

Abstract
The nonpsychoactive phytocannabinoid cannabidiol (CBD) has been shown to have analgesic effects in animal studies but little is known about its mechanism of action. We examined the effects of CBD on intrinsic excitability of primary pain-sensing neurons. Studying acutely dissociated capsaicin-sensitive mouse DRG neurons at 37°C, we found that CBD effectively inhibited repetitive action potential firing, from 15–20 action potentials evoked by 1 s current injections in control to 1–3 action potentials with 2 μm CBD. Reduction of repetitive firing was accompanied by a reduction of action potential height, widening of action potentials, reduction of the afterhyperpolarization, and increased propensity to enter depolarization block. Voltage-clamp experiments showed that CBD inhibited both TTX-sensitive and TTX-resistant (TTX-R) sodium currents in a use-dependent manner. CBD showed strong state-dependent inhibition of TTX-R channels, with fast binding to inactivated channels during depolarizations and slow unbinding on repolarization. CBD alteration of channel availability at various voltages suggested that CBD binds especially tightly [Kd (dissociation constant), ∼150 nm] to the slow inactivated state of TTX-R channels, which can be substantially occupied at voltages as negative as −40 mV. Remarkably, CBD was more potent in inhibiting TTX-R channels and inhibiting action potential firing than the local anesthetic bupivacaine. We conclude that CBD might produce some of its analgesic effects by direct effects on neuronal excitability, with tight binding to the slow inactivated state of Nav1.8 channels contributing to effective inhibition of repetitive firing by modest depolarizations.
SIGNIFICANCE STATEMENT Cannabidiol (CBD) has been shown to inhibit pain in various rodent models, but the mechanism of this effect is unknown. We describe the ability of CBD to inhibit repetitive action potential firing in primary nociceptive neurons from mouse dorsal root ganglia and analyze the effects on voltage-dependent sodium channels. We find that CBD interacts with TTX-resistant sodium channels in a state-dependent manner suggesting particularly tight binding to slow inactivated states of Nav1.8 channels, which dominate the overall inactivation of Nav1.8 channels for small maintained depolarizations from the resting potential. The results suggest that CBD can exert analgesic effects in part by directly inhibiting repetitive firing of primary nociceptors and suggest a strategy of identifying compounds that bind selectively to slow inactivated states of Nav1.8 channels for developing effective analgesics.
Keywords: cannabidiol, Nav1.8, nociceptors, slow inactivation, sodium channels, TTX-resistant
Introduction
Cannabidiol (CBD), a nonpsychoactive phytocannabinoid present in marijuana (Mechoulam et al., 1970), has been developed as an antiepileptic agent (Epidiolex) and shown to be effective in treating Dravet syndrome (Devinsky et al., 2017, 2018b, 2019; Miller et al., 2020) and Lennox–Gastaut syndrome (Devinsky et al., 2018a; Thiele et al., 2019) epilepsies. The molecular mechanism of action of CBD in epilepsy is still unclear (Jones et al., 2010; Rosenberg et al., 2015, 2017; Franco and Perucca, 2019; Gray and Whalley, 2020). Unlike Δ(9)-tetrahydrocannabinol, the other major phytocannabinoid in marijuana, CBD does not act as a direct primary ligand at CB1 or CB2 receptors, the G-protein-coupled receptors activated by endocannabinoids (Pertwee, 2005). However, CBD can inhibit the effects of endocannabinoids naturally released by neurons to modulate synaptic transmission (Straiker et al., 2018) by a negative allosteric effect mediated by cannabidiol binding to CB1 receptors at a site distinct from the primary binding site (Laprairie et al., 2015; Straiker et al., 2018). CBD actions in the CNS may also reflect its activity as an antagonist of the lipid-activated G-protein-coupled receptor GPR55 (Ryberg et al., 2007; Kaplan et al., 2017). In addition, CBD can act at micromolar concentrations to directly inhibit a wide range of ion channels, including voltage-dependent sodium channels (Hill et al., 2014; Patel et al., 2016; Ghovanloo et al., 2018; Mason and Cummins, 2020) and modifies the intrinsic excitability of central neurons (Kaplan et al., 2017; Khan et al., 2018).
Although the efficacy of oral CBD in humans has been demonstrated in rigorous large-scale clinical trials only for specific epilepsies, there are anecdotal reports that oral CBD is effective in inhibiting pain (Argueta et al., 2020; Tran and Kavuluru, 2020). Small clinical trials have shown reduction in myofascial pain by CBD applied in a transdermal cream (Nitecka-Buchta et al., 2019) and relief of peripheral neuropathic pain by topical cannabidiol oil (Xu et al., 2020). In animal experiments, CBD has been shown to relieve pain in rodent models of inflammatory and arthritic pain by oral administration (Malfait et al., 2000; Costa et al., 2004, 2007; Rock et al., 2018), intraperitoneal injection (Malfait et al., 2000; Gallily et al., 2018), intra-articular injection (Philpott et al., 2017), and transdermal application (Hammell et al., 2016). Oral CBD relieved allodynia in a mouse sciatic nerve injury model (Abraham et al., 2020), and reduced a pain index and enhanced locomotion in dogs with osteoarthritis (Verrico et al., 2020).
CBD effects on pain could involve a mixture of effects on the CNS (Maione et al., 2011; De Gregorio et al., 2019) and the periphery. Here, we examined the effects of CBD on the excitability of primary sensory neurons, using a preparation of acutely dissociated neurons in which well defined concentrations of CBD can be applied. We find that submicromolar concentrations of CBD inhibit the excitability of nociceptors by state-dependent binding to voltage-dependent sodium channels, in a manner suggesting especially tight binding to slow inactivated states of Nav1.8 channels.
Materials and Methods
Cell preparation.
Acutely dissociated dorsal root ganglion (DRG) neurons were prepared as previously described (Liu et al., 2017; Zheng et al., 2019). Briefly, DRGs were removed from Swiss Webster mice of either sex [postnatal day 9 (P9) to P14]. Ganglia were removed in ice-cold Ca2+-free, Mg2+-free (CMF) HBSS (Thermo Fisher Scientific) under a dissection microscope. Ganglia were treated for 20 min at 37°C with 20 U/ml papain (Worthington Biochemical) in CMF HBSS and then treated for 20 min at 37°C with 3 mg/ml collagenase (type I) and 4 mg/ml dispase II (Roche Diagnostics) in CMF HBSS. Cells were dispersed by gentle trituration with a fire-polished glass Pasteur pipette in a solution composed of the following two media combined in a 1:1 ratio: Invitrogen Leibovitz's L-15 medium (Thermo Fisher Scientific) supplemented with 5 mm HEPES, and DMEM/F12 medium (Thermo Fisher Scientific) supplemented with 10% Heat-Inactivated Fetal Bovine Serum (Thermo Fisher Scientific); this solution also had added 130 ng/ml nerve growth factor (NGF; Sigma-Aldrich). Cells were then plated on glass coverslips and incubated at 37°C (95% O2, 5% CO2) for 1.5–2 h, after which they were stored at 4°C in Neurobasal medium (Thermo Fisher Scientific) supplemented with 200 ng/ml Invitrogen NGF (Thermo Fisher Scientific), 1% penicillin and streptomycin (Sigma-Aldrich), 2% Invitrogen B-27 supplement (Thermo Fisher Scientific), and 30 mm NaCl. Cells were used within 48 h. Storage at 4°C prevented neurite growth, enabling faster decay of capacity transients, more accurate voltage clamp, and the ability to lift cells in front of flow pipes to facilitate temperature control and to enable rapid solution exchange.
Electrophysiology.
Recordings were performed at 37°C using a Molecular Devices Multiclamp 700B Amplifier. Voltage or current commands were delivered, and signals were recorded, using a Digidata 1322A data acquisition system (Molecular Devices) controlled by pCLAMP 10.3 software (Molecular Devices). Electrodes were pulled on a P-97 puller (Sutter Instruments), and shanks were wrapped with Parafilm (American National Can Company) to allow optimal series resistance compensation without oscillation. The resistances of the pipettes were 3–9 MΩ. Seals were formed in Tyrode's solution consisting of 155 mm NaCl, 3.5 mm KCl, 1.5 mm CaCl2, 1 mm MgCl2, 10 mm HEPES, and 10 mm glucose, at pH 7.4 adjusted with NaOH. After establishing whole-cell recording, cell capacitance was nulled and series resistance was partially (70–80%) compensated. Cells were lifted off the bottom of the recording chamber and placed in front of an array of quartz flow pipes (internal diameter, 250 µm; external diameter, 350 µm; Polymicro Technologies) attached with styrene butadiene glue (Amazing Goop, Eclectic Products) to an aluminum rod (1 × 1 cm) whose temperature was controlled to 38°C by resistive heating elements and a feedback-controlled temperature controller (model TC-344B, Warner Instruments). With the end of the rod and the flow pipes (extending 1 mm from the end of the rod) lowered just below the surface of the bulk chamber solution, the solution exiting from the flow pipes is maintained at 37°C (Carter and Bean, 2009).
Current and voltage records were filtered at 5 or 10 kHz and digitized at 100 or 200 kHz. Analysis was performed with Igor Pro 6.12 (WaveMetrics) using DataAccess (Bruxton Software) to import pClamp data. Voltage-clamp current records were corrected for linear capacitive and leak current by subtracting scaled responses to 5 mV hyperpolarizations delivered from the holding potential. Action potential–clamp current records were corrected by subtracting current elicited by a scaled (0.1) inverted action potential waveform. In analyzing current-clamp experiments, action potentials were defined using criteria of action potential peak >0 mV and height >40 mV.
Current-clamp experiments were performed using cells that had resting potentials negative to −50 mV, which showed robust and reproducible firing. The range of resting potentials for cells used in current-clamp experiments was −50.7 to −86.6 mV (mean, −65.4 ± 1.48 mV; n = 35). Action potentials in control were elicited from the natural resting potential of the cell. CBD and bupivacaine often produced a depolarization of the resting potential, and a steady negative current was injected to bring the membrane potential back to the control resting value to allow better comparison of action potentials.
Solutions.
Current-clamp recordings (and voltage-clamp recordings of overall ionic currents) used external Tyrode's solution and an internal solution consisting of 140 mm K aspartate, 13.5 mm NaCl, 1.8 mm MgCl2, 0.09 mm EGTA, 9 mm HEPES, 14 mm creatine phosphate (Tris salt), 4 mm MgATP, and 0.3 mm Tris-GTP, at pH 7.2 adjusted with KOH. Reported membrane potentials are corrected for a liquid junction potential of −10 mV between the internal solution and the Tyrode's solution in which current was zeroed before sealing onto the cell, measured using a flowing 3 m KCl reference electrode, as described by Neher (1992). Voltage-clamp recordings of sodium current used an internal Cs-based solution [140 mm CsCl, 13.5 mm NaCl, 1.8 mm MgCl2, 0.09 mm EGTA, 9 mm HEPES, 14 mm creatine phosphate (Tris salt), 4 mm MgATP, and 0.3 mm Tris-GTP, at pH adjusted to 7.2 with CsOH] and an external solution of Tyrode's solution with added 10 mm TEACl to inhibit potassium currents and 30 μm CdCl2 to inhibit voltage-dependent calcium currents. Currents through TTX-resistant sodium channels were isolated by including 300 nm TTX in the solution.
CBD was obtained as a 1 mg/ml solution in methanol (Sigma-Aldrich). The methanol was evaporated using a gentle stream of nitrogen and CBD was dissolved in dimethylsulfoxide (DMSO) to make a stock solution of 10 mm, which was aliquoted and stored at 4°C. In all experiments, control solutions were prepared to contain the same concentration of DMSO as the CBD-containing solutions to be tested. Some early experiments used the 1 mg/ml methanol solution as a stock solution, with equivalent methanol added to the control solutions. There was no obvious difference in the effects of CBD seen using methanol and DMSO stock solutions but the higher-concentration DMSO stocks were preferred because previous control experiments have shown that DMSO at >1:1000 dilutions has no obvious effect on electrophysiological behavior of neurons. CBD was applied acutely in all experiments except in those shown in Figure 13, B and C, where cells were preincubated in 300 nm CBD for 1–2 h before recording.
