

Down-regulation of HMBOX1 via METTL3-catalyzed m6A causes genomic instability driven by telomere dysfunction in cancer.
Abstract
The role of RNA methylation on N6-adenosine (m6A) in cancer has been acknowledged, but the underlying mechanisms remain obscure. Here, we identified homeobox containing 1 (HMBOX1) as an authentic target mRNA of m6A machinery, which is highly methylated in malignant cells compared to the normal counterparts and subject to expedited degradation upon the modification. m6A-mediated down-regulation of HMBOX1 causes telomere dysfunction and inactivation of p53 signaling, which leads to chromosome abnormalities and aggressive phenotypes. CRISPR-based, m6A-editing tools further prove that the methyl groups on HMBOX1 per se contribute to the generation of altered cancer genome. In multiple types of human cancers, expression of the RNA methyltransferase METTL3 is negatively correlated with the telomere length but favorably with fractions of altered cancer genome, whereas HMBOX1 mRNA levels show the opposite patterns. Our work suggests that the cancer-driving genomic alterations may potentially be fixed by rectifying particular epitranscriptomic program.
INTRODUCTION
More than 150 different chemical modifications have been identified on RNA molecules and together add a new layer of regulation in gene expression. Methylation of adenosine base at nitrogen-6 position, so-called N6-methyladenosine (m6A), is the most abundant internal modification on messenger RNA (mRNA) (1). These m6A sites are preferentially enriched near stop codons and 3′ untranslated regions (UTRs) and can also be found within long internal exons and at 5′ UTRs (2). They occur mainly within the consensus sequence motif [G>A](m6A)C[A/C/U] (2). The landscape and biology of m6A mark are orchestrated collectively by the dynamic interplay among three classes of proteins: methyltransferases (“writers”), demethylases (“erasers”), and m6A-binding proteins (“readers”). The methyl group is deposited by a multisubunit methyltransferase complex consisting of three core components: methyltransferase like-3 (METTL3), METTL14, and Wilms’ tumor 1-associating protein (WTAP) (3, 4). Meanwhile, m6A mark can be selectively removed by two RNA demethylases: fat mass and obesity-associated protein (FTO) (5) and AlkB homolog 5 (ALKBH5) (6). ALKBH5 is the primary eraser of m6A modification, whereas FTO is reported to demethylate both internal m6A methylation and N6,2′-O-dimethyladenosine at the 5′ cap (m6Am) (7). Last, several m6A-specific interacting proteins have been identified, which bind to distinct groups of m6A-containing mRNAs and mediate the downstream cellular functions of this epitranscriptomic mark (8). Changes in the levels or activities of these three classes of protein enzymes have a profound impact on the intensities, patterns, and biological functions of m6A mark.
It is now well accepted that m6A modification plays a comprehensive role in almost every aspect of mRNA life cycle (9). It regulates mRNA stability, splicing, transportation, and translation. Considering its prevalent role in regulation of mRNA fate, m6A is highly associated with a variety of human diseases including cancer once it goes awry (10). Alterations in m6A machinery may give rise to neoplastic phenotypes, such as uncontrolled proliferation, differentiation blockage, metastasis, and therapeutic resistance. Unfortunately, the roles of this epitranscriptomic mark in human cancer have just been investigated in a limited number of cancer types, and the underlying mechanisms remain quite elusive. In addition, it is still ambiguous whether aberrant m6A levels on particular target mRNAs directly contribute to cancer evolution. Furthermore, such altered m6A signals are usually characterized in cultured cancer cells and have rarely been verified in human tumor tissues. Confirmation in clinical samples will definitely help to elucidate and endorse the function of the epitranscriptomic program in cancer.
