

Abstract
S-Adenosyl-l-methionine (AdoMet) is synthesized by the MAT2A isozyme of methionine adenosyltransferase in most human tissues and in cancers. Its contribution to epigenetic control has made it a target for anticancer intervention. A recent kinetic isotope effect analysis of MAT2A demonstrated a loose nucleophilic transition state. Here we show that MAT2A has a sequential mechanism with a rate-limiting step of formation of AdoMet, followed by rapid hydrolysis of the β–γ bond of triphosphate, and rapid release of phosphate and pyrophosphate. MAT2A catalyzes the slow hydrolysis of both ATP and triphosphate in the absence of other reactants. Positional isotope exchange occurs with 18O as the 5′-oxygen of ATP. Loss of the triphosphate is sufficiently reversible to permit rotation and recombination of the α-phosphoryl group of ATP. Adenosine (α–β or β–γ)-imido triphosphates are slow substrates and the respective imido triphosphates are inhibitors. The hydrolytically stable (α–β, β–γ)-diimido triphosphate (PNPNP) is a nanomolar inhibitor. The MAT2A protein structure is highly stabilized against denaturation by binding of PNPNP. A crystal structure of MAT2A with 5′-methylthioadenosine and PNPNP shows the ligands arranged appropriately in the ATP binding site. Two magnesium ions chelate the α– and γ-phosphoryl groups of PNPNP. The β-phosphoryl oxygen is in contact with an essential potassium ion. Imidophosphate derivatives provide contact models for the design of catalytic site ligands for MAT2A.
Keywords: S-adenosylmethionine, SAM synthetase, enzyme mechanism, positional isotope exchange, iminotriphosphates, MAT2A
INTRODUCTION
Methionine adenosyltransferases (MATs) exist in all metabolically active organisms and function to form S-adenosyl-L-methionine (AdoMet). AdoMet is the universal methyl donor for primary metabolism and for regulatory macromolecule methylation, including gene expression via DNA methylation and regulation of expression by methylation of lysine and arginine groups in histones and other regulatory proteins. MAT enzymes synthesize AdoMet from ATP and methionine in a unique reaction where the sulfur of methionine attacks the 5′-carbon of ATP, resulting in formation of AdoMet and triphosphate.1, 2 Enzyme-bound triphosphate is hydrolyzed into pyrophosphate and orthophosphate in a second step prior to product release (Fig. 1).1-4
Figure 1.

Reaction catalyzed by methionine adenosyltransferase. MAT2A converts ATP and methionine to AdoMet, pyrophosphate and orthophosphate. Triphosphate hydrolysis occurs in a distinct step following formation of AdoMet.
The catalytic mechanism of Escherichia coli MAT is the best-established of the MAT enzymes and has a rate-limiting step of AdoMet formation. ATP and methionine bind in random order with obligatory release of pyrophosphate and orthophosphate before AdoMet.5-9 The reaction requires magnesium ions to orient the ATP triphosphate and a potassium cation to organize the catalytic site.6, 10 A gating loop modulates binding of substrates in the active site and the triphosphate intermediate is positioned by an arginine residue that assists in water activation for triphosphate hydrolysis between the β–γ phosphoryl groups (with respect to ATP).8, 9 Kinetic isotope effect studies on the E. coli MAT reaction revealed an SN2 transition state with approximately equal bond orders from the C5′ to the triphosphate leaving group and the attacking methionine sulfur anion.11
Three human isoforms of MAT include MAT1 and MAT3 expressed in liver tissue, while MAT2A is expressed ubiquitously in human cell types and is the dominant form in human cancers.12 Cancers of hepatocellular origin switch from MAT1 and MAT3 expression to also express MAT2A.13 Crystal structures and mechanistic studies have reported significant differences in the mechanisms of the human MATs, despite their 85% identity in amino acid sequence.5, 14, 15 MAT2A forms a functional homodimer in its purified active form and also associates with a regulatory protein, MAT2B. MAT2B regulates the activity of MAT2A by increasing susceptibility of MAT2A to product inhibition by AdoMet, but does not provide significant rate enhancement.16, 17 Crystallographic studies have compared MAT2A to the MAT2A/MAT2B complex bound to AdoMet and PPNP and show no significant variation in the structure of the catalytic site due to MAT2B binding.3, 4 Kinetic isotope effects with the MAT2A dimer revealed a later, more product-like, SN2 transition-state than found for the E. coli enzyme.18 The transition-state structure for the MAT2A/MAT2B complex was unchanged from MAT2A alone, suggesting no change in the chemical mechanism, despite altered steady-state kinetic properties for the complex.18 MAT2A has been proposed to interact with MafK to form a nuclear complex or to form interactions with Swi/Snf, NuRD, BAF53a, CHD4, PARP1 and histone methyltransferases.19,20 Cellular location studies indicate a presence of MAT2A both in the cytosol and the nucleus.19 Nuclear enrichment of MAT2A has been reported to occur in replication and in the subsequent G2-phase, meeting the high methylation requirement inside the nucleus during S-phase for both DNA and histone methylation processes.21 AdoMet activity in transmethylation reactions has been identified as a rate-limiting factor in the development of lung cancer stem cells, making MAT2A and associated enzymes of the methionine cycle targets for anticancer agents.22
The methionine for AdoMet production by MAT2A comes from dietary sources or from the polyamine cycle, where the 5-methylthio-α-d-ribose 1-phosphate, produced by S-methyl-5'-thioadenosine phosphorylase (MTAP), is recycled to methionine. Enzymes of polyamine synthesis and methionine salvage pathways have also been targets for cancer therapy due to correlation between their upregulation and increased cell division.23-25 Increased MAT2A protein expression is reported in cancers, including colon, liver, gastric, blood, and liver.26-30 Approximately 15% of human cancers exhibit deletion of the MTAP gene and are therefore deficient in the methionine salvage pathway from polyamine synthesis. Deletion of MTAP from chr9p21 usually includes deletion of the CDKN2A tumor suppressor locus.31 Synthetic genetic lethality studies of MTAP−/− cancer lines indicate an increased susceptibility of these cancer cells to inhibition of MAT2A, together with PRMT5 and PRMT1.31-33 This finding has led to studies to identify effective agents to target these enzymes, and several agents have reached clinical trials.34-36
Here we examine the mechanism of MAT2A and provide characterization of its substrate, product and inhibitor binding. The formation of AdoMet in the first chemical step is rate-limiting for the reaction. The MAT2A catalytic mechanism is similar to that for E. coli MAT. For human MAT2A we characterize a nanomolar catalytic site inhibitor by kinetic and crystallographic analysis. Kinetics of internal steps of the reaction mechanism are characterized by positional isotope exchange and by pre-steady state kinetic analysis for MAT2A.
RESULTS AND DISCUSSION
Steady-state kinetics.
Steady-state kinetics of MAT2A with ATP and methionine as substrates monitored orthophosphate produced in the final step of the MAT2A reaction (see SI). Double-reciprocal plots show common x-intercepts of the lines to the left of the y-axis, suggesting sequential binding mechanism, excluding equilibrium ordered, for MAT2A (Figure 2). Maximum reaction rates (kcat values) of 0.27 s−1 were obtained (Table 1). Substrate kinetic constants (Ka values), 23 μM for methionine and 98 μM for ATP, were not strongly dependent on the binding of the second substrate, as the corresponding Kia dissociation constants were 30 and 129 μM, respectively (Table 1). A random equilibrium mechanism is supported with KiATPKMet ≈ KiMetKATP. The slow kcat for purified MAT2A may not reflect its in vivo capacity, due to its presence in the nucleus as a multi-protein complex interacting with nucleosomes, DNA, and multiple regulatory proteins with the potential to activate its function in vivo.
