinow R (2005). NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron 48, 289–301. [DOI] [PubMed] [Google Scholar]
- Bayer KU, and Schulman H (2019). CaM kinase: Still inspiring at 40. Neuron 103, 380–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone C, and Nicoll RA (2007). Rapid bidirectional switching of synaptic NMDA receptors. Neuron 55, 779–785. [DOI] [PubMed] [Google Scholar]
- Biederer T, and Scheiffele P (2007). Mixed-culture assays for analyzing neuronal synapse formation. Nat. Protoc 2, 670–676. [DOI] [PubMed] [Google Scholar]
- Bosch M, and Hayashi Y (2012). Structural plasticity of dendritic spines. Curr. Opin. Neurobiol 22, 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowling KM, Thompson ML, Amaral MD, Finnila CR, Hiatt SM, Engel KL, Cochran JN, Brothers KB, East KM, Gray DE, et al. (2017). Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med. 9, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Loo LS, Atlashkin V, Hanson BJ, and Hong W (2011). Deficiency of sorting nexin 27 (SNX27) leads to growth retardation and elevated levels of W-methyl-D-aspartate receptor 2C (NR2C). Mol. Cell. Biol 31, 1734–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Keramidas A, Harvey RJ, and Lynch JW (2020). Effects of GluN2A and GluN2B gain-of-function epilepsy mutations on synaptic currents mediated by diheteromeric and triheteromeric NMDA receptors. Neurobiol. Dis 140, 104850. [DOI] [PubMed] [Google Scholar]
- Chih B, Gollan L, and Scheiffele P (2006). Alternative splicing controls selective trans-synaptic interactions of the neuroligin-neurexin complex. Neuron 51, 171–178. [DOI] [PubMed] [Google Scholar]
- Chiu AM, Barse L, Hubalkova P, and Sanz-Clemente A (2019). An antibody feeding approach to study glutamate receptor trafficking in dissociated primary hippocampal cultures. J. Vis. Exp 150, e59982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clairfeuille T, Mas C, Chan AS, Yang Z, Tello-Lafoz M, Chandra M, Widagdo J, Kerr MC, Paul B, Mérida I, et al. (2016). A molecular code for endosomal recycling of phosphorylated cargos by the SNX27-retromer complex. Nat. Struct. Mol. Biol 23, 921–932. [DOI] [PubMed] [Google Scholar]
- Cullen PJ, and Korswagen HC (2011). Sorting nexins provide diversity for retromer-dependent trafficking events. Nat. Cell Biol 14, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkach V, Barria A, and Soderling TR (1999). Ca2+/calmodulin-kinase II enhances channel conductance of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc. Natl. Acad. Sci. USA 96, 3269–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon C, Sah P, Lynch JW, and Keramidas A (2014). GABAA receptor α and γ subunits shape synaptic currents via different mechanisms. J. Biol. Chem 289, 5399–5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallon M, Clairfeuille T, Steinberg F, Mas C, Ghai R, Sessions RB, Teasdale RD, Collins BM, and Cullen PJ (2014). A unique PDZ domain and arrestin-like fold interaction reveals mechanistic details of endocytic recycling by SNX27-retromer. Proc. Natl. Acad. Sci. USA 111, E3604–E3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardoni F, Schrama LH, van Dalen JJ, Gispen WH, Cattabeni F, and Di Luca M (1999). αCaMKII binding to the C-terminal tail of NMDA receptor subunit NR2A and its modulation by autophosphorylation. FEBS Lett. 456, 394–398. [DOI] [PubMed] [Google Scholar]
- Gardoni F, Bellone C, Cattabeni F, and Di Luca M (2001). Protein kinase C activation modulates α-calmodulin kinase II binding to NR2A subunit of N-methyl-d-aspartate receptor complex. J. Biol. Chem 276, 7609–7613. [DOI] [PubMed] [Google Scholar]
- Gray JA, Shi Y, Usui H, During MJ, Sakimura K, and Nicoll RA (2011). Distinct modes of AMPA receptor suppression at developing synapses by GluN2A and GluN2B: Single-cell NMDA receptor subunit deletion in vivo. Neuron 71, 1085–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosshans DR, Clayton DA, Coultrap SJ, and Browning MD (2002). LTP leads to rapid surface expression of NMDA but not AMPA receptors in adult rat CA1. Nat. Neurosci 5, 27–33. [DOI] [PubMed] [Google Scholar]