Figure 13.
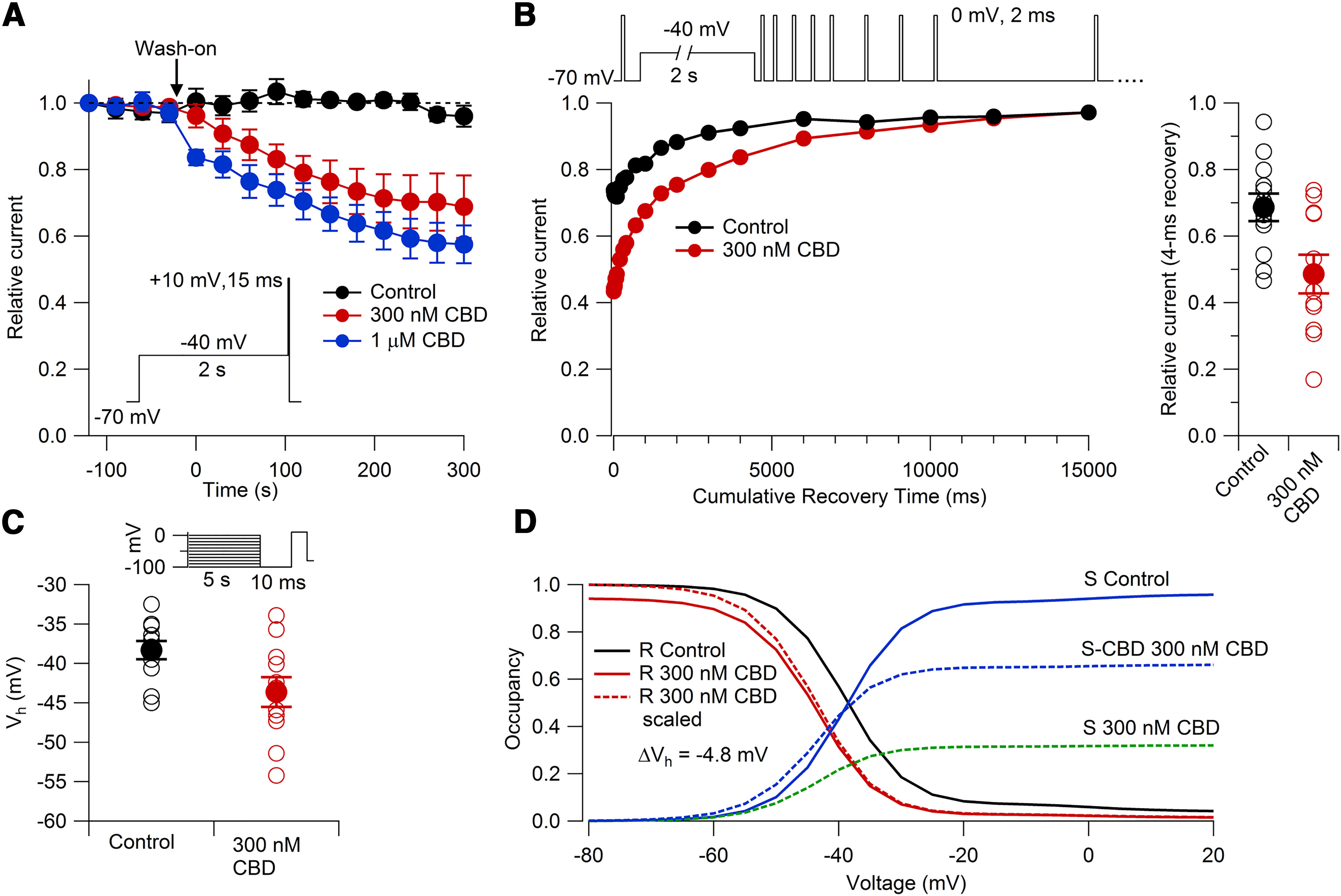
Effects of 300 nm and 1 μm CBD on TTX-R sodium channels. A, Inhibition of TTX-R sodium current by 300 nm CBD (red; n = 6 cells from three animals) and by 1 μm CBD (blue; n = 6 cells from two animals). Currents were evoked by a 15 ms step to +10 mV after a 2 s depolarization at −40 mV, applied once every 30 s. Black symbols: data for cells with the same protocol with extended recording in control solution (n = 6 cells from four animals). p < 0.05 after 60 s between 300 nm CBD and control; p < 0.05 after 0 s between 1 μm CBD and control. B, Effect of 300 nm CBD on the time course of the recovery of availability of TTX-R sodium channels following a 2 s prepulse to −40 mV. Cells were preincubated with 300 nm CBD or control (DMSO-containing) solution for 1–2 h. Representative recovery time course in control (black) and 300 nm CBD (red). Right, Collected results for fraction available after 4 ms recovery at −70 mV, normalized to test pulse before conditioning pulse. Control: relative current = 0.68 ± 0.04; n = 12 cells from three animals; 300 nm CBD: relative current = 0.48 ± 0.05; n = 11 cells from three animals; p = 0.009. C, Availability using the protocol of Figure 11B to measure voltage dependence of slow inactivation, with 5 s conditioning pulses followed by a 10 ms return to −100 mV to allow recovery from fast inactivation followed by the test pulse. Control: Vh = −38.3 ± 1.2 mV, n = 12 from three animals; 300 nm CBD: Vh = −43.6 ± 1.9 mV, n = 11 cells from three animals; p = 0.026. D, Prediction of the model in Figure 12 for channel fraction in R (i.e., available channels) with the protocol in C in control (solid black line) and with 300 nm CBD (solid red line). Dashed red line shows channel availability in CBD normalized to 1 at −100 mV. Blue line shows occupancy of the slow inactivated state in control; in the presence of 300 nm CBD, blue dashed line shows occupancy of CBD-bound slow inactivated state, and green dashed line shows occupancy of CBD-free slow inactivated state.
Experimental design and statistical analyses.
Numbers of cells and animals used for each experiment are given in figure legends. Data are represented as mean ± SEM. p Values were calculated (Excel version 140.7266.5000, Microsoft) using paired or nonpaired Student's t tests, as appropriate. Data are available from the authors on request.
Model.
The model for state-dependent binding of CBD was implemented in IgorPro (WaveMetrics) using fourth-order Runge-Kutta integration with a 200 μs step size. The parameters of the model were chosen in two steps. In the first, voltage-dependent rate constants were assigned for transitions among resting state (R), fast inactivated state (F), and slow inactivated state (S) to approximate the experimental measurements summarized and seen in Figure 11, as well as approximately matching the kinetics of entry into slow and fast inactivated states shown in Figures 7, 8, and 10, along with the more comprehensive measurements of kinetics in a previous study (Blair and Bean, 2003). To match experimental data, it was necessary to allow the entry of channels into the slow inactivated state both directly from the resting state (the primary pathway for small depolarizations in the range from −60 to −40 mV, where fast inactivation is minimal) and also from the fast inactivated state, to describe entry into slow inactivation during prolonged steps to voltages where channels first enter into fast inactivated states (see Fig. 8, steps to 0 mV). The model was constructed to satisfy microscopic reversibility, first choosing the forward and backward rate constants for movement between R and F, those for movement between R and S, and the forward rate constant from F to S, and then setting the rate constant for movement from S to F determined by the other rate constants to preserve microscopic reversibility. After setting the parameters for movement among the resting, fast inactivated, and slow inactivated states, rate constants for binding and unbinding of CBD to each state were chosen by trial and error to match the experimental results for the shift in the midpoint of availability curves determined with a 50 ms conditioning pulse (mostly, but not exclusively, fast inactivation) and with 5 s conditioning pulses followed by 10 ms at −100 mV, which in control conditions measures the voltage dependence of slow inactivation.
Figure 11.

CBD alteration of availability of TTX-R sodium channels measured with protocols to emphasize fast or slow inactivation. A, Voltage-dependent availability of TTX-R sodium channels measured by a 3 ms test pulse to 0 mV following 50 ms conditioning pulses in the absence (black) and presence (red) of 5 μm CBD. Solid lines: Boltzmann equation, IMax/(1 + exp((V – Vh)/k)), where IMax is maximal test pulse current, V is holding voltage, Vh is the voltage of half-maximal availability, and k is the slope factor. For the example cell, control: IMax = −3866 pA, Vh = −21.0 mV, k = 5.0 mV; 5 μm CBD: IMax = −1781 pA, Vh = −29.4 mV, k = 7.1 mV, ΔVh= −8.4 mV. For collected results with 50 ms conditioning pulses: Vh = −22.2 ± 1.4 mV, k = 4.6 ± 0.1 mV; 5 μm CBD: Vh = −29.8 ± 1.7 mV, k = 6.1 ± 0.4 mV; ΔVh = −7.6 ± 0.4 mV. N = 6 cells from three animals. B, Availability using a protocol to measure voltage dependence of slow inactivation, with 5 s conditioning pulses followed by a 10 ms return to −100 mV to allow recovery from fast inactivation followed by the test pulse. Symbols are as in A. For the example cell: control: IMax = −3336 pA, Vh = −36.9 mV, k = 5.5 mV; 5 μm CBD: IMax = −2190 pA, Vh = −52.1 mV, k = 6.0 mV, ΔVh = −15.2 mV. For collected results with 5 s conditioning pulses: control: Vh = −39.2 ± 2.7 mV, k = 5.2 ± 0.4 mV; 5 μm CBD: Vh = −53.0 ± 3.3 mV, k = 5.6 ± 0.3 mV; ΔVh = −13.8 ± 1.5 mV. N = 6 cells from three animals.
Figure 7.

Effect of CBD on the time course of the recovery of availability of TTX-R sodium channels following a 20 ms prepulse to 0 mV. A, Following the 20 ms prepulse to 0 mV, a 3 ms test pulse to 0 mV assayed the fraction of available channels following various times at −70 mV. B, C, Recovery of availability in 10 ms at −70 mV in control and (in the same cell) in the presence of 5 μm CBD. D, E, Collected results for recovery of availability in control (black) and 5 μm CBD (red) for this protocol (mean ± SEM; n = 6 cells from one animal). Recovery in control was fit by a single exponential of 1.03 ms to an asymptote of 0.85; recovery in 5 μm CBD was fit by a double exponential to an asymptote of 0.80, with a fast time constant (a fraction of 28%) set to match the recovery of unmodified channels together with a dominant slower time constant (66 ms).
Figure 8.

Time course of CBD-induced entry into slowly recovering states of TTX-R channels. A, Voltage protocol (top) and currents in control (middle) and after application of 5 μm CBD (bottom), with a prepulse duration of 20 ms. B, Collected results (mean ± SEM; n = 6 cells from one animal) for the fraction of available channels after prepulse conditioning in control (black) and in 5 μm CBD (red). Solid red curve, Single exponential of time constant 8.5 ms.
Figure 10.

Time course of recovery from slow inactivated states in control and with 5 μm CBD measured with a single-sweep protocol. A, Voltage protocol. After a 300 ms or 10 s conditioning pulse to 0 mV, the recovery of availability at −70 mV is assayed by multiple short (2 ms) test pulses (kept short to minimize inactivation during the test pulse) delivered at various times after the voltage returns to −70 mV. For clarity, not all test pulses are shown. B, Time course of recovery of channel availability at −70 mV (assayed by test pulse current relative to the peak current evoked at the start of the conditioning pulse to 0 mV) following a 300 ms conditioning pulse in control (black symbols: mean ± SEM, n = 6 cells from one animal) and with 5 μm CBD (red symbols: mean ± SEM, same 6 cells). Solid lines, Single exponential fits to data points from 60 ms to 6 s of cumulative recovery time at −70 mV. C, Time course of recovery of channel availability at −70 mV following a 10 s conditioning pulse in control (black symbols: mean ± SEM, same 6 cells as in B) and with 5 μm CBD (red symbols: mean ± SEM, same 6 cells). Solid lines, Single exponential fits to data points from 300 ms to 30 s of cumulative recovery time at −70 mV.