Figure 2.

Kinetic patterns are consistent with rapid equilibrium binding of ATP and methionine. The Km values are insensitive to the concentration of the second substrate.
Table 1.
Kinetic constants for MAT2A from fits to the data of Fig. 2. Kia and Ka are from abscissa intercepts at zero and saturating concentrations of the second substrate, respectively.
| Varied Substrate | kcat(s−1) | Kia (μM) | Ka (μM) |
|---|---|---|---|
| ATP | 0.27 ± 0.01 | 129 ± 22 | 98 ± 9 |
| Methionine | 0.27 ± 0.01 | 30 ± 4 | 23 ± 3 |
Energy of activation.
We measured the activation energy of the reaction by varying the temperature under near-saturating substrate concentrations. The experimentally determined activation energy of 17.3 kcal/mol is lower than found for most enzymes, thus the relatively inefficient catalytic rate does not arise from an unusually large energy of activation (Figure S1).
Product inhibition by AdoMet.
Product inhibition has been proposed as a regulatory mechanism for MAT2A, as AdoMet affinity is increased in the MAT2A-MAT2B regulatory complex.15,16 Product inhibition studies indicated AdoMet is a classic non-competitive inhibitor against both ATP and methionine (Figure 3). Thus, AdoMet binds to distinct forms of the enzyme than either ATP or methionine and implicates phosphate or pyrophosphate as the last product to leave the catalytic site. The Ki value for AdoMet with respect to ATP was 136 ± 7 μM, and 81 ± 10 μM with respect to methionine. The Km values for ATP and methionine are independent of AdoMet as a product inhibitor.
Figure 3.

Product inhibition by AdoMet with respect to ATP and methionine. Classic noncompetitive inhibition occurs with both substrates.
Pre-steady-state kinetics of MAT2A.
Pre-steady-state, multiple-turnover experiments were used to determine if a burst phase in AdoMet formation was present prior to steady-state turnovers (Figure 4). A chemical quench-flow experiment was designed to capture product profiles at early reaction times. For example, if triphosphate cleavage or product release of AdoMet, pyrophosphate or phosphate is rate limiting, a burst of AdoMet would be expected in pre-steady-state conditions prior to steady-state formation of products. Pre-steady-state multiple-turnover reactions used 1 mM ATP (with [2,8-3H]-ATP) and 1 mM methionine as substrates with 50 μM MAT2A to monitor the formation of [2,8-3H]-AdoMet. [2,8-3H]-AdoMet was produced at 0.19 ± 0.01 s−1, consistent with the steady-state rate. A single turn-over burst of 50 μM was anticipated if a subsequent step was rate-limiting, but no burst was observed (Figure 4). The absence of an AdoMet burst, supports the chemical step of AdoMet formation as the rate-limiting step in the MAT2A reaction.
Figure 4.
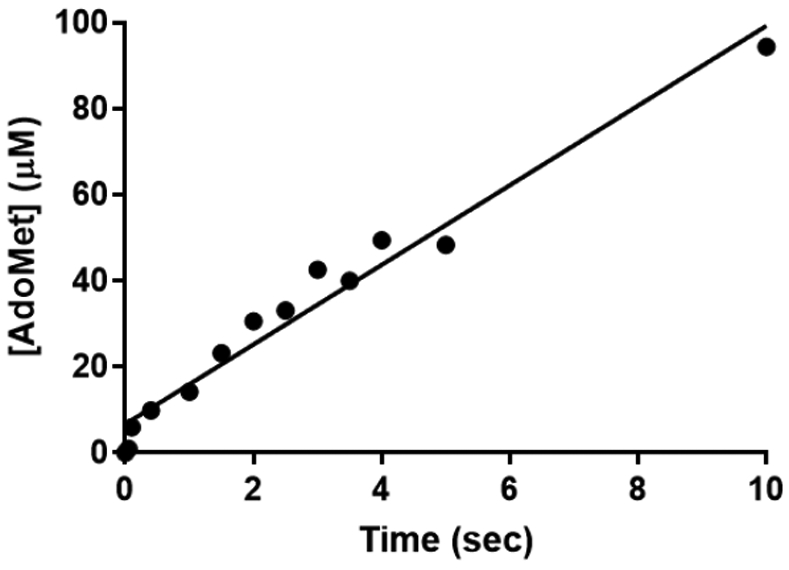
Pre-steady state kinetics of AdoMet formation from MAT2A. AdoMet was formed at kcat rates without a burst from 50 μM MAT2A and excess substrates. AdoMet formation from [2,8-3H]-ATP was used in a chemical quench-flow instrument. Reactions were quenched in 0.03 M sulfuric acid and AdoMet isolated by passage through a DOWEX-1 column and counted in a scintillation counter.
The lack of a burst indicates that the rate-limiting step of the MAT2A reaction lies before product-release and that triphosphate hydrolysis is fast relative to AdoMet formation. Thus, the rate of AdoMet formation in these experiments is similar to the kcat determined by steady-state kinetics. The 32P isotopic label from [γ-32P]-ATP appeared exclusively in inorganic phosphate (Figure S2). Triphosphate hydrolysis occurs exclusively between β-γ phosphates. The result also indicates no triphosphate release, rebinding and hydrolysis occurring even under multiple turnover conditions, as this would create 32P isotopic labels in both phosphate and pyrophosphate.
Triphosphate hydrolysis by MAT2A.
The rate of triphosphate hydrolysis to pyrophosphate and orthophosphate products and the effect of AdoMet on this rate can inform the order of product release when considered with the pre-steady-state multiple-turnover reactions described above. MAT2A hydrolyzes triphosphate without AdoMet present at a rate of 0.020 ± 0.003 s−1 with a Km of 4 ± 2 μM. AdoMet at 150 μM activates hydrolysis 3.5-fold to a rate of 0.07 ± 0.01 s−1 without changing the triphosphate Km (6 ± 2 μM). AdoMet added at a higher concentration (1 mM) decreased the kcat to 0.033 ± 0.003 s−1 without changing the Km, suggesting inhibition of Pi or PPi product release (Figure S3). Notably, MAT2A hydrolyzes triphosphate slowly and added AdoMet enhances the rate but not to the full rate consistent with steady-state and pre-steady state kinetic results. Forming the MAT2A-AdoMet-PPPi complex from the reverse direction creates a complex distinct from that formed in the forward direction, but one that remains catalytically competent, albeit with reduced catalytic potential. The result is consistent with, but does not prove, PPPi hydrolysis, AdoMet release, followed by Pi and PPi release.
Reactivity of imido-ATP analogues in the MAT2A reaction.