In this study, we revealed an unexpected role of m6A modification in regulation of telomere length and genomic integrity in human cancer. Telomere is a stretch of repetitive nucleotide sequences at the ends of chromosomes. It is cumulatively lost during each cycle of DNA replication and replenished by telomerase, a multisubunit ribonucleoprotein complex that consists of two catalytically essential components: the reverse transcriptase protein TERT and the associated short RNA template TERC. In most differentiated somatic cells, TERT is stringently repressed, and therefore the telomere length reaches a critical limit with repetitive cell divisions (11). Severely short telomeres serve as a double-sided sword in cancer development and progression. On the one hand, dysfunctional telomeres can be tumor suppressive, as they are recognized as double-strand DNA breaks and trigger activation of p53-centered DNA damage response network, which causes cell cycle arrest or apoptosis (12). On the other hand, unchecked telomere erosion can be oncogenic when p53 is deficient or nonfunctional, eventually leading to various types of chromosomal aberrations (13). With the assistance of reactivated telomerase or alternative lengthening of telomeres (ALT) mechanism in human malignancies, heavily rearranged genome creates a tumor-permissive environment that drives cancer development and progression (14). We show that METTL3-catalyzed m6A modification has a profound impact on telomere homeostasis and genomic stability. Therefore, we have not only uncovered a previously unrecognized mechanism of how a specific epitranscriptomic program contributes to full malignancy but also laid the foundation for a new model of cancer genome evolution.
RESULTS
m6A mark and the RNA methyltransferase METTL3 are both up-regulated in a broad range of cancer types
To fully elucidate the function of the epitranscriptomic mark m6A that is mainly catalyzed by METTL3 in human cancer, we first examined the abundance of m6A in ribosomal RNA (rRNA)–depleted RNAs in several different types of cancer cells and the corresponding normal cells (Fig. 1A). Compared to the respective benign counterparts, the overall levels of methylated adenosines relative to input RNAs are significantly elevated in cancer cell lines. Expression of METTL3 exhibits very similar patterns to those of overall m6A levels in all the tested cell lines (Fig. 1B). METTL3 has a functional active site for binding with the methyl donor S-adenosyl methionine and therefore is considered as the catalytic subunit of the m6A methyltransferase complex that actually catalyzes the methylation reaction (3). Other enzymes that help determine the cellular m6A quantities, such as METTL14 (fig. S1A), WTAP (fig. S1B), FTO (fig. S1C), and ALKBH5 (fig. S1D), are also misregulated in cancer cells, although the changes of their expression are inconsistent either across different cancer types or to a more moderate extent than METTL3. This observation implies that dysregulation of m6A modification is universal in human cancer, which may be attributed mainly to the abnormal expression of METTL3.
Fig. 1. The m6A methyltransferase METTL3 is up-regulated in human cancer and essential for the malignant properties, which requires the intact enzymatic activity.
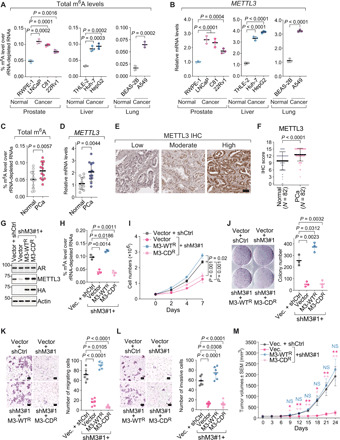
(A to D) Levels of N6-methyladenosine (m6A) in rRNA-depleted RNAs (A and C) or expression of METTL3 by RT-qPCR (B and D) in either the indicated human cell lines (A and B) or 12 pairs of prostate tumors (PCa) and normal adjacent tissues (Normal) (C and D). (E and F) Representative IHC staining images of METTL3 protein (E) and quantification of IHC scores (F). Scale bar, 50 μm. N, numbers of cases. (G) Immunoblotting in LNCaP cells that were infected with either control shRNA (shCtrl) or METTL3-specific shRNA (shM3#1) and then overexpressed control vector (vector), wild-type METTL3 (M3-WTR), or catalytically dead mutant (M3-CDR), which are HA-tagged and resistant to shM3#1. (H to L) Levels of m6A in rRNA-depleted RNAs (H), cell growth (I), colony formation assay (J), in vitro transwell migration (K), and invasion (L) assay in the METTL3 rescue system of LNCaP cells that is described in (G). Scale bars, 100 μm (K and L). (M) Tumor growth of prostate cancer xenografts by inoculating the METTL3 rescue system established in 22Rv1 cells. Data are presented as the mean tumor volume in mm3 ± SEM. *P < 0.05; **P < 0.01; NS, not significant. P values in (C) and (D) were calculated by two-tailed paired t test and in (F) by Wilcoxon signed-rank test.