Results
CBD reduces the excitability of nociceptors
We first tested the effect of CBD on action potential firing of acutely dissociated small-diameter DRG neurons in current clamp. The identity of neurons as nociceptors was confirmed by inward currents at −70 mV induced by 1 μm capsaicin applied at the end of each recording, and only capsaicin-sensitive neurons were included in the analysis. CBD dramatically reduced the number of action potentials during step current injections (Fig. 1). At 2 μm, CBD reduced the number of action potentials evoked by an 80 pA current injection from 17.1 ± 2.9 in vehicle to 2.0 ± 1.0 in 2 μm CBD (n = 14 pairs, p = 0.0002). Remarkably, the effects of CBD were more dramatic than those of bupivacaine, one of the most potent local anesthetics, which reduced firing evoked by an 80 pA current injection from 20.1 ± 6.2 action potentials in vehicle to 10.6 ± 4.0 in 2 μm bupivacaine (n = 7 pairs, p = 0.02; Fig. 1C).
Figure 1.

Inhibition of action potential firing in mouse nociceptors by CBD compared with bupivacaine. A, Effect of 2 μm CBD on action potential firing elicited by 1 s current injections ranging from 15 to 80 pA in a capsaicin-sensitive mouse nociceptor. B, Collected results for the number of action potentials during 1 s current injections in control and 2 μm CBD. Values are the mean ± SEM; n = 14 cells from two animals. C, Number of action potentials during 1 s current injections in control and 2 μm bupivacaine. Values are the mean ± SEM; n = 7 cells from two animals.
Figure 2 shows the effect of CBD on action potential shape. CBD decreased the height of the action potential (from 104 ± 3 mV in vehicle to 71 ± 6 mV in 2 μm CBD; n = 10 pairs, p < 0.0001), increased the width of the action potential (from 2.4 ± 0.1 ms in vehicle to 4.0 ± 0.3 ms in 2 μm CBD; n = 10 pairs, p = 0.0002), and produced a depolarizing shift in the trough of the first action potential during a current injection (from −75.3 ± 2.3 mV in vehicle to −65.8 ± 2.8 mV in 2 μm CBD for a 40 pA current injection; n = 10 pairs, p < 0.0001). Bupivacaine had smaller effects on all of these parameters (Fig. 2C–E).
Figure 2.

Effect of CBD on action potential shape compared with bupivacaine. A, The first action potential evoked by a 1 s 80 pA current injection in control and 2 μm CBD (same cell as in Fig. 1). B, The first action potential evoked by a 1 s 40 pA current injection in control and 2 μm bupivacaine. C, Collected results for the effect of 2 μm CBD (n = 10 cells from two animals) or 2 μm bupivacaine (n = 6 cells from two animals) on the peak of the first action potential elicited by 40 pA current injections. Control versus 2 μm CBD, p < 0.0001; control versus 2 μm bupivacaine, p = 0.07. D, Collected results for the effect of 2 μm CBD (n = 10 cells from two animals) or 2 μm bupivacaine (n = 6 cells from two animals) on the width of the first action potential (measured at half action potential height). Control versus 2 μm CBD, p = 0.0002; control versus 2 μm bupivacaine, p = 0.27. E, Collected results for the effect of 2 μm CBD (n = 10 cells from two animals) or 2 μm bupivacaine (n = 6 cells from two animals) on the membrane potential of the trough after the first action potential. Control versus 2 μm CBD, p < 0.0001; control to 2 μm bupivacaine, p = 0.009.
CBD inhibits voltage-activated inward and outward currents
To explore the action of CBD on particular ion channels in nociceptors, we next did a series of voltage-clamp experiments. Examining overall currents evoked by voltage steps from −80 mV, CBD produced a dose-dependent reduction of both inward and outward components of current (Fig. 3). The inhibition of inward currents was more dramatic than the inhibition of outward currents. In collected results, the peak inward current at −10 mV was reduced by 2 μm CBD to 60 ± 4% of control (n = 13 pairs, p < 0.0001) and by 5 μm CBD to 21 ± 2% of control (n = 7 pairs, p < 0.0001). Peak outward current measured at +20 mV was reduced to 83 ± 3% by 2 μm CBD (n = 13 pairs, p < 0.0001) and to 60 ± 4% by 5 μm CBD (n = 7 pairs, p < 0.0001).
Figure 3.

Effect of CBD on overall voltage-activated currents in nociceptors. A, Currents evoked by depolarization before and after 2 μm CBD. The 200 ms voltage steps were applied at 0.2 Hz from a holding voltage of −80 mV. The first 90 μs of the test pulse currents are blanked to remove capacity artifacts. Series resistance compensation for 80% of series resistance of 7 MΩ (measured from the time constant of the uncompensated capacity transient). B, Effect of 2 μm CBD on peak inward and peak outward currents as a function of test pulse voltage. Same cell as in A. C, Collected results for the inhibition of peak currents evoked by voltage steps from −80 to −10 mV (inward current) and +20 mV (outward current). Values are the mean ± SEM; n = 13 cells from two animals for 2 μm CBD and n = 7 cells from one animal for 5 μm CBD.
CBD inhibition of TTX-sensitive and TTX-resistant sodium currents
Previous work has shown the ability of CBD to inhibit a number of different types of heterologously expressed sodium channels (Hill et al., 2014; Patel et al., 2016; Ghovanloo et al., 2018; Mason and Cummins, 2020) as well as native sodium currents in cultured cortical neurons (Hill et al., 2014). Quantifying the concentration-dependent effects of CBD is less straightforward than for many agents because the approach to steady state can be very slow. For example, Hill et al. (2014) found that 10 μm CBD inhibited sodium current in cultured cortical neurons slowly at room temperature, requiring ∼20 min to approach an average steady-state inhibition by ∼70%. Interestingly, Ghovanloo et al. (2018) found that CBD inhibited heterologously expressed Nav1.2 channels in HEK293 cells more slowly and less potently at 33°C than at 20°C, suggesting the importance of testing on native neurons at physiological temperature. Figure 4 shows the time course of inhibition of sodium current in mouse nociceptors at 37°C. The time course of inhibition by 5 μm CBD could be fit by an exponential function with an average time constant of 76 ± 8.7 ms (n = 8), so that steady state was approached in 4–5 min. On washout of CBD, there was progressive reversal of its effects (Fig. 4), but this was so slow that it was incomplete even after >10 min of washing, and we did not attempt to quantify the recovery kinetics.
Figure 4.

Time course of CBD inhibition of voltage-activated sodium current. A, Inhibition by 5 μm CBD of sodium current evoked by a 15 ms step from −70 to 0 mV applied every 5 s. Red line, Fit of a single exponential curve (τ = 65.2 s); inset, current in control and in 5 μm CBD. B, Collected results for the effect of 5 μm CBD on total sodium current after inhibition reached steady state. N = 8 cells from two animals. Red symbol, Mean ± SEM. C, Collected results for the time constant of inhibition. N = 8 cells from two animals. Red symbol, Mean ± SEM.
We were particularly interested in characterizing the effects of CBD on native TTX-resistant sodium channels, because Nav1.8-mediated TTX-resistant sodium current is prominent in small-diameter DRG neurons (Roy and Narahashi, 1992; Rush et al., 1998), carries the majority of the sodium current during action potentials in rat primary nociceptors (Renganathan et al., 2001; Blair and Bean, 2002), and is critical for supporting repetitive firing of small-diameter DRG neurons (Renganathan et al., 2001; Blair and Bean, 2002; Rush et al., 2007). The effects of CBD on this current have not been previously described. Figure 5 shows the results of experiments characterizing the effect of CBD on the TTX-sensitive and TTX-resistant components of sodium current in a mouse nociceptor, taking advantage of the rapid onset and reversibility of TTX inhibition to isolate TTX-S and TTX-R components of current before and after CBD inhibition. Studied with test pulses delivered from a holding potential of −70 mV, CBD inhibited both components of current with similar potency, with 5 μm CBD inhibiting TTX-S current to an average of 9 ± 2% of control and TTX-R current to an average of 15 ± 2% of control (n = 7, p < 0.001 for TTX-S and TTX-R currents).
Figure 5.
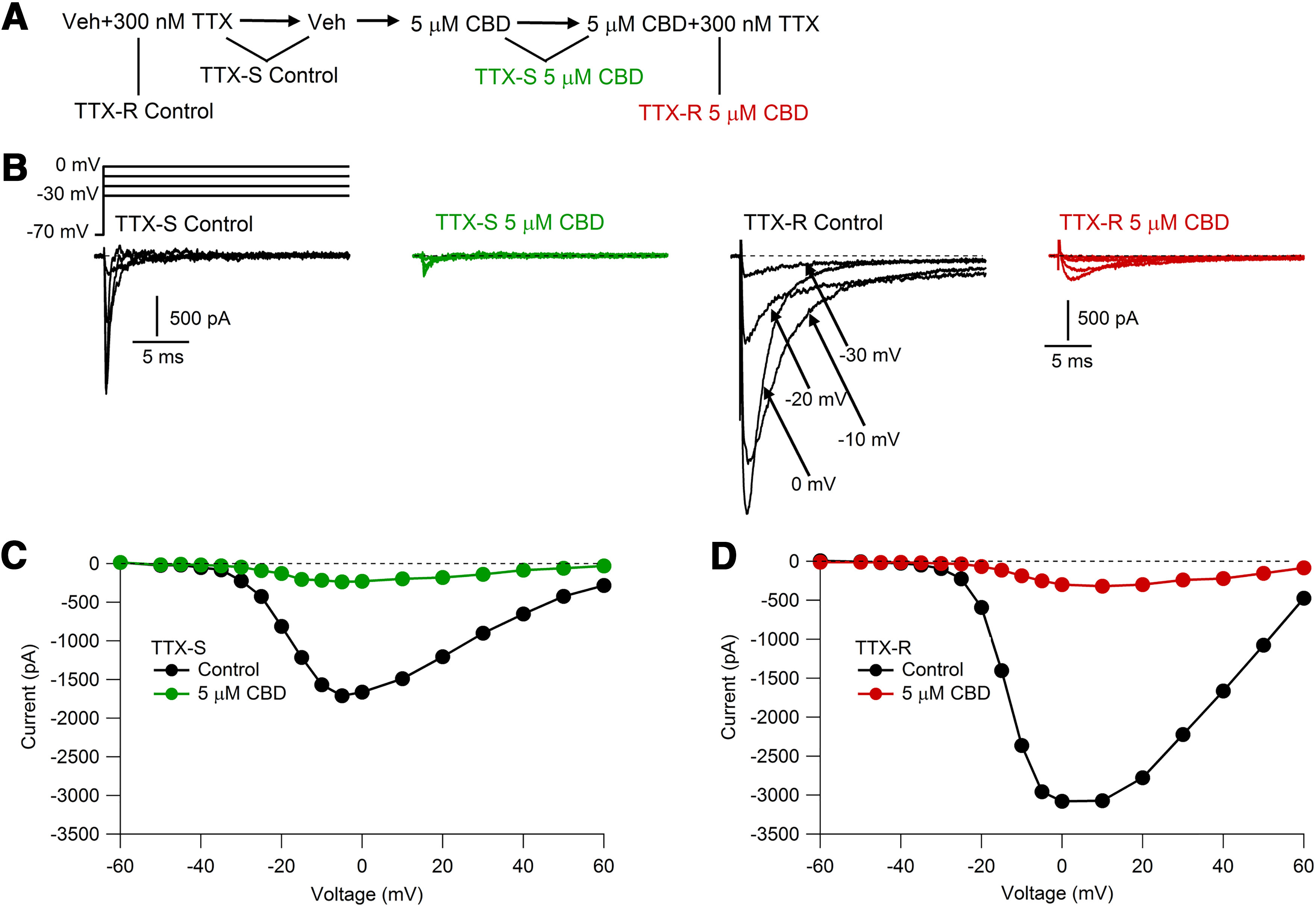
CBD inhibition of TTX-S and TTX-R components of sodium current. A, Solution sequence for isolating TTX-S and TTX-R components before and after CBD. B, Families of TTX-S and TTX-R currents recorded before and after 5 μm CBD application. Depolarizing steps (100 ms) were applied every 5 s from a holding voltage of −70 mV. C, Voltage dependence of peak TTX-S currents before and after 5 μm CBD. D, Voltage dependence of peak TTX-R currents before and after 5 μm CBD application.