Imido-ATP analogues were used to explore AdoMet and phosphate formation when adenosine 5′-(α,β-imido)triphosphate (AMPNPP), adenosine 5′-(β,γ-imido)triphosphate (AMPPNP) and methionine were used as substrates (Figure 5). The reaction of AMPNPP and methionine showed the steady-state production of AdoMet with a rate of 1.9± 0.4 x 10−3 s−1, approximately 100-fold slower than with ATP as substrate (Figure 5A). With AMPPNP as substrate, AdoMet formation occurred in a pre-steady-state burst equal to MAT2A concentration at 0.021 ± 0.007 s−1, ten-fold slower than the maximal rate with ATP (Figure 5B). This phase is followed by slower steady-state rate of AdoMet formation of 2.2 ± 0.3x10−4 s−1, 1,000-fold slower than kcat with ATP. The β,γ-imido blocks the hydrolysis of the β,γ-imido-triphosphate, and the slow rate reflects a slow rate of β,γ-imido-triphosphate release with this hydrolytically stable analogue.
Figure 5.
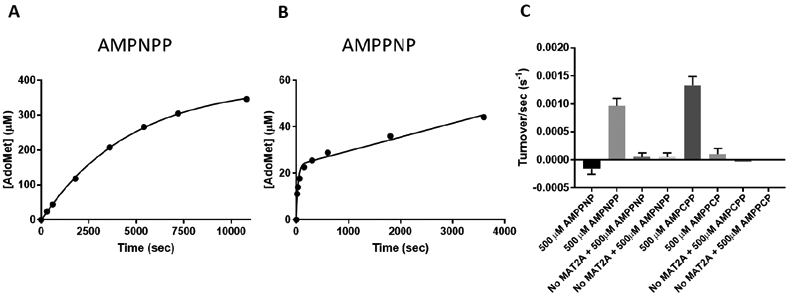
ATP analogues as alternative substrates of MAT2A. Adenylyl-imidodiphosphate analogue reactivity with methionine and 25 μM MAT2A to form AdoMet and phosphate. (A) AdoMet formation from AMPNPP and methionine produces multiple catalytic turnovers with no pre-steady state burst and an initial rate of 1.9x10−3 ± 3.8x10−4 s−1. (B) AMPPNP produces a pre-steady state burst of AdoMet formation followed by an even slower steady state rate. The AdoMet burst equals the MAT2A concentration (24 ± 3 μM). The burst occurs at 0.021 ± 0.007 s−1 and the steady-state rate at 2.2x10−4 ± 2.5x10−5 s−1. (C) The production of orthophosphate by MAT2A from methionine and ATP analogues was monitored and showed phosphate formation only with AMPNPP and AMPCPP.
With AMPNPP and AMPPNP as substrates, phosphate formation occurred with AMPNPP but not with AMPPNP (Figure 5C). Hydrolysis of imido-triphosphate occurs when the imido group is α,β-imido triphosphate with respect to ATP but not when it is in the β,γ-imido, the normal site of hydrolysis. This specificity also establishes that the imido-triphosphates cannot release and rebind to reposition the phosphoryl group for hydrolysis. The pre-steady-state burst with AMPPNP demonstrates facile displacement of the β,γ-imido-triphosphate by the attacking methionine sulfur. The subsequent step of β,γ-imido-triphosphate hydrolysis does not occur (no phosphate formed); therefore, the very slow subsequent rate of AdoMet formation provides a rate of imido-triphosphate release from the MAT2A-AdoMet-PPNP complex.
A parallel specificity occurs with α,β-CH2-triphosphate and β,γ-CH2-triphosphate when phosphate product formation is measured (Figure 5C). Phosphate is formed from α,β-CH2-triphosphate at a rate equivalent to that when α,β-imido-triphosphate is the substrate. Substitution of the β,γ- bridge with either imino or −CH2- groups prevents triphosphate hydrolysis. Hydrolytic stability of the positionally substituted triphosphate establishes that these analogues cannot dissociate and rebind to permit positional swapping of the α,β and β,γ-positions to permit hydrolysis of the triphosphate analogues. Triphosphate derived from the normal reaction acts in the same manner, evidenced by the isotope label from γ-32P-ATP appearing only in the monophosphate product (see above).
ATPase of MAT2A.
Triphosphate hydrolysis in the natural reaction sequence raises the possibility of hydrolysis of the β,γ-triphosphate bond of ATP in the absence of methionine. MAT2A catalyzes a slow ATPase activity of (1.8 ± 0.09) x 10−3 s−1, to form ADP and phosphate, approximately 100-fold slower than kcat for the native reaction (Figure S4). The reaction was inhibited by AdoMet but not by vanadate, a non-specific phosphatase inhibitor, supporting ATP hydrolysis as an intrinsic property of MAT2A.
Positional isotope exchange.
Positional isotope exchange experiments detect reversible enzyme reaction steps to help define mechanism37, 38. The slow kcat of MAT2A raised the possibility that the formation of AdoMet and triphosphate is reversible before product release. Alternatively, if triphosphate as a leaving group has freedom to rotate the α-phosphoryl group, then reform ATP and methionine, in catalytic site microscopic reversibility, positional isotope exchange would be expected for the 5′-oxygen of ATP. Positional isotope exchange experiments used 5′-18O-ATP, where the bridging oxygen to the triphosphate is isotopically labelled. The presence of the 18O label at the bridging oxygen causes an NMR shift in the 5′-carbon signal from 67.96 to 67.93 ppm (Figure 6, S5). If formation of AdoMet and triphosphate is reversible, or if the triphosphate group is dissociated sufficiently to permit rotation of the α-phosphoryl group, the 18O-label will scramble from the bridging to a non-bridging position, with 16O replacing the bridging position 18O-label.
Figure 6.

Positional isotope exchange experiments for the MAT2A reaction. (A) 2D 1H-13C HSQC NMR experiments overlayed showing the upfield doublet of the 5′-carbon of ATP. The ATP used in PIX reactions was 0.25 in 5′-16O-ATP / 5′-18O-ATP isotopic label. HSQC spectra for the reaction are 0 (blue), 10 (red), 30 min (green). (B) The overlay next to the HSQC shows the slice through the 13C dimension of the HSQC spectra at the 5′-C of ATP along with a 5′-16O-ATP standard (pink). An upfield shift in the spectra is observed in the presence of the 18O label. (C) This ratio changed as the reaction proceeded. (D) PIX exchange rates are calculated from the approximately linear rate of exchange during the first 18% of the reaction.
With excess 5′-18O-ATP and methionine, reactions were sampled at intervals to 18% conversion of ATP to AdoMet. Early sampling was selected to capture PIX in the event that PIX > catalysis. The 13C peaks quantitating the 5′-16O-ATP / 5′-18O-ATP ratio changed steadily throughout the reaction for a ratio change from 0.25 to 0.31, a change of 0.06 (Figure 6). With the original 5′-16O-ATP / 5′-18O-ATP ratio of 0.25, the initial fraction containing 5′-16O-ATP is silent. The fraction exchanged is therefore 0.06 x 1.25 = 0.075 during the 0.18 fractional formation of AdoMet. This represents a PIX rate / catalytic rate of 0.42. In this assay, the reactive ternary complex forms for 0.18 + 0.075 = 0.255 catalytic events. Of the 0.255 reactive complexes formed, 0.075 experience PIX and 0.18 form product. Each ATP that reaches a chemically competent ternary complex (E-ATP-Met ↔ E-AdoMet-PPP) has a 0.075/0.255 = 0.29 probability of the 5′-bridging oxygen of ATP being replaced by the neighboring non-bridging oxygen. Oxygen exchange at the 5′-18O-ATP position can occur by reversible formation of enzyme-bound AdoMet and triphosphate A less likely possibility is an expanded transition state-like intermediate where triphosphate has lost its covalent bond prior to full bond formation from the attacking methionine sulfur. Rotational reorientation of the α-phosphoryl group of the triphosphate and reformation of ATP could also accomplish the positional isotope exchange.