To corroborate the observations in human cell lines, we collected 12 pairs of tumor samples and normal adjacent tissues from patients with primary prostate cancer and quantified the total m6A levels (Fig. 1C) as well as METTL3 expression (Fig. 1D) in these clinical specimens. Notably, m6A mark itself is significantly elevated in prostate cancer compared to the matched benign tissues, and so is METTL3. Not only in our cohort, up-regulation of METTL3 expression is also constantly seen across a wide spectrum of cancers (fig. S1E) in The Cancer Genome Atlas (TCGA) data and separate cohorts of patients with prostate (fig. S1F) (15, 16), liver (fig. S1G) (17, 18), and lung (fig. S1H) (19, 20) cancer. Furthermore, we investigated the protein levels of METTL3 by immunohistochemistry (IHC) analysis in tissue microarrays (TMAs) containing 82 pairs of prostate tumors and matched benign tissues. Specific nuclear staining of METTL3 was detected (Fig. 1E), which displays much stronger intensities in the cancer samples than in the tumor-adjacent normal tissues (Fig. 1F). Together, these results suggest that the RNA methyltransferase METTL3 and its enzymatic readout m6A mark are both aberrantly up-regulated in human cancer.
Methyltransferase activity of METTL3 is essential for the malignant phenotypes of cancer cells
We carried out a series of functional studies to confirm the oncogenic function of METTL3. We knocked down METTL3 using two specific short hairpin RNAs (shRNAs) in prostate adenocarcinoma LNCaP cells, hepatocarcinoma Huh-7 cells, and lung adenocarcinoma A549 cells, which resulted in an efficient depletion of METTL3 protein (fig. S2A). Knockdown of METTL3 significantly abrogated the anchorage-independent growth of all tested cancer cells in the soft agar colony formation assay (fig. S2B), indicating an essential role of METTL3 for the tumorigenic potentials of cancer cells.
To elucidate whether the oncogenic effect of METTL3 is mediated through RNA methylation, we replaced the endogenous METTL3 in LNCaP cells with either the wild-type protein or the catalytically dead mutant, whose adenine-binding motif DPPW is mutated into APPA (Fig. 1G) (21). Overall m6A levels were reduced upon silencing of METTL3, which was reversed by reintroduction of the enzymatically competent METTL3 but not the inactive mutant (Fig. 1H). Concomitantly, cancer cell proliferation (Fig. 1I), colony formation capacity (Fig. 1J), migration (Fig. 1K), and invasion (Fig. 1L), which were blocked by METTL3-targeting shRNA, were rescued only when the enzymatically intact METTL3 was exogenously expressed.
To confirm the oncogenic function of METTL3 in vivo, we established the xenograft models by inoculating the androgen-independent prostate cancer 22Rv1 cells into the castrated male nude mice. Again, overexpression of the wild-type METTL3, but not the catalytically dead mutant, could recover the inhibitory effect of METTL3 knockdown on xenograft tumor growth (Fig. 1M and fig. S2C). Weights of the animals in all groups were comparable and stable throughout the entire experimental period (fig. S2D). Together, our results affirm that METTL3 plays a very important role in promoting the malignancy of transformed cells, which is dependent on its methyltransferase activity.
HMBOX1 is the bona fide target of m6A modification in cancer cells
To understand the mechanism underlying the protumorigenic effect of METTL3-catalyzed m6A modification, we mapped the m6A landscapes in both immortalized benign prostate epithelial cells RWPE-1 and prostate adenocarcinoma cells LNCaP using MeRIP/m6A-seq (methylated RNA immunoprecipitation sequencing). Classical consensus motif “GGAC” was enriched at the methylated sites in both cell lines (fig. S3A). As reported (2), m6A marks are accumulated predominantly at the stop codons and 3′ UTRs as well as the coding sequence regions (fig. S3, B and C). These features provide the validation that our MeRIP/m6A-seq worked as intended. We noticed that the m6A signals at certain loci are quite disparate between RWPE-1 and LNCaP, so we categorized all the m6A-marked sites identified in MeRIP/m6A-seq into three clusters based on their intensities between these two cell lines (Fig. 2A): The “LNCaP” and the “RWPE-1″ cluster respectively represent the group of m6A modifications that are statistically stronger in LNCaP and RWPE-1, while the “common” cluster includes all the m6A sites that display very comparable intensities between these two types of cells. The m6A-modified mRNAs in each cluster are enriched with distinct biological functions (fig. S3D).