Use-dependent inhibition of action potential-evoked sodium currents
We next examined the effects of CBD on TTX-S and TTX-R currents flowing during physiological action potential waveforms, performing action potential clamp experiments with a previously recorded action potential waveform recorded at 37°C. As expected from the ability of TTX-S channels to be activated by smaller depolarizations and with more rapid kinetics, the TTX-S component of overall sodium current flowed earlier in the action potential than the TTX-R current (Fig. 6A,B). To explore possible use-dependent effects on CBD inhibition, the action potential waveform was delivered at frequencies of either 5 or 20 Hz. Both TTX-S and TTX-R current showed substantial frequency-dependent reduction in control solutions, with TTX-S current during the 60th action potential reduced to 72 ± 5% of that evoked by the first action potential during 5 Hz stimulation and to 52 ± 5% during 20 Hz stimulation (n = 8). TTX-R current showed slightly less frequency-dependent reduction, with TTX-R current during the 60th action potential reduced to 80 ± 5% of that evoked by the first action potential during 5 Hz stimulation and to 56 ± 7% during 20 Hz stimulation (n = 8). CBD inhibited both TTX-S and TTX-R components of current in a use-dependent manner. Application of 2 μm CBD reduced the TTX-S current evoked by the first action potential to 45 ± 5% of control (n = 8) and TTX-R current to 67 ± 8% of control (n = 8). After stimulation at 20 Hz, 2 μm CBD reduced TTX-S current during the 60th action potential to 37 ± 6% compared with the current evoked by the 60th action potential in control (and to 18 ± 2% compared with the current evoked by the first action potential in control), and CBD reduced TTX-R current during the 60th action potential to 40 ± 7% compared with the current evoked by the 60th action potential in control (and to 25 ± 7% compared with the current evoked by the first action potential in control). Use-dependent reduction of both TTX-R and TTX-S current was also seen to a lesser extent with 5 Hz stimulation (Fig. 6C). Thus, 2 μm CBD produces both tonic and use-dependent reduction of both TTX-S and TTX-R currents during stimulation by physiological waveforms at physiological frequencies at physiological temperature.
Figure 6.
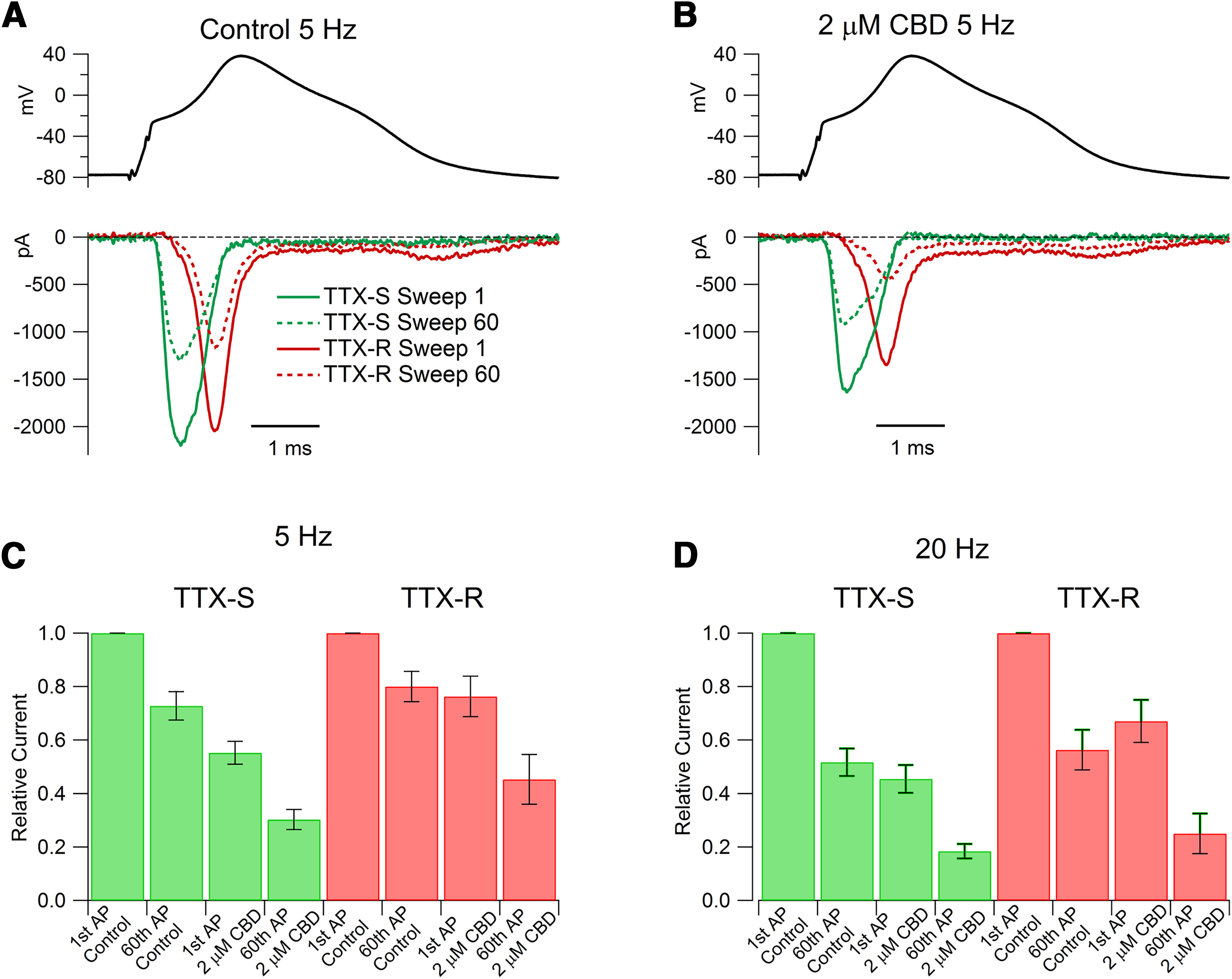
Use-dependent inhibition by CBD of TTX-S and TTX-R currents evoked by action potential waveforms. A, TTX-S and TTX-R sodium currents evoked by action potential waveforms delivered at 5 Hz in a nociceptor in control. A previously recorded action potential (AP) waveform evoked by a 0.2 ms current injection was used as command voltage, with membrane potential held at −80 mV for 10 s before delivery of action potential waveforms repeated at 5 Hz. Solid lines, Currents elicited by the first action potential waveform; dashed lines, currents elicited by the 60th action potential waveform. B, Currents in the same cell recorded in 2 μm CBD. C, Collected results showing the change in action potential-evoked TTX-R and TTX-S sodium currents produced by 5 Hz stimulation in the absence and presence of 2 μm CBD. Values are the mean ± SEM. N = 8 cells from one animal. For TTX-S current: p = 0.0014 for the first AP control and 2 μm CBD; p < 0.0001, between first AP 2 μm CBD and 60th AP 2 μm CBD; for TTX-R current: p = 0.01 for first AP control and 2 μm CBD; p < 0.0001 between first AP 2 μm CBD and 60th AP 2 μm CBD. D, Collected results with 20 Hz stimulation. Values are the mean ± SEM. N = 8 cells from one animal. For TTX-S current: p < 0.0001 for first AP control and 2 μm CBD; p = 0.00,013 between first AP 2 μm CBD and 60th AP 2 μm CBD; for TTX-R current: p = 0.0006 for first AP control and 2 μm CBD; p < 0.0001 between first AP 2 μm CBD and 60th AP 2 μm CBD.
CBD slowing of recovery of availability of TTX-R channels
The use-dependent inhibition of TTX-R current by CBD suggests that channel inhibition is increased during repeated depolarizations and that the extra inhibition does not recover completely in between depolarizations. To explore how CBD alters the recovery of channel availability of TTX-R channels, we used a two-pulse experiment (Fig. 7). A 20 ms conditioning pulse to 0 mV induced inactivation, and after a variable time at −70 mV a test pulse assayed the recovery of channel availability. In control, ∼85% of the channels recovered quickly from inactivation, with a time constant (fit to collected data) of 1.03 ms. Interestingly, however, ∼10% of the channels did not fully recover even in 300 ms, the longest time tested in this protocol. CBD reduced the sodium current evoked by the first pulse to 0 mV and also dramatically slowed the recovery of channel availability of the remaining channels after they experienced the 20 ms depolarization to 0. In the presence of CBD, recovery of availability occurred with two phases, a fast component similar to recovery of channels in control (making up 28% of the total extent of recovery in 300 ms) and a larger, much slower, component that could be fit reasonably well by a time constant of 66 ms.
A reasonable interpretation of this result is that during the 20 ms conditioning step to 0 mV, CBD binds to a fraction of channels that were initially unbound at the holding voltage of −70 mV, presumably reflecting tighter binding to inactivated channels than resting channels, and that the recovery of availability in CBD occurs by the CBD-bound inactivated channels converting to CBD-bound resting channels followed by unbinding of CBD from the channels now in the lower-affinity resting state. With this sequence, the recovery of availability in CBD is slower than in control because it requires an extra unbinding step. It is also possible that the recovery of CBD-bound inactivated channels occurs by CBD first unbinding from the inactivated state followed by recovery from the unbound inactivated state to the unbound resting state. With either route, CBD must unbind for channels to become available, and the rate of recovery is slowed because of this additional step. (In the model depicted in Fig. 12, CBD-bound inactivated channels recover to the unbound resting state mainly by the first route, with additional slowing of recovery because the movement of CBD-bound inactivated states to CBD-bound resting states is slower than the movement of CBD-free inactivated channels to resting channels.)
Figure 12.

Model for CBD interaction with TTX-R Na channels. A, CBD is assumed to bind weakly to R (KR = 5 μm), tightly to F (KF = 0.60 μm), and more tightly to S (KS = 0.15 μm). B–D, Channels move among R, F, and S states with voltage-dependent rate constants chosen to approximate voltage-dependent occupancy in the different states with conditioning pulses of various durations. Channels move from R to F with a forward rate constant of 3/(1 + exp(–(V + 8.7)/6.5) and a backward rate constant of 0.12 + 0.9/(1 + exp(V + 23.7/6.5). Channels move from R to S with a forward constant of 0.004/(1 + exp(–(V + 14.3)/9.3)) and a backward constant of 0.004/(1 + exp((V + 64.3)/9.3)). Channels move from F to S with a forward rate constant of 0.005 and a backward rate constant determined by other rate constants to preserve microscopic reversibility. CBD binds to R with an on rate (KonR) of 0.02 ms−1µm−1 and unbinds with off rate (koffR) of 0.1 ms−1. CBD binds to F with an on rate (KonF) of 0.02 ms−1µm−1 and unbinds with off rate (koffF) of 0.012 ms−1. CBD binds to S with an on rate (KonS) of 0.02 ms−1µm−1 and unbinds with an off rate (koffS) of 0.003 ms−1. The rate constants for movements from R-CBD to F-CBD and F-CBD to S-CBD are the same as for movements from R to F and from F to S. To preserve microscopic reversibility, the rate constant for movement from F-CBD to R-CBD is multiplied by a factor of (koffF/koffR) * (konR/konF) relative to that for F to R, and the rate constant for movement from S-CBD to F-CBD is multiplied by a factor of (koffS/koffF) * (konF/konS) relative to that for S to F. For preserving microscopic reversibility in the cycle of unbound and bound R and S states, the rate constant from R-CBD to S-CBD is the rate constant from R to S divided by the square root of (koffS/koffR) * (konR/konS) and the rate constant S-CBD to R-CBD is the rate constant from S to R multiplied by the square root of (koffS/koffR) * (konR/konS). B, Prediction of voltage dependence of fast inactivation (green) and slow inactivation (blue) at equilibrium if each occurred in the absence of the other. C, Left, Prediction of channel fraction in R state (i.e., available channels) after 50 ms conditioning pulses in control (solid black line) and with 5 μm CBD (solid red line). Dashed red line shows channel availability in CBD normalized to 1 at −100 mV. Right, Occupancy of channels in fast and slow inactivated states in control (solid lines) and occupancy of channels in the corresponding CBD-bound states with 5 μm CBD (dashed lines). D, Left, Prediction of channel fraction in R with the indicated protocol for measuring slow inactivation, with 5 s conditioning pulses followed by 10 ms at −100 mV to allow recovery of channels in the fast inactivated state. Right, Occupancy of channels in fast and slow inactivated states in control (solid lines) and occupancy of channels in the corresponding CBD-bound states with 5 μm CBD (dashed lines).