Thermal stabilization of MAT2A by reactants and inhibitors.
Thermal melting of MAT2A was conducted in the presence of Sypro Orange and potential catalytic site ligands. A slow temperature increase, observing the fluorescence change that accompanies protein denaturation, gives a thermal melt profile for the protein. The ATP and methionine substrates shifted the thermal melt temperature (Tm) by 1-3 °C compared to MAT2A alone (Figure 7). Likewise, AdoMet and phosphate did little to increase MAT2A stability. However, large shifts in the Tm were seen with pyrophosphate and triphosphate, indicating stabilization upon binding of these products. The triphosphate shift was larger than that for ATP. The adenosine moiety of ATP therefore decreases the ability of the triphosphate to stabilize MAT2A. The triphosphate analogue inhibitor PNPNP, discussed below, stabilized the protein to the 95 °C limit of the analysis.
Figure 7.

Thermal melt curves showing stabilization of MAT2A by substrates, products and analogues of the reaction. A quantitative PCR instrument and Sypro Orange protein dye were used to follow the denaturation of MAT2A alone and with the indicated ligands. Protein denaturation exposes hydrophopic regions and the dye binds with increased fluorescence. Dashed lines are experimental data and solid lines are fits to the data. All assays contained buffered 10 mM MgCl2.
Kinetic summary of MAT2A.
The slow catalytic step, significant positional isotope exchange and kinetic patterns for MAT2A can be summarized (Scheme 1). Steady-state kinetics with methionine and ATP reveal Ka and Kia values for ATP and methionine that are largely independent of the second substrate, suggesting independent, random binding. MAT2A complexes with ATP or methionine alone do not provide thermal stabilization, indicating structurally loose binary complexes. Slow catalysis suggests that the MAT2A-ATP-Met complex equilibrates prior to the chemical reaction (rapid-equilibrium random). Steady-state and stopped flow rates of AdoMet formation are equivalent, supporting AdoMet production as the rate limiting step. Product release (k3) is > 0.27 s−1 accounting for the absence of a burst in AdoMet formation. The reverse reaction from triphosphate and AdoMet and/or Pi, PPi and AdoMet was not observed under multiple conditions (not shown), indicating that reversal (k4) does not occur, or is experimentally far smaller than 0.27 s−1. The observation of positional isotope exchange (k2 and/or k5) at PIX / k3 of 0.42 requires that k1 must occur at a rate that also accounts for the internal return of k2 (or k5), and is therefore approximately 0.33 s−1. Essential steps following formation of the MAT2A-AdoMet-PPP complex include PPP hydrolysis, release of Pi and of PPi. As there is no burst of AdoMet or Pi in stopped flow studies, PPP hydrolysis and release of PPi and Pi are all faster than 0.27 s−1.
Scheme 1.
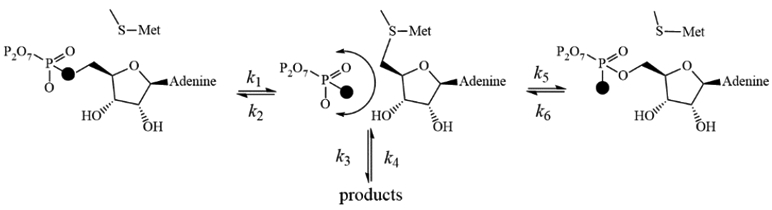
Catalytic mechanism of MAT2A. The filled oxygen represents 18O. Note that k2 and k5 are the same mechanistic step with distinct stereochemical outcomes.
Pyrophosphate analogue inhibitors of MAT2A.
Pyrophosphate and triphosphate stabilized MAT2A from thermal melt assays, establishing their ability to form binary complexes and suggesting their analogues as inhibitors. Likewise, the slow reaction rates with non-hydrolyzable ATP analogues, permitted their analysis as inhibitors. Imidodiphosphate was a classic non-competitive inhibitor of both ATP and methionine with Ki values of 14 ± 1 μM toward ATP and 17 ± 2 μM toward methionine (Figure 8A-B). Imidotriphosphate, PPNP, was also a classical non-competitive inhibitor towards both ATP, binding more tightly to give Ki values of 0.86 ± 0.06 μM μM toward ATP and 0.76 ± 0.07 μM toward methionine (Figure 8C-D).
Figure 8.

Non-hydrolyzable triphosphate analogues as inhibitors of MAT2A. Anion exchange chromatography was used to monitor AdoMet formation from MAT2A in the presence of PNP or PPNP. The data were fit to the equation for non-competitive inhibition.
Kinetic patterns.
Product (AdoMet) and dead-end imidophosphate inhibitors all generate non-competitive patterns. Inhibitors give competitive patterns when substrate and inhibitor compete for the same enzyme form. In a fully rapid-equilibrium random mechanism all products and substrates are competitive unless dead-end complexes form. Uncompetitive patterns occur when inhibitor binds to an enzyme form distinct from substrate. Non-competitive patterns are from inhibitor binding to two enzyme forms separated by irreversible steps. Scheme 2 indicates potential non-competitive binding combinations for the MAT2A reaction. The two enzyme forms to which inhibitor (AdoMet, PNP and PPNP) bind must not be in rapid equilibrium, or a competitive pattern would result. In the MAT2A mechanism, irreversible steps are imposed by the hydrolysis of triphosphate, indicated by single arrows (Scheme 2). Dead-end complex candidates to explain the non-competitive inhibition patterns for AdoMet are therefore E-AdoMet and E-AdoMet-P or E-AdoMet-PP. For PNP, the complexes are E-PNP and E-PNP-Met, and for PPNP similar complexes of E-PPNP and E-PPNP-Met can give rise to competitive inhibition (Scheme 2).
Scheme 2.
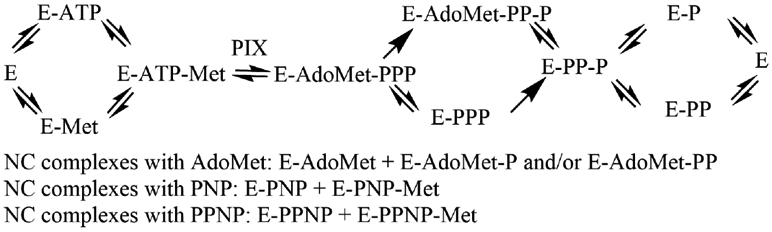
Kinetic mechanism giving rise to kinetic patterns of AdoMet and imidophosphates inhibition. All steps are reversible except the hydrolysis of triphosphate (PPP). AdoMet is assumed to release prior to phosphate (P) or pyrophosphate (PP) as it is weakly stabilizing in thermal protection and its release in unhindered based on the crustal structure. There is no direct proof for this assumption.
ATP analogue inhibitors of MAT2A.