Fig. 2. HMBOX1 mRNA is modified with m6A mark and down-regulated by catalytically active METTL3 in multiple types of cancer cells.
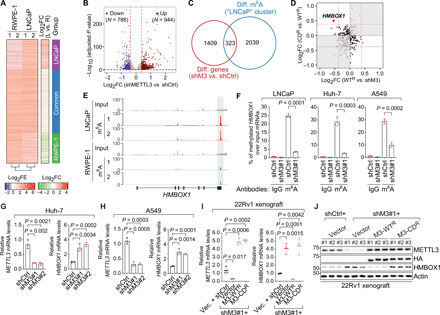
(A) Heatmaps for differential m6A patterns between RWPE-1 and LNCaP. FE, fold enrichment of m6A signals over input; FC, fold change of m6A signals in LNCaP versus in RWPE-1 (L vs. R). (B) Volcano plot of differentially expressed genes upon knockdown of METTL3 in LNCaP cells. N, numbers of significantly changed genes [log2 fold change (FC) = ±log2(1.3) indicated by the red dashed lines and P = 0.05 by the black dashed line]. (C) Venn diagram showing the overlap between differentially expressed genes upon METTL3 knockdown (shM3 versus shCtrl) and mRNAs with stronger m6A intensity in LNCaP cells (LNCaP cluster). (D) Scatterplot showing the expression changes of the overlapped genes in (C) in the METTL3 rescue system. (E) IGV browser tracks of MeRIP/m6A-seq data at the genomic location of HMBOX1. (F) MeRIP-qPCR analysis of m6A signals on HMBOX1 mRNAs in the specified cells, which were infected with control shRNA (shCtrl) or shRNA targeting METTL3 (shM3#1). Normal rabbit IgG was included as the negative control. (G and H) Expression of METTL3 and HMBOX1 in the control (shCtrl) and METTL3 knockdown (shM3#1 and shM3#2) cells established in Huh-7 (G) and A549 (H). (I and J) mRNA (I) and protein (J) levels of METTL3 and HMBOX1 in the 22Rv1 xenograft model of prostate cancer. P values in (B) were calculated by the Wald test with Benjamini-Hochberg adjustment.
Because the total quantities of m6A were noticeably elevated in LNCaP than in RWPE-1 (Fig. 1A), we sought to investigate how the up-regulated m6A modification leads to changes in the levels of target mRNAs and may subsequently cause carcinogenesis. Therefore, we first performed the gene expression profiling in LNCaP cells upon knockdown of METTL3, which gave us a list of differentially expressed genes (Fig. 2B). Then, we overlapped these METTL3-dependent genes as identified in RNA sequencing (RNA-seq) and the genes in the LNCaP cluster as indicated in MeRIP/m6A-seq, which gave rise to 323 transcripts representing the potential functioning targets of m6A in human cancer (Fig. 2C). Last, we examined the expression changes of these 323 candidate target mRNAs in the METTL3 rescue system expressing competent or inactive RNA methyltransferase. In this way, HMBOX1 stood out, as reintroduction of the wild-type METTL3 and the enzymatically dead mutant has the opposite effect on the differential expression of HMBOX1 upon METTL3 silencing (Fig. 2D).