We then explored how quickly inactivated channels bind CBD to enter slowly recovering states (Fig. 8) by varying the duration of a prepulse to 0 mV followed by 6 ms at −70 mV, a duration that allows nearly complete recovery of unmodified inactivated channels but little recovery of CBD-bound channels. In the presence of CBD, the slowly recovering fraction of channels began to develop within a few milliseconds during depolarization, and the development of slowly recovering channels could be fit by a time constant of 8.5 ms. We interpret this as reflecting the time course with which CBD binds to inactivated states of the channels to produce “extra” inhibition over that present at the holding voltage of −70 mV.
Interestingly, even in control conditions a small but significant fraction of channels entered slowly recovering states during prepulse durations greater than ∼5 ms. In control, the fraction of slowly recovering channels reached an average of 24 ± 1% with a 40 ms prepulse. This can be interpreted as reflecting the entry of unmodified TTX-R channels into a slow inactivated state from which recovery is slow (Roy and Narahashi, 1992; Rush et al., 1998; Blair and Bean, 2002; Choi et al., 2006, 2007; Tripathi et al., 2006; Binshtok et al., 2008). Even with a prepulse of 20 ms, ∼10% of channels enter the slow inactivated state.
The main purpose of the experiments shown in Figures 7 and 8 was to illustrate the action of CBD to place TTX-R channels into a state from which channel availability recovers far more slowly than in control. A secondary observation in these experiments was that the currents in the presence of CBD had slower decay kinetics than in control. For example, the currents evoked by the 20 ms conditioning pulse in Figure 7 decay with a time constant of 0.87 ms in control and with a time constant of 1.40 ms in the presence of 5 μm CBD. The slower decay of currents in the presence of CBD was seen systematically (Fig. 8A), and the difference did not result from the different sizes of the currents (such as might be produced by a series resistance error) because the decay kinetics of currents in control showed little or no change as the size of the test pulse current evoked by a pulse to 0 mV changed over a 10-fold range with different degrees of recovery from inactivation. Whether the slower decay of TTX-R currents reflects differential inhibition by CBD of multiple populations of TTX-R channels with different kinetics or an effect of CBD on kinetics of a homogeneous population of channels remains to be determined and might be best approached using cloned channels.
Altered availability of TTX-R sodium channels with CBD
To determine quantitatively how tightly CBD binds to inactivated channels, we characterized the effect of CBD using conditioning pulses to alter the distribution of channels between resting and inactivated states. With such protocols, tighter binding of drug to inactivated states compared with resting states is manifested by a change in the voltage dependence of channel availability and the shift in the midpoint of channel availability can be used to estimate the Kd (dissociation constant) for drug binding to the inactivated state (Bean et al., 1983). Figure 9A shows results from experiments using 50 ms conditioning pulses, long enough to reach steady state for conventional fast inactivation (Blair and Bean, 2002). With this protocol, 5 μm CBD produced a shift in the midpoint of channel availability of −7.6 ± 0.4 mV (n = 6). Because of the evidence for an important physiological role of slow inactivated states of Nav1.8 channels for controlling nociceptor excitability (Rush et al., 1998; Blair and Bean, 2003; Tripathi et al., 2006; Choi et al., 2007), we also examined longer conditioning pulses. We found that the shift of channel availability became progressively larger with longer conditioning prepulses, with a shift of midpoint of −10.6 ± 0.5 mV (n = 5) for 200 ms conditioning pulses (Fig. 9B) and −15.7 ± 0.6 mV (n = 4) for 5 s conditioning pulses (Fig. 9C). Bupivacaine produced similar but smaller effects on channel availability, with 5 μm bupivacaine producing a −10.9 ± 0.8 mV (n = 6) shift in the midpoint of availability with 5 s conditioning pulses. Thus, the stronger effects of CBD on nociceptor firing compared with bupivacaine are paralleled by stronger effects on channel availability, suggesting tighter binding of CBD to inactivated states of TTX-R channels compared with bupivacaine.
Figure 9.

Alteration of availability of TTX-R sodium channels by CBD. A, Voltage-dependent availability of TTX-R sodium channels measured by a 3 ms test pulse to 0 mV following 50 ms conditioning pulses in the absence (black) and presence (red) of 5 μm CBD. Availability is normalized to the maximal test pulse current measured in each condition. Symbols, Mean ± SEM. Solid line, Boltzmann equation, 1/(1 + exp((V – Vh)/k)), where V is holding voltage, Vh is voltage of half-maximal availability, and k is the slope factor. Control: Vh = −22.2 ± 1.4 mV, k = 4.6 ± 0.1 mV; 5 μm CBD: Vh = −29.8 ± 1.7 mV, k = 6.1 ± 0.4 mV. Values of Vh and k are mean values of fits to data from individual cells (n = 6 cells from three animals) and ΔVh is the mean ± SEM for measurements in individual cells. B, Availability measured using 200 ms conditioning pulses. Symbols and statistics as in A. Control: Vh = −32.3 ± 0.4 mV, k = 4.7 ± 0.4 mV; 5 μm CBD: Vh = −43.0 ± 0.4 mV, k = 7.6 ± 0.4 mV (n = 5 cells from three animals). C, Availability measured using 5 s conditioning pulses. Symbols and statistics as in A. Control: Vh = −36.7 ± 2.2 mV, k = 4.4 ± 0.2 mV; 5 μm CBD: Vh = −52.5 ± 2.1 mV, k = 5.0 ± 0.0 mV (n = 4 cells from three animals).
Time course of recovery from slow inactivated states
The larger shifts in midpoint of channel availability with longer conditioning prepulses raises the possibility that CBD binds tightly to slow inactivated channels, which would be increasingly populated during longer conditioning pulses. We next designed protocols to test this idea. First, we tested the effect of CBD on the recovery of availability using conditioning pulses designed to put channels into slow inactivated states. Figure 10 shows the effect of CBD on the recovery of availability following long (300 ms or 10 s) depolarizations to 0 mV. To decrease the time required to assay the time course of recovery following long conditioning pulses, we used a pulse protocol in which the time course of recovery was determined in a single sweep, assaying availability by multiple short test pulses presented at a range of times after the long conditioning pulse. After a conditioning pulse of 300 ms under control conditions, channels recovered availability with a dominant time constant of 730 ms, far more slowly than the dominant time constant of 1 ms for the recovery after a conditioning pulse of 20 ms (Fig. 7). Following the 300 ms conditioning pulse, the time constant of 730 ms accounted for ∼93% of the recovery of availability, with ∼7% of the channels recovering within 2–3 ms. Thus, it appears that ∼93% of channels end up in a slow inactivated state after a 300 ms pulse to 0 mV. In the presence of 5 μm CBD, the recovery of availability after a 300 ms conditioning pulse was slower, with a dominant time constant of ∼2 s. This suggests that CBD binds to slow inactivated channels and that the recovery of availability of CBD-bound slow inactivated states is even slower than recovery from normal CBD-free slow inactivated states.
The protocol incorporating the conditioning pulse and multiple test pulses into a single sweep made it feasible to extend the analysis to even longer conditioning pulses. Figure 10C shows an experiment examining the recovery of availability following a 10 s conditioning pulse. Interestingly, the recovery of availability in control was substantially slower following a 10 s conditioning pulse (dominant time constant, 2.72 s) than after a 300 ms conditioning pulse (dominant time constant, 0.73 s). This suggests that there are actually multiple slow inactivated states, from which recovery has different time courses. With the 10 s conditioning pulses, the recovery in CBD was even slower, with a dominant time constant of 9.57 s, consistent with the idea that the slow inactivated states produced by a 10 s conditioning pulse also bind CBD and that these CBD-bound slow inactivated states are also stabilized relative to the CBD-free slow inactivated states.
CBD binding to slow versus fast inactivated states
We next attempted to estimate quantitatively the binding affinity of CBD to fast and slow inactivated states. Figure 11 shows the results of an experiment designed to compare the voltage dependence of fast inactivation and slow inactivation in the same cell. The voltage dependence of fast inactivation was assayed using 50 ms prepulses to various voltages followed by a test pulse to +10 mV. The voltage dependence of channel availability could be fit well by a Boltzmann function with a midpoint of −21 and a slope factor of 5.0 mV (Fig. 11A). Figure 11B shows the results in the same cell of a protocol designed to measure the voltage dependence of slow inactivation, using 5 s conditioning pulses to various voltages followed by 10 ms at −100 mV (long enough for complete recovery from fast inactivation but little recovery from slow inactivation) and then a test pulse. The voltage dependence of slow inactivation could be fit well by a Boltzmann function (going to zero) with a midpoint of –36.9 and a slope factor of 5.5 mV. Thus, slow inactivation has a midpoint ∼16 mV hyperpolarized to that of fast inactivation (in collected results, −17 ± 1.4 mV more negative; n = 6). It should be noted that the measurement of fast inactivation is not perfect because there is almost certainly some entry of channels into slow inactivation during the 50 ms conditioning pulse, because with 40 ms steps to 0 mV ∼20% of channels enter slow inactivation (Fig. 6). Thus, if pure fast inactivation could be measured, it would likely have a slightly depolarized midpoint compared with that measured with 50 ms conditioning pulses.
In these experiments, we also measured the effect of CBD on the voltage dependence of channel availability measured with the two protocols. Consistent with the results in the protocols in Figure 9, CBD altered the midpoint of channel availability much more (−13.8 ± 1.5 mV) with the slow inactivation protocol than with the fast inactivation protocol (−7.6 ± 0.4 mV, measured in the same six cells).
Model for state-dependent CBD binding to Nav1.8 channels
These results suggest substantially tighter binding of CBD to slow inactivated states of the channel than to fast inactivated states. However, the pulse protocols do not cleanly separate fast inactivated from slow inactivated states. And although the shift in the midpoint of availability is smaller in the protocol using 50 ms conditioning pulses, this could in principle be because 50 ms is too little time for CBD to reach steady-state binding (Karoly et al., 2010). To better quantify the affinity of CBD to the different states, we developed a model incorporating state-dependent CBD binding that gave a reasonable approximation of the experimental results (Fig. 12) in terms of both voltage dependence and time dependence of the state-dependent effects, which can better distinguish binding to fast versus slow inactivated states (Karoly et al., 2010). Voltage-dependent rate constants for movement of channels between resting (R), fast inactivated (F), and slow inactivated (S) channels were chosen to approximate experimental data for the voltage-dependent occupancy of the various states with conditioning pulses of various durations. Then, rate constants for CBD binding and unbinding to each state were developed that could approximately predict the shifts in the midpoint of availability seen experimentally with short or long prepulses. The best matches to experimental data were obtained by relatively weak binding to R (KR = 5 μm), much tighter binding to F (KF = 0.60 μm), and even tighter binding to S (KS = 0.15 μm). The model is clearly oversimplified, because detailed kinetic models of fast inactivation would require multiple fast inactivated states corresponding to channels with different positions of the S4 voltage-sensing regions of the channel (distinguishing, for example, inactivation from various closed states and the open state). The assumption of a single slow inactivated state is also clearly oversimplified, because experiments like those in Figure 10 examining the rate of recovery from slow inactivated states show different rates depending on the conditioning pulse duration; this is similar to previous observations in Nav1.1 and Nav1.2 channels (Toib et al., 1998) and suggests the existence of multiple slow inactivated states (Vilin and Ruben, 2001). Nevertheless, the general result from the model that CBD binding to slow inactivated states must occur with higher affinity than binding to fast inactivated states seems unlikely to be changed by more detailed models incorporating multiple fast and slow inactivated states.