- Hussain NK, Diering GH, Sole J, Anggono V, and Huganir RL (2014). Sorting Nexin 27 regulates basal and activity-dependent trafficking of AMPARs. Proc. Natl.Acad. Sci. USA 111, 11840–11845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Incontro S, Díaz-Alonso J, Iafrati J, Vieira M, Asensio CS, Sohal VS, Roche KW, Bender KJ, and Nicoll RA (2018). The CaMKII/NMDA receptor complex controls hippocampal synaptic transmission by kinase-dependent and independent mechanisms. Nat. Commun 9, 2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood A, Rioult MC, and Bear MF (1996). Experience-dependent modification of synaptic plasticity in visual cortex. Nature 381, 526–528. [DOI] [PubMed] [Google Scholar]
- Kristensen AS, Jenkins MA, Banke TG, Schousboe A, Makino Y, Johnson RC, Huganir R, andTraynelis SF (2011). Mechanism of Ca2+/calmodulin-dependent kinase II regulation of AMPA receptor gating. Nat. Neurosci 14, 727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffer BE, Melero C, Temkin P, Lei C, Hong W, Kortemme T, and von Zastrow M(2010). SNX27 mediates PDZ-directed sorting from endosomes to the plasma membrane. J. Cell Biol 190, 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavezzari G, McCallum J, Dewey CM, and Roche KW (2004). Subunit-specific regulation of NMDA receptor endocytosis. J. Neurosci 24, 6383–6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhang J, Tang W, Mizu RK, Kusumoto H, XiangWei W, Xu Y, Chen W, Amin JB, Hu C, et al. (2019). De novo GRIN variants in NMDA receptor M2 channel pore-forming loop are associated with neurological diseases. Hum. Mutat 40, 2393–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo LS, Tang N, Al-Haddawi M, Dawe GS, and Hong W (2014). A role for sorting nexin 27 in AMPA receptor trafficking. Nat. Commun 5, 3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, and Wang YT (2001). Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 29, 243–254. [DOI] [PubMed] [Google Scholar]
- Lussier MP, Sanz-Clemente A, and Roche KW (2015). Dynamic regulation of N-methyl-d-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors by posttranslational modifications. J. Biol. Chem 290, 28596–28603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan KJ, Banks PJ, Hellel FLN, Carmichael RE, Clairfeuille T, Evans AJ, Heesom KJ, Lewis P, Collins BM, Bashir ZI, et al. (2020). Sorting nexin-27 regulates AMPA receptor trafficking through the synaptic adhesion protein LRFN2. bioRxiv. 10.1101/2020.04.27.063248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, and Seeburg PH (1994). Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12, 529–540. [DOI] [PubMed] [Google Scholar]
- Morris RG (2013). NMDA receptors and memory encoding. Neuropharmacology 74, 32–40. [DOI] [PubMed] [Google Scholar]
- Mota Vieira M, Nguyen TA, Wu K, Badger JD 2nd, Collins BM, Anggono V, Lu W, and Roche KW (2020). An epilepsy-associated GRIN2A rare variant disrupts CaMKIIa phosphorylation of GluN2A and NMDA receptor trafficking. Cell Rep. 32, 108104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers SJ, Yuan H, Kang JQ, Tan FCK, Traynelis SF, and Low CM (2019). Distinct roles of GRIN2A and GRIN2B variants in neurological conditions. F1000Res. 8, F1000 Faculty Rev–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolai C, and Sachs F (2013). Solving ion channel kinetics with the QuB software. Biophys. Rev. Lett 8, 191–211. [Google Scholar]
- Nicoll RA, and Roche KW (2013). Long-term potentiation: Peeling the onion. Neuropharmacology 74, 18–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opazo P, Labrecque S, Tigaret CM, Frouin A, Wiseman PW, De Koninck P, and Choquet D (2010). CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron 67, 239–252. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, and Zhou Q (2013). NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci 14, 383–400. [DOI] [PubMed] [Google Scholar]
- Qin XF, An DS, Chen IS, and Baltimore D (2003). Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc. Natl. Acad. Sci. USA 100, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan EM, Philpot BD, Huganir RL, and Bear MF (1999). Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat. Neurosci 2, 352–357. [DOI] [PubMed] [Google Scholar]