HMBOX1 (homeobox containing 1, also known as HOT1) was originally found as a protein that directly and specifically binds to the double-stranded telomeric DNA (22). It is required for telomerase chromatin binding and maintenance of telomere length (23). In our MeRIP/m6A-seq data, we detected a stronger m6A peak at the 3′ UTR of HMBOX1 in the prostate cancer cell LNCaP than in the normal epithelial cell RWPE-1 (Fig. 2E). The same location was methylated in several other epithelial cancer cell lines as well, including the hepatocellular carcinoma cells Huh-7 (24) and HepG2 (2) and the lung cancer cell A549 (fig. S3E) (25). We validated that HMBOX1 transcripts were enriched in the immunoprecipitates from the m6A-specific antibody compared to the pull-down from control immunoglobulin G (IgG), which was diminished when METTL3 is silenced in LNCaP, Huh-7, or A549 cells (Fig. 2F). These data suggest that methylation of HMBOX1 is not a cell type–specific event and that HMBOX1 may represent a general target mRNA of m6A machinery in human cancer. HMBOX1 was significantly up-regulated when METTL3 was knocked down in two additional cancer cell lines, Huh-7 (Fig. 2G) and A549 (Fig. 2H). We further confirmed our observations in the xenograft mouse models of prostate cancer. Concordant with the in vitro data, both mRNA (Fig. 2I) and protein (Fig. 2J) levels of HMBOX1 were markedly elevated in xenograft tumors where METTL3 was either knocked down or functionally deficient and reversed back to the control levels when the wild-type METTL3 was expressed. Together, we conclude that HMBOX1 is a bona fide target of m6A machinery in several types of cancers, which is down-regulated by this epitranscriptomic modification.
The m6A mark conjugated to HMBOX1 facilitates mRNA degradation in cancer cells
The next question we asked is how m6A attachment results in reduction of HMBOX1 expression. When we silenced METTL3 in LNCaP cells, we found that the mature mRNA level of HMBOX1 was clearly increased, whereas its pre-mRNA was barely changed (Fig. 3A). Similar results were obtained when METTL14, another key subunit of m6A methyltransferase complex, was knocked down (fig. S4A). Consistently, when the RNA demethylase ALKBH5 was depleted, the steady-state level of HMBOX1 was significantly diminished, while its pre-mRNA level did not demonstrate this trend or was even slightly increased (fig. S4B). Consequently, HMBOX1 protein levels were robustly increased upon depletion of METTL3 and METTL14 but reduced by the small interfering RNA (siRNA) of ALKBH5 (fig. S4C). Considering the fact that one of the best-established functions for m6A is to cause mRNA destabilization (26), we speculated that the methyl groups on HMBOX1 transcript act to degrade the mRNA. To prove this hypothesis, we treated LNCaP cells with the transcription inhibitor actinomycin D (ActD) and compared the half-life of HMBOX1 mRNA between control cells and METTL3- or ALKBH5-knockdown cells. While silencing of METTL3 markedly stabilized HMBOX1 mRNA (Fig. 3B), perturbation of ALKBH5 led to faster decay rate (Fig. 3C). Degradation of HMBOX1 pre-mRNA was unaffected by knockdown of METTL3 (fig. S4D).
Fig. 3. The m6A modification expedites the degradation of HMBOX1 mRNA per se, which is mediated by the reader protein YTHDF2.
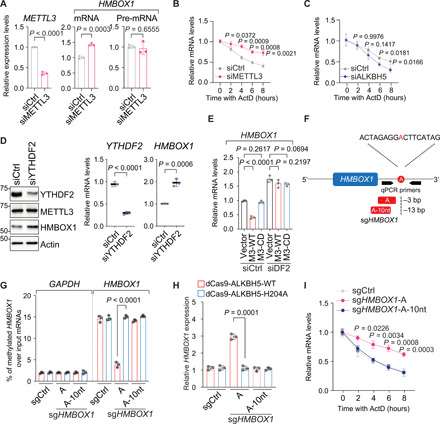
(A) Levels of specified mRNA molecules in LNCaP cells transfected with control siRNA (siCtrl) or siRNA targeting METTL3 (siM3) for 72 hours. (B and C) Half-life of HMBOX1 mRNA in LNCaP cells upon silencing METTL3 (B) or ALKBH5 (C) with the treatment of ActD (5 μg/ml) before total mRNAs were collected at indicated time points. (D) Levels of indicated molecules upon YTHDF2 knockdown in LNCaP. (E) Expression of HMBOX1 in the control (siCtrl) and YTHDF2-knockdown (siDF2) cells of LNCaP stably expressing empty backbone (vector), wild-type METTL3 (M3-WT), or catalytically dead mutant (M3-CD). (F) Schematic illustration of effective (A) and noneffective (A-10nt) sgRNA targeting HMBOX1 transcript (sgHMBOX1). (G) MeRIP-qPCR analysis of m6A signals on HMBOX1 in LNCaP cells expressing dCas9 fused to the wild-type ALKBH5 (dCas9-ALKBH5-WT) or to the incompetent demethylase (dCas9-ALKBH5-H204A) together with control sgRNA (sgCtrl), sgHMBOX1-A, or sgHMBOX1-A-10nt. GAPDH was included as a negative control. (H) Expression of HMBOX1 in the CRISPR-dCas9–based, m6A-editing system that is described in (G). (I) Half-life of HMBOX1 mRNA in LNCaP cells expressing dCas9-ALKBH5-WT together with specified sgRNAs and treated with ActD as described in (B) and (C).