ATP-imidotriphosphates were micromolar inhibitors of MAT2A, while their ATP-methylenetriphosphate counterparts did not inhibit significantly (Table 2). Triphosphate was a 3.45 μM inhibitor and its di-imidotriphosphate variant, PNPNP, was a 115 nM inhibitor. The namomolar affinity of PNPNP is consistent with its remarkable thermal stabilization of >50 °C for MAT2A, as shown by the Tm of over 95 °C for the complex (Figure 7). The nearly 10-fold increased affinity of PNPNP compared to PPNP suggests a substantial difference based on the geometry or electrostatics of binding. We sought to explore these properties in crystal structures of MAT2A in complexes with PNPNP.
Table 2.
Inhibition of MAT2A by ATP and triphosphate analogues. Inhibition assays contained 1 mM methionine and 200 μM ATP. NO = no significant inhibition observed.
| Inhibitor | Ki (μM) |
|---|---|
| AMPNPP | 0.9 ± 0.1 |
| AMPPNP | 8.6 ± 1.9 |
| AMPCPP | High mM |
| AMPPCP | NO |
| Triphosphate | 3.4 ± 0.6 |
| PNPNP | 0.12 ± 0.03 |
Structure of MAT2A bound to AdoMet and PNPNP.
MAT2A was crystallized in the presence of AdoMet, PNPNP, potassium and magnesium ions. A structure was refined at 1.25 Å resolution (Table S1) with full occupancy of the MAT2A active site (Figures 9,10). Consistent with previous reports,4 the structure reveals that the active site is composed of the residues from two monomers related by 2-fold crystallographic symmetry axis (Figure 10). During crystallization, the AdoMet substrate is chemically degraded to 5′-methylthioadenosine (MTA) and the methionine site is occupied by a group structurally resolved as alanine, (possibly a homoserine, disordered in the sidechain). The overall structural fold of MAT2A is similar to that previously published for MAT2A co-crystallized with AdoMet, adenosine, methionine, and PPNP; superimposable with an R.M.S.D. of 0.1 Å over 381 aligned Cα atoms.4
Figure 9.

Omit map of ligands at the MAT2A active site. The MAT2A active site consists of contacts from two monomers (stick representation; green (A) and grey (B)). PNPNP (red, yellow, blue) is coordinated (potential contacts, black dashes) to protein residues, magnesium ions (MG, blue spheres), a potassium ion (K, purple sphere), and water molecules (red spheres). The AdoMet site is occupied by 5′-methylthioadenosine (MTA, purple) and a ligand that can be modeled as alanine (ALA, pink). A potential nucleophilic water molecule for the hydrolysis step is indicated as Wat*.
Figure 10.

MAT2A dimer structure and catalytic site contacts for PNPNP. A) The dimer (ribbon representation; arrows for β-strands and ribbons for α-helices) formed by the two MAT2A subunits A (green) and B (grey) and constitutes two equivalent catalytic sites (green and grey), shown with the MTA, PNPNP, Mg2+, K+ and alanine ligands at the sites. B) A two-dimensional ligand distance map reveals multiple contacts from both subunits to the triphosphate analogue (color coded as panel A). Magnesium ions, potassium ion and water oxygens are in blue, purple and red, respectively. Putative nucleophilic water molecule for triphosphate hydrolysis is indicates by Wat*. Potential atomic contacts are indicated by dashed lines.
The adenosyl moiety is held in the active site by π-π stacking interactions between Phe250A (of monomer A) and Ile117B (of monomer B) and by hydrogen bonds with Arg249A, Ser247A and a structural water molecule (Figure 9). The PNPNP is bound in the active site by two magnesium ions, a potassium ion, and hydrogen bonds to protein residues, and to structural waters. Both magnesium ions in the active site are hexacoordinated with each binding an end phosphoryl group of PNPNP, creating a symmetric bent geometry for the bound PNPNP. The potassium ion contacts the central phosphate of PNPNP, bridging to protein residues Asp258A and Ala259A. Direct interactions of catalytic site residues with PNPNP include residues His29A, Asp134B, Lys181A, Lys265A, Ala281B, and Asp291B. The bridged-ligand nature of the PNPNP interaction explains its thermal stabilizing properties for the MAT2A protein and its tight binding. A stereo view of the catalytic site of the complex is provided in the supporting information (Figure S6). The high affinity of PNPNP compared to triphosphate and other mono-iminotriphosphate analogues might be explained by catalytic site contacts to phosphoryl groups and to both bridging imido groups of PNPNP. Thus, Asp291B contacts the nitrogen bridging β and γ phosphates and Asp134B contacts the nitrogen bridging α and β phosphates, contributing to the binding energy. The catalytic mechanism for triphosphate hydrolysis is also suggested by this structure. The proposed nucleophilic water molecule is positioned between Arg264A and the γ-phosphoryl group, in a position suitable to attack the terminal phosphate of the triphosphate, 2.9 Å from Arg264 and 2.7 Å from the γ-phosphoryl (Figures 9 and 10B).
CONCLUSIONS
Human MAT2A binds ATP and methionine followed by a rate-limiting catalytic step of AdoMet formation. As each molecule of AdoMet and triphosphate are formed at the catalytic site, they have a high probability of going forward to triphosphate hydrolysis and release but also a significant possibility to be reformed to ATP and methionine for re-equilibration with unreacted substrates. With AMPPNP as substrate, the burst of AdoMet formation followed by a very slow reaction of methionine and AMPPNP indicates that triphosphate hydrolysis is required for release of AdoMet. Like other MAT enzymes,7 MAT2A requires both catalytic events to occur before efficient release of products, consistent with the finding that AdoMet activates triphosphatase activity of MAT2A. Triphosphate hydrolysis occurs exclusively between the β and γ positions, establishing that once triphosphate is formed at the catalytic site, it cannot release and rebind for subsequent hydrolysis. Despite the positional isotope exchange with 5′-18O-ATP, the overall reaction, initiated with products, was experimentally irreversible. These catalytic features suggest that positional isotope exchange occurs from a novel catalytic complex that cannot be efficiently reformed by the addition of AdoMet and triphosphate. The transition state for the MAT2A dimer revealed a product-like, SN2 structure where the bond to the leaving triphosphate was largely lost at the transition-state,18 a condition that could possibly explain the positional isotope exchange (PIX), although transition state lifetimes are generally too short to permit atomic rotational motions.
Alternative ATP substrate analogue studies indicated that steady-state formation of AdoMet occurs with AMPNPP, as the resulting PNPP triphosphate analogue can be hydrolyzed at the β–γ bond with respect to ATP, leading to more efficient product release than with AMPPNP. Non-hydrolyzable ATP analogues containing methylene groups in the α–β or β–γ positions were not significant inhibitors of MAT2A, distinguishing it from the E. coli enzyme. Experiments with phosphate analogues indicated that triphosphate was a significant inhibitor, but the imidotriphosphates were more potent. Diimidotriphosphate acted as a nanomolar inhibitor and its binding was not enhanced by the addition of AdoMet, supporting action as the binary E-PNPNP complex. Inhibition of E. coli MAT by dimidotriphosphate differs by requiring AdoMet, to form a ternary inhibitory complex.39 Hence, AdoMet is implicated as the last product to dissociate from E. coli MAT, but Pi and PPi the last dissociate from MAT2A.