- Sanz-Clemente A, Nicoll RA, and Roche KW (2013). Diversity in NMDA receptor composition: many regulators, many consequences. Neuroscientist 19, 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott S, Lynch JW, and Keramidas A (2015). Correlating structural and energetic changes in glycine receptor activation. J. Biol. Chem 290, 5621–5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN, and Jan LY (1994). Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368, 144–147. [DOI] [PubMed] [Google Scholar]
- Shipton OA, and Paulsen O (2013). GluN2A and GluN2B subunit-containing NMDA receptors in hippocampal plasticity. Philos. Trans. R. Soc. Lond. B Biol. Sci 369, 20130163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Stevens CF, Tonegawa S, and Wang Y (1992). Deficient hippocampal long-term potentiation in α-calcium-calmodulin kinase II mutant mice. Science 257, 201–206. [DOI] [PubMed] [Google Scholar]
- Steinberg F, Gallon M, Winfield M, Thomas EC, Bell AJ, Heesom KJ, Tavaré JM, and Cullen PJ (2013). A global analysis of SNX27-retromer assembly and cargo specificity reveals a function in glucose and metal ion transport. Nat. Cell Biol 15, 461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroebel D, Casado M, and Paoletti P (2018). Triheteromeric NMDA receptors: From structure to synaptic physiology. Curr. Opin. Physiol 2, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanger SA, He YA, Richter JD, and Bassell GJ (2013). Dendritic GluN2A synthesis mediates activity-induced NMDA receptor insertion. J. Neurosci 33, 8898–8908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan MC, Widagdo J, Chau YQ, Zhu T, Wong JJ, Cheung A, and Anggono V (2017). The activity-induced long non-coding RNA Meg3 modulates AMPA receptor surface expression in primary cortical neurons. Front. Cell. Neurosci 11, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temkin P, Morishita W, Goswami D, Arendt K, Chen L, and Malenka R (2017). The retromer supports AMPA receptor trafficking during LTP. Neuron 94, 74–82.e5. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, and Dingledine R (2010). Glutamate receptorion channels: Structure, regulation, and function. Pharmacol. Rev 62, 405–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira M, Yong XLH, Roche KW, and Anggono V (2020). Regulation of NMDA glutamate receptor functions by the GluN2 subunits. J. Neurochem 154, 121–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao Y, Zhang X, Badie H, Zhou Y, Mu Y, Loo LS, Cai L, Thompson RC, Yang B, et al. (2013). Loss of sorting nexin 27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down’s syndrome. Nat. Med 19, 473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widagdo J, Chai YJ, Ridder MC, Chau YQ, Johnson RC, Sah P, Huganir RL, and Anggono V (2015). Activity-dependent ubiquitination of GluA1 and GluA2 regulates AMPA receptor intracellular sorting and degradation. Cell Rep. 70, 783–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie DJ, Béhé P, and Colquhoun D (1998). Single-channel activations and concentration jumps: Comparison of recombinant NR1a/NR2A and NR1a/NR2D NMDA receptors. J. Physiol 510, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie DJ, Livesey MR, and Hardingham GE (2013). Influence of GluN2 subunit identity on NMDA receptor function. Neuropharmacology 74, 4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XiangWei W, Jiang Y, and Yuan H (2018). De novo mutations and rare variants occurring in NMDA receptors. Curr. Opin. Physiol 2, 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro K, and Philpot BD (2008). Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology 55, 1081–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong XLH, Cousin MA, and Anggono V (2020). PICK1 controls activity-dependent synaptic vesicle cargo retrieval. Cell Rep. 33, 108312. [DOI] [PubMed] [Google Scholar]
- Zhang XM, Yan XY, Zhang B, Yang Q, Ye M, Cao W, Qiang WB, Zhu LJ, Du YL, Xu XX, et al. (2015). Activity-induced synaptic delivery of the GluN2A-containing NMDA receptor is dependent on endoplasmic reticulum chaperone Bip and involved in fear memory. Cell Res. 25, 818–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, and Sheng M (2013). NMDA receptors in nervous system diseases. Neuropharmacology 74, 69–75. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets/code.