Recent studies have revealed that two YTH domain–containing, m6A-recognizing proteins, YTHDC2 and YTHDF2, facilitate the degradation of modified mRNAs (27, 28). When we knocked down YTHDF2, both mRNA and protein levels of HMBOX1 are increased (Fig. 3D). However, knockdown of YTHDC2 had very minimal effect on the expression of HMBOX1 (fig. S5A). Moreover, when YTHDF2 was depleted (fig. S5B), HMBOX1 can no longer be suppressed by the wild-type METTL3 and is constantly expressed at relatively higher level (Fig. 3E). These results signify the critical role of YTHDF2 in m6A-dependent decay of HMBOX1 mRNAs.
To further confirm that the attachment of the methyl groups per se elicits the instability of HMBOX1 mRNA, we used the CRISPR-Cas9–based m6A-editing tool. This system uses the endonuclease-dead Cas9 protein (also known as dCas9) that is fused to the RNA demethylase ALKBH5 to exert single-guide RNA (sgRNA)–guided RNA demethylation at designated transcriptomic locus (29). As an enzymatically inactive control, the iron-binding histidine 204 (H204) on ALKBH5, which is required for the competency of the enzyme (30), was mutated to alanine (A). We overexpressed the dCas9 fusion protein that was attached to either the wild-type ALKBH5 (A5-WT) or the H-to-A mutant (A5-HA) in LNCaP cells (fig. S5C). On the basis of the strategy outlined in the original study (29), we designed two sgRNAs and corresponding protospacer adjacent motif (PAM)-presenting oligonucleotides targeting an adenosine that falls within the consensus GGAC motif and is methylated according to our MeRIP/m6A-seq data in LNCaP (Fig. 3F). One sgRNA is located at a distance of 3 base pairs (bp) to the targeted adenosine (sgHMBOX1-A), and the other is 13 bp away, which serves as an ineffective control (sgHMBOX1-A-10nt). M6A-specific antibody pulled down much less HMBOX1 transcripts upon the introduction of sgHMBOX1-A than sgHMBOX1-A-10nt, which was only detected when the wild-type ALKBH5, but not the H204A mutant, was overexpressed (Fig. 3G). Concomitantly, the mature mRNA of HMBOX1 was significantly up-regulated only in the presence of both functional sgRNA and enzymatically intact ALKBH5 (Fig. 3H), which was accompanied by a longer lifetime (Fig. 3I). Again, sgRNAs targeting another methylated adenosine at 3′ UTR of HMBOX1 (fig. S5D), which is also flanked by the most common m6A consensus sequences, showed very similar results: The m6A intensity on HMBOX1 was significantly decreased (fig. S5E), and expression of HMBOX1 mRNA was subsequently increased (fig. S5F) only when functional sgHMBOX1 and enzymatically intact ALKBH5 were simultaneously introduced. Under this condition, HMBOX1 protein levels were noticeably augmented as well (fig. S5G). In summary, we demonstrated that the m6A mark is the executive factor that causes decay of HMBOX1 transcript in cancer cells.