Although the model is designed mainly to interpret quasi-steady-state occupancy of the various states of the channel and not detailed kinetics, the chosen rate constants provide generally reasonable simulations of the kinetics of development and recovery of availability in different conditions (e.g., slowing of the recovery of availability from CBD-bound states). In the model, rate constants for the binding and unbinding of CBD from various states are much faster than the kinetics of development of CBD inhibition and recovery from CBD inhibition when CBD is washed on and washed off cells (Fig. 4). We believe the slow kinetics of wash-on and wash-off likely reflect slow equilibration of CBD between the aqueous solution and the membrane. Because of the highly lipophilic nature of CBD, the membrane concentration of CBD is expected to be far higher than the aqueous concentration once steady state is reached, which takes 4–5 min (Fig. 5). The kinetics in experiments using voltage changes to alter CBD binding to the various states of the channel, which are much faster, likely reflect state-dependent binding and unbinding of CBD to the channel after a steady-state CBD concentration has been achieved in the membrane. The model uses the CBD concentrations in the external aqueous solution since these are known exactly. A more detailed model might use the CBD concentration in the membrane, but this is unknown. In the simplest case, the membrane concentration might scale linearly with the concentration in the external aqueous solution, but it seems possible that the relationship might be more complicated.
Altered availability of TTX-R sodium channels and reduced firing with submicromolar CBD
Although the model in Figure 12 suggests CBD binding to slow inactivated states with a Kd of ∼150 nm, it is based on experiments using much higher (5 μm) concentrations of CBD. To test the model more rigorously, we did a series of voltage-clamp experiments with lower concentrations of CBD. First, we examined inhibition of the TTX-R sodium current using a voltage protocol designed to put a substantial fraction of channels into slow inactivated states, with a 2 s conditioning pulse at −40 mV (expected from the results shown in Fig. 11B to put approximately one-third of the channels into the slow inactivated state) followed by a test pulse to +10 mV. We saw effective inhibition of current by both 300 nm CBD and 1 μm CBD using this protocol (Fig. 13A). Because the action of these concentrations of CBD was slow, we interspersed recordings examining CBD inhibition with “sham-application” cells in which control (DMSO-containing) solution was applied for the same amount of time. With 5 min applications of CBD, currents were reduced to 0.69 ± 0.09 (n = 6) of predrug current by 300 nm CBD and to 0.58 ± 0.06 (n = 6) by 1 μm CBD, with minimal effects of equivalent sham application of control solution (0.96 ± 0.03 of the initial value after 5 min; n = 6; p = 0.02 for comparison between 300 nm CBD and sham application; p = 0.003 for comparison between 1 μm CBD and sham application).
A somewhat surprising feature of these experiments was that the rate of inhibition by 1 μm CBD was not much different from that by 300 nm CBD. Although the reasons for this are not clear, it suggests that the rate-limiting step is not CBD binding to the channel but some other process, perhaps partitioning of CBD into the membrane or into the cell. Because of the possibility that a 5 min application was not enough to reach steady state, especially with low concentrations of CBD, we also did a series of experiments in which we preincubated cells with 300 nm CBD for 1–2 h. In these population-study experiments, the substantial cell-to-cell variation in the size of the TTX-R sodium current precluded making a simple comparison of current sizes. Therefore, we used protocols similar to those in Figures 10 and 11B that were designed to assay the fraction of channels that enters slowly recovering states during long depolarizations to −40 mV (slow inactivated states in control, and with CBD present, a combination of CBD-free slow inactivated states and CBD-bound inactivated states). Figure 13B shows results with a protocol in which a 2 s step to −40 mV was followed by a series of 2 ms steps to 0 mV delivered at various times after the return to −70 mV to assay the time course of recovery. As expected, in control a substantial fraction of channels (on average, 32 ± 4%; n = 12) recovered slowly. The fraction of slowly recovering channels was significantly larger in cells incubated in 300 nm CBD (52 ± 5%, n = 11), consistent with substantial binding of 300 nm CBD to slow inactivated states. We also measured the effect of 300 nm CBD incubation on the voltage dependence of channel availability using a protocol to assay slow inactivation as in Figure 11B, with a 5 s conditioning pulses to various voltages followed by 10 ms at −100 mV and then a test pulse. As illustrated in Figure 13C, in these experiments there was an approximately −5 mV shift in the midpoint of curves in cells incubated with 300 nm CBD (Vh = −43.6 ± 1.9 mV; n = 11) compared with control (Vh = −38.3 ± 1.2 mV; n = 12; p = 0.02). We found that this shift is remarkably close to the predictions of the model in Figure 12, which predicts Vh = −39.1 for control and Vh = −43.9 with 300 nm CBD (Fig. 13D).
We next examined whether submicromolar concentrations of CBD can modify the firing of nociceptors. Figure 14 shows the effects of 500 nm CBD on nociceptor firing. These experiments showed that 500 nm CBD had little effect on firing evoked by small current injections but significantly reduced the ability of cells to fire repetitively throughout 1 s current injections with current injections of 90 pA and above. In collected results (Fig. 14B), 500 nm CBD applied for 4 min reduced the number of action potentials evoked by 1 s 100 pA current injections from 23.1 ± 5.6 in control to 7.6 ± 2.5 in 500 nm CBD (n = 7, p = 0.01). This effect was much greater than the small changes seen during sham applications of control solution for the same period of time (Fig. 14C), where the number of action potentials changed from 22.4 ± 5.8 to 19.7 ± 5.9 during the 4 min sham application (p = 0.3). The effect of 500 nm CBD to reduce maintained firing during moderately strong current injections seems consistent with high-affinity binding to slow inactivated states reached during depolarizations to near −40 mV and above, although there may well be additional effects of 500 nm CBD on TTX-S channels or other channels influencing firing.
Figure 14.

Inhibition of action potential firing in mouse nociceptors by 500 nM CBD. A, Action potential firing elicited by 1 s current injections at 40 and 90 pA before and after 4 min wash-on of 500 nM CBD. B, Collected results for number of action potentials during 1 s current injections before and after 4 min in 500 nM CBD. N = 7 cells from three animals; p = 0.047 for 90 pA; p = 0.01 for 100 pA. C, Same for sham application in cells in which current was recorded for >4 min in control solution. N = 7 cells from three animals. Data are shown as the mean ± SEM.
Discussion
These results show that CBD at concentrations as low as 500 nm significantly reduces the excitability of mouse nociceptors, with a concentration of 2 μm inhibiting firing very effectively, producing more effective inhibition than the same concentration of bupivacaine, one of the most potent local anesthetics. CBD inhibits both TTX-sensitive and TTX-resistant sodium channels, which play complementary roles in controlling nociceptor excitability (Cummins et al., 2007; Bennett et al., 2019).
We find that CBD binds tightly to slow inactivated states of TTX-R channels. TTX-R channels are unusual in that the voltage dependence of slow inactivation is hyperpolarized compared with fast inactivation (Blair and Bean, 2003; Fig. 11), the opposite of most other sodium channels (Vilin and Ruben, 2001; Ulbricht, 2005; Ghovanloo et al., 2016). In the neurons we studied, ∼30% of channels enter slow inactivated states at −40 mV (Fig. 11); in some other subtypes of small DRG neurons, 20–60% of channels can be in the slow inactivated state even at −60 mV (Cardenas et al., 2006) and up to 90% at −50 mV (Tripathi et al., 2006). Also, entry into slow inactivated states of TTX-R channels is rapid compared with slow inactivation in other types of sodium channels, so that with repeated 5 ms depolarizations or action potentials, cumulative inactivation can reduce currents substantially at frequencies of 1–10 Hz (Fazan et al., 2001; Tripathi et al., 2006; Fig. 6). As a result, slow inactivation of TTX-R current can limit repetitive firing induced by depolarizing stimuli like capsaicin (Blair and Bean, 2003) and can slow axonal conduction mediated by TTX-R sodium channels (De Col et al., 2008). Conversely, some excitation-enhancing agents like interleukin-1β and tumor necrosis factor activate second messenger pathways that disrupt slow inactivation of TTX-R channels (Binshtok et al., 2008; Gudes et al., 2015). The significance of the slow inactivation of TTX-R channels under physiological conditions suggests that pharmacological agents targeting these states may be especially effective in reducing repetitive firing of nociceptors. In fact, carbamazepine, which is effective for treating neuropathic pain in a subset of patients, also interacts with higher affinity with slow-inactivated than fast-inactivated states of TTX-R channels (Rush and Elliott, 1997; Cardenas et al., 2006), although with far weaker slow-inactivated state binding (estimated Kd, 30 μm; Cardenas et al., 2006) than for CBD (estimated Kd, 0.15 μm).
The molecular mechanism of slow inactivation has been studied extensively in Nav1.4 and Nav1.5 channels, where it appears to involve movements in the outer pore region (Townsend and Horn, 1997; Vilin et al., 1999) and immobilization of voltage sensor regions (Silva and Goldstein, 2013). However, the distinctive properties of slow inactivation in Nav1.8 channels suggest that its structural basis might be different from those of other sodium channels. So far, the only structurally relevant information on slow inactivation in Nav1.8 channels is that the extent of slow inactivation occurring during strong depolarizations can be regulated by calmodulin (Choi et al., 2006, 2007), which may underlie the variable properties in different types of DRG neurons. Our results suggest that structures of CBD-bound Nav1.8 channels would likely be in the slow inactivated state, which would be stabilized relative to other states by tight CBD binding. A high-resolution x-ray crystallography study of CBD bound to a bacterial voltage-dependent sodium channel showed a binding site near the pore, located in a hydrophobic “fenestration” that can plausibly account for entry of the hydrophobic CBD molecule from the membrane phase (Sait et al., 2020). Entry of hydrophobic molecules into the pore through such lateral fenestrations in the pore was previously proposed for lidocaine (Gamal El-Din et al., 2018), providing a concrete molecular mechanism for the proposal by Hille (1977) for such a pathway for the binding of hydrophobic local anesthetic molecules to mammalian sodium channels. A similar location for CBD binding in mammalian sodium channels seems plausible based on homology modeling (Sait et al., 2020), although CBD inhibition of the bacterial sodium channel is significantly weaker (IC50, ∼17 μm) than for mammalian sodium channels, so it is unclear how good a model the CBD binding to the bacterial channel will be for high-affinity binding to inactivated states of mammalian channels.
Further work will be needed to determine the relative importance of CBD inhibition of TTX-S and TTX-R channels for inhibiting nociceptor firing. With 500 nm CBD, there was little effect on firing of the first few action potentials, and the main effect was the reduction of maintained firing during relatively large current injections. While this is consistent with CBD binding to slow inactivated states of TTX-R channels, the application of TTX can also reduce maintained firing (our unpublished observations), so combined action of CBD on both TTX-R and TTX-S channels is likely important. Studies on cloned Nav1.1–Nav1.7 channels show that CBD binds to inactivated states with higher affinity than resting states (Ghovanloo et al., 2018), but studies on native TTX-S channels in nociceptors have not yet been done. It remains to be seen whether particularly tight binding of CBD to slow inactivated states also occurs in TTX-S channels. In general, the inhibition of both TTX-S and TTX-R channels may be a desirable characteristic for an effective analgesic (Goodwin and McMahon, 2021).
Similar to other antiepileptic drugs like phenytoin and carbamazepine, CBD inhibits not only sodium channels but also voltage-dependent potassium channels and calcium channels (Ross et al., 2008; Hill et al., 2014; Patel et al., 2016; Ghovanloo et al., 2018; Le Marois et al., 2020; Orvos et al., 2020), and has effects on a wide variety of other signaling molecules (Gray and Whalley, 2020; Watkins, 2019; Senn et al., 2020), including TRPV1 (Iannotti et al., 2014), TRPV2 (Qin et al., 2008; Neumann-Raizel et al., 2019), and various G-protein-coupled receptors (de Almeida and Devi, 2020). Although CBD affects many channels and receptors, the fact that well defined binding sites can be seen in structures of CBD-bound proteins (Pumroy et al., 2019; Sait et al., 2020), along with the dramatic state dependence of binding, suggests that its effects are mediated by specific effects on the operation of particular channels and receptors and not by completely nonspecific effects on membranes and membrane proteins. It is difficult to know which of these effects, or combination of effects, is most important for the action of CBD on either epilepsy and pain. However, given the primary importance of voltage-dependent sodium channels in controlling neuronal excitability, it seems likely that effects on sodium channels are of major importance in pain. As for other antiepileptic sodium channel inhibitors like carbamazepine, phenytoin, and lamotrigine, and amitriptyline, used to treat neuropathic pain, the clinical effects and tolerability of CBD likely depend critically on relatively small differences in actions on particular channel types together with the importance of each affected channel for the excitability of particular neuronal types and other excitable cells. For example, it may be significant that the IC50 for CBD inhibition of cardiac Nav1.5 channels is several-fold higher than for neuronal TTX-sensitive channels (Ghovanloo et al., 2018) or Nav1.8 channels (this study). The strong dependence of CBD binding on the gating state of sodium channels and the strong dependence of gating states on membrane voltage are likely to be key factors in determining the effects on different types of neurons, as well as on other excitable cells like heart muscle. Recent experiments showed that CBD is much more effective in inhibiting resurgent sodium current generated by Nav1.2 channels than for inhibiting transient current through these or other channels (Mason and Cummins, 2020), which may help to explain potency in epilepsy. Such strong state-dependent actions are impossible to predict a priori, but, once known, can help in designing compounds with improved therapeutic ratios. The particularly strong interaction of CBD with the slow inactivated state of Nav1.8 channels may offer such an opportunity.