Crystallization of MAT2A in the presence of AdoMet, Mg2+, K+ and PNPNP, provided a detailed view of this complex catalytic site. Contacts between PNPNP, ions and structural waters involves more than a dozen ionic or hydrogen bond interactions of 3.0 Å or less, explaining both its tight binding and why triphosphate cannot be released at a catalytically significant rate from the catalytic site until the terminal phosphate is hydrolyzed. The PNPNP-Mg2+-K+ ternary cluster acts as a molecular bridge to stabilize both subunits that form the catalytic site, thus providing strong protein thermal stabilization to the complex. Direct interactions to both imido groups of the PNPNP provides a structural explanation of its tighter binding than triphosphate. A well-localized water, coordinated and potentially activated by Arg264, is the candidate for hydrolysis of the terminal phosphate by attack on the γ-phosphoryl group. Triphosphate hydrolysis and subsequent release of all products is faster than formation of AdoMet, as no burst of AdoMet or phosphate is seen in pre-steady-state kinetic analysis. Prodrug approaches or additional methods to utilize the PNPNP ligand suggest a potential route for therapeutic development and a new avenue for targeting MAT2A in cancer cells.
EXPERIMENTAL METHODS
Complete methods can be found in the supplemental information and the methods here are central to interpretation of the experimental results.
Pre-Steady-State Quench Flow Reactions
Quench-flow reactions (at 25 °C) monitored AdoMet formation on a Kintek Chemical Quench-Flow instrument. Syringe A contained: 50 mM Tris-HCl pH 8.0, 50 mM KCl, 10 mM MgCl2, 2 mM ATP (including [2,8-3H]-ATP), and 2 mM methionine. Syringe B contained: 50 mM Tris-HCl pH 8.0, 50 mM KCl, 10 mM MgCl2, and 100 μM MAT2A. Drive syringes A and B were filled with 50 mM Tris-HCl pH 8.0, 50 mM KCl, 10 mM MgCl2 and the quench syringe filled with 0.03 M sulfuric acid and the system flushed before samples were injected. Final concentrations within the quench-flow loops were 50 mM Tris-HCl pH 8.0, 50 mM KCl, 10 mM MgCl2, 1 mM ATP (including [2,8-3H]-ATP), 1 mM methionine and 50 μM MAT2A. Times samples were collected from the instrument and placed on ice. A slurry of Dowex 1x2 (chloride form, 50-100 mesh, Sigma-Aldrich) was prepared in 1:2 with Milli-Q water. 500 μL of the slurry was added to small gravity-flow columns from Bio-Rad (5 cm height, 1.2 mL bed volume), one for each timepoint, and washed with 5 mL of Milli-Q water. Half the reaction was loaded onto the column and the resin washed with 5 mL of Milli-Q water and flow-through collected. The flow-through was mixed with 10 mL of Ultima Gold (Perkin Elmer) scintillation fluid and mixed before counting in a Perkin Elmer Tricarb 2910 TR scintillation counter to obtain the number of cpm (counts per minute) in channel A (3H) for 1 min/sample. In addition to the purified samples containing AdoMet, samples of the total reaction at each timepoint were analyzed in parallel to measure the total number of counts in the sample. The fraction of ATP converted to AdoMet was determined by this ratio and plotted as a function of time and fit to a straight line in GraphPad Prism.
Origin of the Orthophosphate in the MAT2A Reaction
Standards of 32P-phosphate were prepared in 50 mM Tris-HCl pH 8, 50 μM MgCl2, 50 μM ZnCl2, and 0.5 μM alkaline phosphatase were combined and the reaction started with 400 μM ATP (with [γ-32P]-ATP from Perkin Elmer). Alkaline phosphatase was expressed and purified as described previously.40 Reactions were incubated at 25 °C for 30 min and quenched to a final concentration of 0.01 M sulfuric acid. For a 32P-pyrophosphate standard, 25 mM Tris-HCl pH 8, 10 mM MgCl2, 50 mM CaCl2, 6 ng/mL adenylate cyclase (R&D Systems), and 0.02 μg/μL calmodulin (Sigma Aldrich) were combined and the reaction started with 400 μM ATP (with [γ-32P]-ATP from Perkin Elmer). The reaction was incubated at 25 °C for 30 min and quenched in a final concentration of 0.01 M sulfuric acid. Standards were treated with activated charcoal to remove ATP as below.
Reactions contained 50 mM Tris-HCl pH 8.0, 50 mM KCl, 10 mM MgCl2, 200 μM ATP (with [γ-32P]-ATP), and 200 μM methionine. Reactions at 25 °C were started with the addition of 1 μM MAT2A and incubated for 30 min. Timed samples were quenched with 0.01 M sulfuric acid. 50 μL samples were mixed with 1 mg of activated charcoal and incubated at room temperature for 20 min to remove unreacted ATP. The sample was then spun down and the supernatant saved. Samples were spotted on PEI cellulose TLC plates (Sigma) and developed in a solvent system of 0.3 M LiCl for 5 min, 1 M LiCl for 15 min, and 1.6 M LiCl until the solvent front reached about 1 inch from the top of the plate. The plate was air dried and exposed to a phosphor screen overnight before scanning in phosphorimager.
HPLC Assay to Monitor ATPase Activity of MAT2A
ATP hydrolysis by MAT2A was analyzed in reactions with 50 mM Tris-HCl pH 8.0, 50 mM KCl, 10 mM MgCl2, and 200 μM ATP. Control reactions contained 10 μM ammonium vanadate (Sigma) and those with AdoMet contained 80 μM AdoMet (Sigma). Reactions were performed from 25 °C on a heat-block and were started with the addition of 5 μM MAT2A and timepoints were taken from 0-4 hrs and quenched in 0.01 M sulfuric acid and placed on ice. Samples were briefly centrifuged to pellet the precipitated protein. Samples were analyzed on a Waters HPLC using Waters C18 column 300 mm x 3.9 mm. The buffer system was buffer A: 12.5 mM KH2PO4 pH 6 + 2 mM tetrabutylammonium bisulfate and buffer B: 50% buffer A + 50 % acetonitrile. Samples were injected to a gradient program with 100% buffer A to 100% B. The amount of ADP formed for each timepoint was calculated based on the area under the curve of the ADP and ATP peaks. The concentration of ADP in each sample was determined by the ratio of the ADP peak compared to the total amount of ATP and ADP in the sample.