METTL3-mediated degradation of HMBOX1 leads to telomere dysfunction in cancer cells
HMBOX1 has been shown to modulate telomere length in various types of tumor cells (22, 23, 31, 32). It is localized at telomeres that are actively extended (23), and knockdown of HMBOX1 led to attrition of telomeres (32). Therefore, we sought to examine whether the telomere length in cancer cells can be controlled by the METTL3-HMBOX1 axis. To answer this question, we established stable clones in LNCaP and A549 cells, which overexpress METTL3 alone or together with HMBOX1 (fig. S6A). As expected, HMBOX1 protein levels were markedly diminished upon overexpression of METTL3. We then measured the average telomere length in both early-passage [3 to 5 population doublings (PDs)] and late-passage (>30 PDs) cells (fig. S6B) by quantitative polymerase chain reaction (qPCR) and used a single-copy gene in human genome, HBG, as a reference for data normalization. Although the quality and quantity of input genomic DNA from all the engineered cells were comparable (fig. S6C), telomeres in METTL3-overexpressing cells were relatively shorter than those in the control LNCaP cells (Fig. 4A). The suppressive effect of METTL3 on maintenance of telomere length was alleviated upon simultaneous overexpression of HMBOX1. Similar results were obtained in A549 cells (Fig. 4B). Regulation of telomere length by the METTL3-HMBOX1 axis was only seen in cells at high passage numbers, further implying that changes in the telomere length are progressively accumulated with propagation of these cells. The changes in telomere length can be visualized using fluorescence in situ hybridization (FISH) analysis of metaphase chromosomal spreads in the late-passage A549 cells expressing METTL3, either alone or with HMBOX1 (Fig. 4C). Quantitative analysis confirmed that eroded or lost telomeres are more prevalent in METTL3-overexpressing cells compared to control cells or cells coexpressing HMBOX1 (Fig. 4D).
Fig. 4. METTL3-catalyzed m6A modification on HMBOX1 leads to cumulative telomere shortening by impeding telomerase recruitment, which ultimately generates TIF in cancer cells.
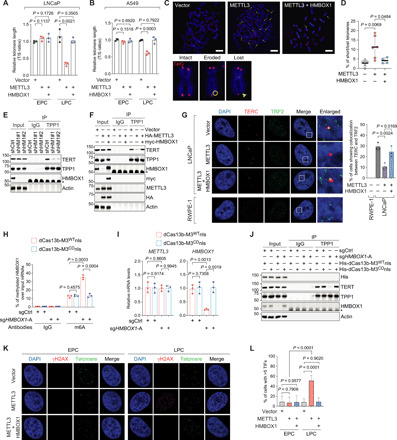
(A and B) Relative telomere length measurement by qPCR in the specified LNCaP (A) and A549 (B) cells. EPC (LPC), early (late) passage cells. (C and D) Representative images (C) and quantitative analysis (D) of metaphase FISH in the specified A549 cells at late passages. Telomere was detected by TelG-Cy3 (red), and DNA was stained with DAPI (blue). Arrows, shortened telomeres; circle, eroded telomeres; arrowhead, lost telomeres. Scale bars, 10 μm. (E and F) IP in LNCaP upon knockdown of HMBOX1 (shHM1#1 and #2) (E) or stably expressing the indicated plasmid DNAs (F). (G) Representative images and quantification of TERC RNA FISH combined with TRF2 immunostaining in the specified cells. At least 60 nuclei were counted in each biological replicate to quantify the colocalization. (H) MeRIP-qPCR analysis of m6A intensities on HMBOX1 in LNCaP cells expressing dCas13b proteins with a nuclear localization signal (nls) fused to either the wild-type METTL3 (dCas13b-M3WTnls) or the catalytically dead mutant (dCas13b-M3CDnls), together with control sgRNA (sgCtrl) or sgHMBOX1-A. (I and J) Expression of METTL3 and HMBOX1 (I) or IP (J) in dCas13b-based, m6A-editing system that is described in (H). (K and L) Representative images (K) and quantification (L) of the TIF assays in A549 cells with either low (EPC) or high (LPC) passage numbers. Cells with five or more colocalization of γH2AX foci and telomeres were scored as TIF positive. Asterisks in (E), (F), and (J), heavy chain of IgG.