The intrinsic potency of CBD for inhibiting nociceptor firing relative to bupivacaine highlights the importance of understanding the complex pharmacokinetics and bioavailability of CBD when applied in vivo by various routes (Millar et al., 2019). Because CBD is highly lipid soluble (Ghovanloo et al., 2018), it likely partitions strongly into membranes and lipid-rich environments like the brain and myelinated nerves, and the concentrations actually seen by channels in central neurons or peripheral neurons with in vivo application are presently essentially impossible to estimate. Possibly local application (e.g., by dermal application) could be an effective route for producing localized analgesia. However, while our results show effective CBD inhibition of firing in nociceptor cell bodies, the excitability of peripheral terminals and axons may well depend on different combinations of ion channels (Goodwin and McMahon, 2021), and CBD inhibition of excitability in these physiologically important cell regions remains to be characterized. Also, while use of dissociated cells allows the application of well defined concentrations of CBD and detailed analysis of effects on ion channels, the process of dissociation and maintenance in vitro (including exposure to NGF) almost certainly alters the electrical properties of the neurons, which may have characteristics more like those in damaged or inflamed tissue than normal tissue.
Footnotes
This work was supported by National Institutes of Health Grants NS-36855 and NS-110860, and the Charles R. Broderick III Phytocannabinoid Research Initiative.
The authors declare no competing financial interests.
References
- Abraham AD, Leung EJY, Wong BA, Rivera ZMG, Kruse LC, Clark JJ, Land BB (2020) Orally consumed cannabinoids provide long-lasting relief of allodynia in a mouse model of chronic neuropathic pain. Neuropsychopharmacol 45:1105–1114. 10.1038/s41386-019-0585-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argueta DA, Ventura CM, Kiven S, Sagi V, Gupta K (2020) A balanced approach for cannabidiol use in chronic pain. Front Pharmacol 11:561. 10.3389/fphar.2020.00561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP, Cohen CJ, Tsien RW (1983) Lidocaine block of cardiac sodium channels. J Gen Physiol 81:613–642. 10.1085/jgp.81.5.613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DL, Clark AJ, Huang J, Waxman SG, Dib-Hajj SD (2019) The role of voltage-gated sodium channels in pain signaling. Physiol Rev 99:1079–1151. 10.1152/physrev.00052.2017 [DOI] [PubMed] [Google Scholar]
- Binshtok AM, Wang H, Zimmermann K, Amaya F, Vardeh D, Shi L, Brenner GJ, Ji R-R, Bean BP, Woolf CJ, Samad TA (2008) Nociceptors are interleukin-1β sensors. J Neurosci 28:14062–14073. 10.1523/JNEUROSCI.3795-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair NT, Bean BP (2002) Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J Neurosci 22:10277–10290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair NT, Bean BP (2003) Role of tetrodotoxin-resistant Na+ current slow inactivation in adaptation of action potential firing in small-diameter dorsal root ganglion neurons. J Neurosci 23:10338–10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas CA, Cardenas CG, de Armendi AJ, Scroggs RS (2006) Carbamazepine interacts with a slow inactivation state of NaV1.8-like sodium channels. Neurosci Lett 408:129–134. 10.1016/j.neulet.2006.08.070 [DOI] [PubMed] [Google Scholar]
- Carter BC, Bean BP (2009) Sodium entry during action potentials of mammalian neurons: incomplete inactivation and reduced metabolic efficiency in fast-spiking neurons. Neuron 64:898–909. 10.1016/j.neuron.2009.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J-S, Hudmon A, Waxman SG, Dib-Hajj SD (2006) Calmodulin regulates current density and frequency-dependent inhibition of sodium channel Nav1.8 in DRG neurons. J Neurophysiol 96:97–108. 10.1152/jn.00854.2005 [DOI] [PubMed] [Google Scholar]
- Choi J-S, Dib-Hajj SD, Waxman SG (2007) Differential slow inactivation and use-dependent inhibition of Nav1.8 channels contribute to distinct firing properties in IB4+ and IB4- DRG neurons. J Neurophysiol 97:1258–1265. 10.1152/jn.01033.2006 [DOI] [PubMed] [Google Scholar]
- Costa B, Colleoni M, Conti S, Parolaro D, Franke C, Trovato AE, Giagnoni G (2004) Oral anti-inflammatory activity of cannabidiol, a non-psychoactive constituent of cannabis, in acute carrageenan-induced inflammation in the rat paw. Naunyn Schmiedebergs Arch Pharmacol 369:294–299. 10.1007/s00210-004-0871-3 [DOI] [PubMed] [Google Scholar]
- Costa B, Trovato AE, Comelli F, Giagnoni G, Colleoni M (2007) The non-psychoactive cannabis constituent cannabidiol is an orally effective therapeutic agent in rat chronic inflammatory and neuropathic pain. Eur J Pharmacol 556:75–83. 10.1016/j.ejphar.2006.11.006 [DOI] [PubMed] [Google Scholar]
- Cummins TR, Sheets PL, Waxman SG (2007) The roles of sodium channels in nociception: implications for mechanisms of pain. Pain 131:243–257. 10.1016/j.pain.2007.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida DL, Devi LA (2020) Diversity of molecular targets and signaling pathways for CBD. Pharmacol Res Perspect 8:e00682. 10.1002/prp2.682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Col R, Messlinger K, Carr RW (2008) Conduction velocity is regulated by sodium channel inactivation in unmyelinated axons innervating the rat cranial meninges. J Physiol 586:1089–1103. 10.1113/jphysiol.2007.145383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Gregorio D, McLaughlin RJ, Posa L, Ochoa-Sanchez R, Enns J, Lopez-Canul M, Aboud M, Maione S, Comai S, Gobbi G (2019) Cannabidiol modulates serotonergic transmission and reverses both allodynia and anxiety-like behavior in a model of neuropathic pain. Pain 160:136–150. 10.1097/j.pain.0000000000001386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R, Scheffer IE, Thiele EA, Wright S (2017) Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N Engl J Med 376:2011–2020. 10.1056/NEJMoa1611618 [DOI] [PubMed] [Google Scholar]
- Devinsky O, Patel AD, Cross JH, Villanueva V, Wirrell EC, Privitera M, Greenwood SM, Roberts C, Checketts D, VanLandingham KE, Zuberi SM (2018a) Effect of cannabidiol on drop seizures in the Lennox-Gastaut syndrome. N Engl J Med 378:1888–1897. 10.1056/NEJMoa1714631 [DOI] [PubMed] [Google Scholar]
- Devinsky O, Patel AD, Thiele EA, Wong MH, Appleton R, Harden CL, Greenwood S, Morrison G, Sommerville K (2018b) Randomized, dose-ranging safety trial of cannabidiol in Dravet syndrome. Neurology 90:e1204–e1211. 10.1212/WNL.0000000000005254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O, Nabbout R, Miller I, Laux L, Zolnowska M, Wright S, Roberts C (2019) Long-term cannabidiol treatment in patients with Dravet syndrome: an open-label extension trial. Epilepsia 60:294–302. 10.1111/epi.14628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazan RJ, Whiteis CA, Chapleau MW, Abboud FM, Bielefeldt K (2001) Slow inactivation of sodium currents in the rat nodose neurons. Auton Neurosci 87:209–216. 10.1016/S1566-0702(00)00281-2 [DOI] [PubMed] [Google Scholar]
- Franco V, Perucca E (2019) Pharmacological and therapeutic properties of cannabidiol for epilepsy. Drugs 79:1435–1454. 10.1007/s40265-019-01171-4 [DOI] [PubMed] [Google Scholar]
- Gallily R, Yekhtin Z, Hanuš LO (2018) The anti-inflammatory properties of terpenoids from cannabis. Cannabis Cannabinoid Res 3:282–290. 10.1089/can.2018.0014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamal El-Din TM, Lenaeus MJ, Zheng N, Catterall WA (2018) Fenestrations control resting-state block of a voltage-gated sodium channel. Proc Natl Acad Sci U S A 115:13111–13116. 10.1073/pnas.1814928115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghovanloo M-R, Aimar K, Ghadiry-Tavi R, Yu A, Ruben PC (2016) Physiology and pathophysiology of sodium channel inactivation. Curr Top Membr 78:479–509. 10.1016/bs.ctm.2016.04.001 [DOI] [PubMed] [Google Scholar]
- Ghovanloo M-R, Shuart NG, Mezeyova J, Dean RA, Ruben PC, Goodchild SJ (2018) Inhibitory effects of cannabidiol on voltage-dependent sodium currents. J Biol Chem 293:16546–16558. 10.1074/jbc.RA118.004929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin G, McMahon SB (2021) The physiological function of different voltage-gated sodium channels in pain. Nat Rev Neurosci 22:263–274. 10.1038/s41583-021-00444-w [DOI] [PubMed] [Google Scholar]
- Gray RA, Whalley BJ (2020) The proposed mechanisms of action of CBD in epilepsy. Epileptic Disord 22:10–15. 10.1684/epd.2020.1135 [DOI] [PubMed] [Google Scholar]
- Gudes S, Barkai O, Caspi Y, Katz B, Lev S, Binshtok AM (2015) The role of slow and persistent TTX-resistant sodium currents in acute tumor necrosis factor-α-mediated increase in nociceptors excitability. J Neurophysiol 113:601–619. 10.1152/jn.00652.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammell DC, Zhang LP, Ma F, Abshire SM, McIlwrath SL, Stinchcomb AL, Westlund KN (2016) Transdermal cannabidiol reduces inflammation and pain-related behaviours in a rat model of arthritis. Eur J Pain 20:936–948. 10.1002/ejp.818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AJ, Jones NA, Smith I, Hill CL, Williams CM, Stephens GJ, Whalley BJ (2014) Voltage-gated sodium (NaV) channel blockade by plant cannabinoids does not confer anticonvulsant effects per se. Neurosci Lett 566:269–274. 10.1016/j.neulet.2014.03.013 [DOI] [PubMed] [Google Scholar]
- Hille B (1977) Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol 69:497–515. 10.1085/jgp.69.4.497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannotti FA, Hill CL, Leo A, Alhusaini A, Soubrane C, Mazzarella E, Russo E, Whalley BJ, Di Marzo V, Stephens GJ (2014) Nonpsychotropic plant cannabinoids, cannabidivarin (CBDV) and cannabidiol (CBD), activate and desensitize transient receptor potential vanilloid 1 (TRPV1) channels in vitro: potential for the treatment of neuronal hyperexcitability. ACS Chem Neurosci 5:1131–1141. 10.1021/cn5000524 [DOI] [PubMed] [Google Scholar]
- Jones NA, Hill AJ, Smith I, Bevan SA, Williams CM, Whalley BJ, Stephens GJ (2010) Cannabidiol displays antiepileptiform and antiseizure properties in vitro and in vivo. J Pharmacol Exp Ther 332:569–577. 10.1124/jpet.109.159145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan JS, Stella N, Catterall WA, Westenbroek RE (2017) Cannabidiol attenuates seizures and social deficits in a mouse model of Dravet syndrome. Proc Natl Acad Sci U S A 114:11229–11234. 10.1073/pnas.1711351114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karoly R, Lenkey N, Juhasz AO, Vizi ES, Mike A (2010) Fast- or slow-inactivated state preference of Na+ channel inhibitors: a simulation and experimental study. PLoS Comput Biol 6:e1000818. 10.1371/journal.pcbi.1000818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AA, Shekh-Ahmad T, Khalil A, Walker MC, Ali AB (2018) Cannabidiol exerts antiepileptic effects by restoring hippocampal interneuron functions in a temporal lobe epilepsy model. Br J Pharmacol 175:2097–2115. 10.1111/bph.14202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprairie RB, Bagher AM, Kelly MEM, Denovan-Wright EM (2015) Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br J Pharmacol 172:4790–4805. 10.1111/bph.13250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Marois M, Ballet V, Sanson C, Maizières M-A, Carriot T, Chantoiseau C, Partiseti M, Bohme GA (2020) Cannabidiol inhibits multiple cardiac ion channels and shortens ventricular action potential duration in vitro. Eur J Pharmacol 886:173542. 10.1016/j.ejphar.2020.173542 [DOI] [PubMed] [Google Scholar]