Positional Isotope Exchange Experiments
Reactions contained 50 mM Tris-HCl pH 8, 50 mM KCl, 10 mM MgCl2, 1 mM methionine, 1 mM 5′-18O-ATP, and 1 μM MAT2A. At each timepoint, a portion of the reaction was taken and quenched with 30 mM EDTA. Before placing into NMR, the reaction was diluted by 37% in D2O, DSS was added as an internal chemical shift reference and placed into a Shigemi tube. All NMR data was acquired at 25 °C using a Bruker AVIII 600 MHz spectrometer running TopSpin 3.6 and equipped with a 5 mm H/F-TCI cryogenic probe. To determine the fraction of substrate reacted, 1D 1H spectra were collected with 32 scans, a 20 ppm sweep width sampled with 64 k points and a 3 s recycle delay and integration was done using the H1′ doublet for ATP at 6.16-6.14 ppm and the H1′ doublet for AdoMet at 6.13-6.11. The C5′ coupling of ATP was determined in a high resolution 2D 1H-13C HSQC to separate the cross peak belonging to the C5′ of 18O-ATP from that of 16O-ATP. Each HSQC timepoint was run over 16 hours with 40 scans acquired for each increment using a spectral width of 16 ppm and 15 ppm for 1H and 13C, respectively and a recycle delay of 1.1 s. The data sets were collected using 4096 and 512 complex points for 1H and 13C, respectively and the time-domain NMR data were multiplied with a shifted sine-bell function in each dimension prior to Fourier transformation. To quantify the amount of 16O and 18O 5’-ATP at each timepoint, slices were taken through the cross peaks of the HSQC data and the area under the peaks was integrated to get an 16O/18O ratio at each timepoint. Experiments with 31P NMR to observe the PIX reaction gave the expected 18O/16O shifts (shifted 0.0165 ppm upfield relative to 16O) to a non-bridgehead 18O (shifted 0.0281 ppm upfield relative to 16O). However, these could not be adequately resolved at the concentration limits to prevent precipitation and on our instrumentation (Bruker 300MHz with 5mm BBFO probe). We achieved better resolution using 13C on a Bruker 600MHz with 5mm TCI cryoprobe. Experimental time points were selected to enable detection of PIX > kcat, and were adequate to measure the PIX reported here.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by NIH research grant R01 GM041916 to VLS and training grant F32 CA225149 to CNN. We thank Dr. Lars Nordstroem in the Chemical Biology core facility at Albert Einstein College of Medicine for synthesis of the triphosphate analogues PPNP and PNPNP and for helpful chemistry discussions throughout this work. The Bruker 600 MHz NMR instrument in the Einstein Structural NMR Resource was purchased using funds from NIH award 1S10OD016305 and is supported by the Albert Einstein College of Medicine. We than Drs. Jeffrey Bonanno, Elena Fedorov and Steven Almo for their help in initiating the structural studies with MAT2A. We are thankful to the 31-ID beamline staff at the Advanced Photon Source (APS), Argonne National Laboratory for their assistance with X-ray diffraction data collection. Shared resource facilities at the Albert Einstein College of Medicine are supported in part by cancer center grant P30 CA013330.
Footnotes
ACCESSION CODE
Structure factors and coordinates are deposited in the RCSB Protein Data Bank with accession codes 7L1A
SUPPORTING INFORMATION
The supporting information supplement contains all materials and methods for the experiments described here. It also contains additional results on the kinetic properties and crystal structure of MAT2A.
REFERENCES
- [1].Mudd SH, and Cantoni GL (1958) Activation of methionine for transmethylation. III. The methionine-activating enzyme of Bakers' yeast, J Biol Chem 231, 481–492. [PubMed] [Google Scholar]
- [2].Takusagawa F, Kamitori S, and Markham GD (1996) Structure and function of S-adenosylmethionine synthetase: crystal structures of S-adenosylmethionine synthetase with ADP, BrADP, and PPi at 28 angstroms resolution, Biochemistry 35, 2586–2596. [DOI] [PubMed] [Google Scholar]
- [3].Murray B, Antonyuk SV, Marina A, Van Liempd SM, Lu SC, Mato JM, Hasnain SS, and Rojas AL (2014) Structure and function study of the complex that synthesizes S-adenosylmethionine, IUCrJ 1, 240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Murray B, Antonyuk SV, Marina A, Lu SC, Mato JM, Hasnain SS, and Rojas AL (2016) Crystallography captures catalytic steps in human methionine adenosyltransferase enzymes, Proc Natl Acad Sci U S A 113, 2104–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Markham GD, and Pajares MA (2009) Structure-function relationships in methionine adenosyltransferases, Cell Mol Life Sci 66, 636–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mato JM, Alvarez L, Ortiz P, and Pajares MA (1997) S-adenosylmethionine synthesis: molecular mechanisms and clinical implications, Pharmacol Ther 73, 265–280. [DOI] [PubMed] [Google Scholar]
- [7].Markham GD, Hafner EW, Tabor CW, and Tabor H (1980) S-Adenosylmethionine synthetase from Escherichia coli, J Biol Chem 255, 9082–9092. [PubMed] [Google Scholar]
- [8].Reczkowski RS, Taylor JC, and Markham GD (1998) The active-site arginine of S-adenosylmethionine synthetase orients the reaction intermediate, Biochemistry 37, 13499–13506. [DOI] [PubMed] [Google Scholar]
- [9].Fu Z, Hu Y, Markham GD, and Takusagawa F (1996) Flexible loop in the structure of S-adenosylmethionine synthetase crystallized in the tetragonal modification, J Biomol Struct Dyn 13, 727–739. [DOI] [PubMed] [Google Scholar]
- [10].McQueney MS, and Markham GD (1995) Investigation of monovalent cation activation of S-adenosylmethionine synthetase using mutagenesis and uranyl inhibition, J Biol Chem 270, 18277–18284. [DOI] [PubMed] [Google Scholar]
- [11].Markham GD, Parkin DW, Mentch F, and Schramm VL (1987) A kinetic isotope effect study and transition state analysis of the S-adenosylmethionine synthetase reaction, J Biol Chem 262, 5609–5615. [PubMed] [Google Scholar]
- [12].Kotb M, and Geller AM (1993) Methionine adenosyltransferase: structure and function, Pharmacol Ther 59, 125–143. [DOI] [PubMed] [Google Scholar]
- [13].Simile MM, Peitta G, Tomasi ML, Brozzetti S, Feo CF, Porcu A, Cigliano A, Calvisi DF, Feo F, and Pascale RM (2019) MicroRNA-203 impacts on the growth, aggressiveness and prognosis of hepatocellular carcinoma by targeting MAT2A and MAT2B genes, Oncotarget 10, 2835–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gonzalez B, Pajares MA, Hermoso JA, Guillerm D, Guillerm G, and Sanz-Aparicio J (2003) Crystal structures of methionine adenosyltransferase complexed with substrates and products reveal the methionine-ATP recognition and give insights into the catalytic mechanism, J Mol Biol 331, 407–416. [DOI] [PubMed] [Google Scholar]