- Liu PW, Blair NT, Bean BP (2017) Action potential broadening in capsaicin-sensitive DRG neurons from frequency-dependent reduction of Kv3 current. J Neurosci 37:9705–9714. 10.1523/JNEUROSCI.1703-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maione S, Piscitelli F, Gatta L, Vita D, De Petrocellis L, Palazzo E, de Novellis V, Di Marzo V (2011) Non-psychoactive cannabinoids modulate the descending pathway of antinociception in anaesthetized rats through several mechanisms of action. Br J Pharmacol 162:584–596. 10.1111/j.1476-5381.2010.01063.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malfait AM, Gallily R, Sumariwalla PF, Malik AS, Andreakos E, Mechoulam R, Feldmann M (2000) The nonpsychoactive cannabis constituent cannabidiol is an oral anti-arthritic therapeutic in murine collagen-induced arthritis. Proc Natl Acad Sci U S A 97:9561–9566. 10.1073/pnas.160105897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason ER, Cummins TR (2020) Differential inhibition of human Nav1.2 resurgent and persistent sodium currents by cannabidiol and GS967. Int J Mol Sci 21:2454. 10.3390/ijms21072454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R, Shani A, Edery H, Grunfeld Y (1970) Chemical basis of hashish activity. Science 169:611–612. 10.1126/science.169.3945.611 [DOI] [PubMed] [Google Scholar]
- Millar SA, Stone NL, Bellman ZD, Yates AS, England TJ, O'Sullivan SE (2019) A systematic review of cannabidiol dosing in clinical populations. Br J Clin Pharmacol 85:1888–1900. 10.1111/bcp.14038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller I, Scheffer IE, Gunning B, Sanchez-Carpintero R, Gil-Nagel A, Perry MS, Saneto RP, Checketts D, Dunayevich E, Knappertz V (2020) Dose-ranging effect of adjunctive oral cannabidiol vs placebo on convulsive seizure frequency in Dravet syndrome: a randomized clinical trial. JAMA Neurol 77:613–621. 10.1001/jamaneurol.2020.0073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E (1992) Correction for liquid junction potentials in patch clamp experiments. Methods Enzymol 207:123–231. 10.1016/0076-6879(92)07008-c [DOI] [PubMed] [Google Scholar]
- Neumann-Raizel H, Shilo A, Lev S, Mogilevsky M, Katz B, Shneor D, Shaul YD, Leffler A, Gabizon A, Karni R, Honigman A, Binshtok AM (2019) 2-APB and CBD-mediated targeting of charged cytotoxic compounds into tumor cells suggests the involvement of TRPV2 channels. Front Pharmacol 10:1198. 10.3389/fphar.2019.01198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitecka-Buchta A, Nowak-Wachol A, Wachol K, Walczyńska-Dragon K, Olczyk P, Batoryna O, Kempa W, Baron S (2019) Myorelaxant effect of transdermal cannabidiol application in patients with TMD: a randomized, double-blind trial. J Clin Med 8:1886. 10.3390/jcm8111886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orvos P, Pászti B, Topal L, Gazdag P, Prorok J, Polyák A, Kiss T, Tóth-Molnár E, Csupor-Löffler B, Bajtel Á, Varró A, Hohmann J, Virág L, Csupor D (2020) The electrophysiological effect of cannabidiol on hERG current and in guinea-pig and rabbit cardiac preparations. Sci Rep 10:16079. 10.1038/s41598-020-73165-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RR, Barbosa C, Brustovetsky T, Brustovetsky N, Cummins TR (2016) Aberrant epilepsy-associated mutant Nav1.6 sodium channel activity can be targeted with cannabidiol. Brain 139:2164–2181. 10.1093/brain/aww129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG (2005) Pharmacological actions of cannabinoids. Handb Exp Handbook of Experimental Pharmacology, pp 1–151. Springer-Verlag: Heidelberg. [DOI] [PubMed] [Google Scholar]
- Philpott HT, O'Brien M, McDougall JJ (2017) Attenuation of early phase inflammation by cannabidiol prevents pain and nerve damage in rat osteoarthritis. Pain 158:2442–2451. 10.1097/j.pain.0000000000001052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pumroy RA, Samanta A, Liu Y, Hughes TE, Zhao S, Yudin Y, Rohacs T, Han S, Moiseenkova-Bell VY (2019) Molecular mechanism of TRPV2 channel modulation by cannabidiol. Elife 8:e48792. 10.7554/eLife.48792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin N, Neeper MP, Liu Y, Hutchinson TL, Lubin ML, Flores CM (2008) TRPV2 is activated by cannabidiol and mediates CGRP release in cultured rat dorsal root ganglion neurons. J Neurosci 28:6231–6238. 10.1523/JNEUROSCI.0504-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renganathan M, Cummins TR, Waxman SG (2001) Contribution of Na(v)1.8 sodium channels to action potential electrogenesis in DRG neurons. J Neurophysiol 86:629–640. 10.1152/jn.2001.86.2.629 [DOI] [PubMed] [Google Scholar]
- Rock EM, Limebeer CL, Parker LA (2018) Effect of cannabidiolic acid and Δ(9)-tetrahydrocannabinol on carrageenan-induced hyperalgesia and edema in a rodent model of inflammatory pain. Psychopharmacology (Berl) 235:3259–3271. 10.1007/s00213-018-5034-1 [DOI] [PubMed] [Google Scholar]
- Rosenberg EC, Tsien RW, Whalley BJ, Devinsky O (2015) Cannabinoids and epilepsy. Neurotherapeutics 12:747–768. 10.1007/s13311-015-0375-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg EC, Patra PH, Whalley BJ (2017) Therapeutic effects of cannabinoids in animal models of seizures, epilepsy, epileptogenesis, and epilepsy-related neuroprotection. Epilepsy Behav 70:319–327. 10.1016/j.yebeh.2016.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross HR, Napier I, Connor M (2008) Inhibition of recombinant human T-type calcium channels by Delta9-tetrahydrocannabinol and cannabidiol. J Biol Chem 283:16124–16134. 10.1074/jbc.M707104200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy ML, Narahashi T (1992) Differential properties of tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels in rat dorsal root ganglion neurons. J Neurosci 12:2104–2111. 10.1523/JNEUROSCI.12-06-02104.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Elliott JR (1997) Phenytoin and carbamazepine: differential inhibition of sodium currents in small cells from adult rat dorsal root ganglia. Neurosci Lett 226:95–98. 10.1016/S0304-3940(97)00258-9 [DOI] [PubMed] [Google Scholar]
- Rush AM, Bräu ME, Elliott AA, Elliott JR (1998) Electrophysiological properties of sodium current subtypes in small cells from adult rat dorsal root ganglia. J Physiol 511:771–789. 10.1111/j.1469-7793.1998.771bg.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Cummins TR, Waxman SG (2007) Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J Physiol 579:1–14. 10.1113/jphysiol.2006.121483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryberg E, Larsson N, Sjögren S, Hjorth S, Hermansson N-O, Leonova J, Elebring T, Nilsson K, Drmota T, Greasley PJ (2007) The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol 152:1092–1101. 10.1038/sj.bjp.0707460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sait LG, Sula A, Ghovanloo M-R, Hollingworth D, Ruben PC, Wallace BA (2020) Cannabidiol interactions with voltage-gated sodium channels. Elife 9:e58593. 10.7554/eLife.58593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senn L, Cannazza G, Biagini G (2020) Receptors and channels possibly mediating the effects of phytocannabinoids on seizures and epilepsy. Pharmaceuticals (Basel) 13:174. 10.3390/ph13080174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva JR, Goldstein SAN (2013) Voltage-sensor movements describe slow inactivation of voltage-gated sodium channels I: wild-type skeletal muscle Na(V)1.4. J Gen Physiol 141:309–321. 10.1085/jgp.201210909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiker A, Dvorakova M, Zimmowitch A, Mackie K (2018) Cannabidiol inhibits endocannabinoid signaling in autaptic hippocampal neurons. Mol Pharmacol 94:743–748. 10.1124/mol.118.111864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele E, Marsh E, Mazurkiewicz-Beldzinska M, Halford JJ, Gunning B, Devinsky O, Checketts D, Roberts C (2019) Cannabidiol in patients with Lennox-Gastaut syndrome: interim analysis of an open-label extension study. Epilepsia 60:419–428. 10.1111/epi.14670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toib A, Lyakhov V, Marom S (1998) Interaction between duration of activity and time course of recovery from slow inactivation in mammalian brain Na+ channels. J Neurosci 18:1893–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend C, Horn R (1997) Effect of alkali metal cations on slow inactivation of cardiac Na+ channels. J Gen Physiol 110:23–33. 10.1085/jgp.110.1.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran T, Kavuluru R (2020) Social media surveillance for perceived therapeutic effects of cannabidiol (CBD) products. Int J Drug Policy 77:102688. 10.1016/j.drugpo.2020.102688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi PK, Trujillo L, Cardenas CA, Cardenas CG, de Armendi AJ, Scroggs RS (2006) Analysis of the variation in use-dependent inactivation of high-threshold tetrodotoxin-resistant sodium currents recorded from rat sensory neurons. Neuroscience 143:923–938. 10.1016/j.neuroscience.2006.08.052 [DOI] [PubMed] [Google Scholar]
- Ulbricht W (2005) Sodium channel inactivation: molecular determinants and modulation. Physiol Rev 85:1271–1301. 10.1152/physrev.00024.2004 [DOI] [PubMed] [Google Scholar]
- Verrico CD, Wesson S, Konduri V, Hofferek CJ, Vazquez-Perez J, Blair E, Dunner KJ, Salimpour P, Decker WK, Halpert MM (2020) A randomized, double-blind, placebo-controlled study of daily cannabidiol for the treatment of canine osteoarthritis pain. Pain 161:2191–2202. 10.1097/j.pain.0000000000001896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilin YY, Ruben PC (2001) Slow inactivation in voltage-gated sodium channels: molecular substrates and contributions to channelopathies. Cell Biochem Biophys 35:171–190. 10.1385/CBB:35:2:171 [DOI] [PubMed] [Google Scholar]
- Vilin YY, Makita N, George ALJ, Ruben PC (1999) Structural determinants of slow inactivation in human cardiac and skeletal muscle sodium channels. Biophys J 77:1384–1393. 10.1016/S0006-3495(99)76987-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins AR (2019) Cannabinoid interactions with ion channels and receptors. Channels (Austin) 13:162–167. 10.1080/19336950.2019.1615824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu DH, Cullen BD, Tang M, Fang Y (2020) The effectiveness of topical cannabidiol oil in symptomatic relief of peripheral neuropathy of the lower extremities. Curr Pharm Biotechnol 21:390–402. 10.2174/1389201020666191202111534 [DOI] [PubMed] [Google Scholar]
- Zheng Y, Liu P, Bai L, Trimmer JS, Bean BP, Ginty DD (2019) Deep sequencing of somatosensory neurons reveals molecular determinants of intrinsic physiological properties. Neuron 103:598–616. e7. 10.1016/j.neuron.2019.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]