- [15].Komoto J, Yamada T, Takata Y, Markham GD, and Takusagawa F (2004) Crystal structure of the S-adenosylmethionine synthetase ternary complex: a novel catalytic mechanism of S-adenosylmethionine synthesis from ATP and Met, Biochemistry 43, 1821–1831. [DOI] [PubMed] [Google Scholar]
- [16].Halim AB, LeGros L, Geller A, and Kotb M (1999) Expression and functional interaction of the catalytic and regulatory subunits of human methionine adenosyltransferase in mammalian cells, J Biol Chem 274, 29720–29725. [DOI] [PubMed] [Google Scholar]
- [17].LeGros HL Jr., Halim AB, Geller AM, and Kotb M (2000) Cloning, expression, and functional characterization of the beta regulatory subunit of human methionine adenosyltransferase (MAT II), J Biol Chem 275, 2359–2366. [DOI] [PubMed] [Google Scholar]
- [18].Firestone RS, and Schramm VL (2017) The Transition-State Structure for Human MAT2A from Isotope Effects, J Am Chem Soc 139, 13754–13760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Katoh Y, Ikura T, Hoshikawa Y, Tashiro S, Ito T, Ohta M, Kera Y, Noda T, and Igarashi K (2011) Methionine adenosyltransferase II serves as a transcriptional corepressor of Maf oncoprotein, Mol Cell 41, 554–566. [DOI] [PubMed] [Google Scholar]
- [20].Kera Y, Katoh Y, Ohta M, Matsumoto M, Takano-Yamamoto T, and Igarashi K (2013) Methionine adenosyltransferase II-dependent histone H3K9 methylation at the COX-2 gene locus, J Biol Chem 288, 13592–13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Herr P, Bostrom J, Rullman E, Rudd SG, Vesterlund M, Lehtio J, Helleday T, Maddalo G, and Altun M (2020) Cell Cycle Profiling Reveals Protein Oscillation, Phosphorylation, and Localization Dynamics, Mol Cell Proteomics 19, 608–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang Z, Yip LY, Lee JHJ, Wu Z, Chew HY, Chong PKW, Teo CC, Ang HY, Peh KLE, Yuan J, Ma S, Choo LSK, Basri N, Jiang X, Yu Q, Hillmer AM, Lim WT, Lim TKH, Takano A, Tan EH, Tan DSW, Ho YS, Lim B, and Tam WL (2019) Methionine is a metabolic dependency of tumor-initiating cells, Nat Med 25, 825–837. [DOI] [PubMed] [Google Scholar]
- [23].Wallace HM (1996) Polyamines in human health, Proc Nutr Soc 55, 419–431. [DOI] [PubMed] [Google Scholar]
- [24].Basu I, Cordovano G, Das I, Belbin TJ, Guha C, and Schramm VL (2007) A transition state analogue of 5'-methylthioadenosine phosphorylase induces apoptosis in head and neck cancers, J Biol Chem 282, 21477–21486. [DOI] [PubMed] [Google Scholar]
- [25].Singh V, Shi W, Evans GB, Tyler PC, Furneaux RH, Almo SC, and Schramm VL (2004) Picomolar transition state analogue inhibitors of human 5'-methylthioadenosine phosphorylase and X-ray structure with MT-immucillin-A, Biochemistry 43, 9–18. [DOI] [PubMed] [Google Scholar]
- [26].Chen H, Xia M, Lin M, Yang H, Kuhlenkamp J, Li T, Sodir NM, Chen YH, Josef-Lenz H, Laird PW, Clarke S, Mato JM, and Lu SC (2007) Role of methionine adenosyltransferase 2A and S-adenosylmethionine in mitogen-induced growth of human colon cancer cells, Gastroenterology 133, 207–218. [DOI] [PubMed] [Google Scholar]
- [27].Liu Q, Chen J, Liu L, Zhang J, Wang D, Ma L, He Y, Liu Y, Liu Z, and Wu J (2011) The X protein of hepatitis B virus inhibits apoptosis in hepatoma cells through enhancing the methionine adenosyltransferase 2A gene expression and reducing S-adenosylmethionine production, J Biol Chem 286, 17168–17180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhang T, Zheng Z, Liu Y, Zhang J, Zhao Y, Liu Y, Zhu H, Zhao G, Liang J, Li Q, and Xu H (2013) Overexpression of methionine adenosyltransferase II alpha (MAT2A) in gastric cancer and induction of cell cycle arrest and apoptosis in SGC-7901 cells by shRNA-mediated silencing of MAT2A gene, Acta Histochem 115, 48–55. [DOI] [PubMed] [Google Scholar]
- [29].Yang H, Li TW, Peng J, Mato JM, and Lu SC (2011) Insulin-like growth factor 1 activates methionine adenosyltransferase 2A transcription by multiple pathways in human colon cancer cells, Biochem J 436, 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Attia RR, Gardner LA, Mahrous E, Taxman DJ, Legros L, Rowe S, Ting JP, Geller A, and Kotb M (2008) Selective targeting of leukemic cell growth in vivo and in vitro using a gene silencing approach to diminish S-adenosylmethionine synthesis, J Biol Chem 283, 30788–30795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mavrakis KJ, McDonald ER 3rd, Schlabach MR, Billy E, Hoffman GR, deWeck A, Ruddy DA, Venkatesan K, Yu J, McAllister G, Stump M, deBeaumont R, Ho S, Yue Y, Liu Y, Yan-Neale Y, Yang G, Lin F, Yin H, Gao H, Kipp DR, Zhao S, McNamara JT, Sprague ER, Zheng B, Lin Y, Cho YS, Gu J, Crawford K, Ciccone D, Vitari AC, Lai A, Capka V, Hurov K, Porter JA, Tallarico J, Mickanin C, Lees E, Pagliarini R, Keen N, Schmelzle T, Hofmann F, Stegmeier F, and Sellers WR (2016) Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5, Science 351, 1208–1213. [DOI] [PubMed] [Google Scholar]
- [32].Marjon K, Cameron MJ, Quang P, Clasquin MF, Mandley E, Kunii K, McVay M, Choe S, Kernytsky A, Gross S, Konteatis Z, Murtie J, Blake ML, Travins J, Dorsch M, Biller SA, and Marks KM (2016) MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis, Cell reports 15, 574–587. [DOI] [PubMed] [Google Scholar]
- [33].Gao G, Zhang L, Villarreal OD, He W, Su D, Bedford E, Moh P, Shen J, Shi X, Bedford MT, and Xu H (2019) PRMT1 loss sensitizes cells to PRMT5 inhibition, Nucleic Acids Res 47, 5038–5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Quinlan CL, Kaiser SE, Bolanos B, Nowlin D, Grantner R, Karlicek-Bryant S, Feng JL, Jenkinson S, Freeman-Cook K, Dann SG, Wang X, Wells PA, Fantin VR, Stewart AE, and Grant SK (2017) Targeting S-adenosylmethionine biosynthesis with a novel allosteric inhibitor of Mat2A, Nat Chem Biol 13, 785–792. [DOI] [PubMed] [Google Scholar]
- [35].Study of AG-270 in Participants With Advanced Solid Tumors or Lymphoma With MTAP Loss, https://ClinicalTrials.gov/show/NCT03435250.
- [36].A Study of JNJ-64619178, an Inhibitor of PRMT5 in Participants With Advanced Solid Tumors, NHL, and Lower Risk MDS, https://ClinicalTrials.gov/show/NCT03573310.
- [37].Rose IA (1978) Enzyme reactions of ATP studied by positional isotope exchange, Fed Proc 37, 2775–2782. [PubMed] [Google Scholar]
- [38].Rose IA (1979) Positional isotope exchange studies of enzyme mechanisms, Adv Enzymol Relat Areas Mol Biol 50, 361–395. [DOI] [PubMed] [Google Scholar]
- [39].Reczkowski RS, and Markham GD (1999) Slow binding inhibition of S-adenosylmethionine synthetase by imidophosphate analogues of an intermediate and product, Biochemistry 38, 9063–9068. [DOI] [PubMed] [Google Scholar]
- [40].O'Brien PJ, and Herschlag D (2001) Functional interrelationships in the alkaline phosphatase superfamily: phosphodiesterase activity of Escherichia coli alkaline phosphatase, Biochemistry 40, 5691–5699. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.