Because it has been reported that HMBOX1 assists with telomere elongation by facilitating the recruitment of active telomerase holoenzyme to double-strand telomeric DNA (23), we surmised that the METTL3-HMBOX1 axis actually dictates the association of telomerase with the telomere repeats so as to control the telomere length in cancer cells. We first confirmed that the intrinsic activity of TERT, the catalytic protein subunit of the telomerase complex (11), is barely affected by METTL3 or METTL3 plus HMBOX1 (fig. S6D). We next showed that neither the protein content nor the mRNA expression of TERT was altered upon HMBOX1 depletion, although they were significantly reduced by TERT-specific siRNA (fig. S6E). Expression of the RNA component of the telomerase holoenzyme, TERC, was unaffected under these circumstances. We corroborated these findings in A549 cells (fig. S6F). All the data suggest that regulation of the telomere length by the METTL3-HMBOX1 axis is unlikely due to modulation of the reverse transcriptase activity or the levels of key components of the ribonucleoprotein enzyme complex.
Last, we detected the interaction between TERT and TPP1, a crucial component of the shelterin complex. The telomere-binding protein TPP1 is a well-documented telomerase recruiter, as it directly binds with TERT and is required for recruiting the whole functional telomerase ribonucleoprotein complex to the chromosome ends (33). When we knocked down HMBOX1, the interaction between TPP1 and TERT was noticeably disrupted, confirming an essential role of HMBOX1 in TPP1-assisted recruitment of telomerase to the telomeres (Fig. 4E). Although similar levels of TPP1 were pulled down, much less amounts of TERT were detected in the immunoprecipitates from METTL3-overexpressing cells compared to those from the control cells, which could be recovered by reintroduction of HMBOX1 (Fig. 4F).
To visualize the effect of the METTL3-HMBOX1 axis on the association between telomerase complex and telomeres, we performed RNA FISH for endogenous TERC and immunostaining with anti-TRF2 antibody in LNCaP cells that overexpress METTL3 with or without HMBOX1 (Fig. 4G). Meanwhile, we carried out the same analysis in the normal counterpart RWPE-1 cells. The percentages of cells harboring colocalization between TERC and TRF2 were significantly diminished by overexpression of METTL3 compared to the control LNCaP cells, which could be reversed when HMBOX1 was reintroduced. Such colocalized foci were much more prevalent in LNCaP than in RWPE-1, where there were almost no overlapping signals, consistent with the fact that telomerase activity is extinguished in most human normal cells and has to be reactivated in cancer cells to enable unlimited replication (11). Similar results were obtained in the lung cancer cell A549 and the matched benign cell BEAS-2B (fig. S6G). These data confirm that functional telomerase complex is reconstituted in cancer cells to promote malignant transformation. They further indicate that up-regulation of METTL3 in tumors leads to telomere shortening by retarding the telomerase recruitment to telomeres, which predisposes the cancer cells to tumorigenic genomic instability and therefore drives full malignancy and even progression to a more aggressive phenotype.
To further demonstrate the direct involvement of METTL3-catalyzed methylation on HMBOX1 in regulation of telomerase recruitment, we set up another programmable m6A-editing platform using the ribonuclease (RNase)–defective Cas13b (dCas13b) (34). In this system, dCas13b is fused with METTL3 that has been engineered to depend on the RNA-guided dCas13b for substrate binding and tagged with two copies of nuclear localization signals for nucleus-localized methylation (dCas13-M3WTnls). Meanwhile, we substituted METTL3 in the dCas13b fusion protein with the methyltransferase-impaired mutant (dCas13-M3CDnls), which serves as a negative control. Compared to the control sgRNAs, introduction of sgHMBOX1-A into cells overexpressing dCas13-M3WTnls significantly elevated the m6A signals on HMBOX1 (Fig. 4H) and concordantly decreased its mRNA levels, although METTL3 expression was quite stable (Fig. 4I). Under this particular condition, TPP1-TERT interaction was disrupted (Fig. 4J). In contrast, there were no differences between control sgRNA and sgHMBOX1-A in cells overexpressing the methyltransferase-dead dCas13 fusion protein (dCas13-M3CDnls). All these data support that the intact activity of METTL3 is required for m6A modification on HMBOX1, which suffices to reduce the expression of HMBOX1 and impede loading of the telomerase onto the telomeres. Together, our data suggest that overabundance of METTL3 causes gradual loss of telomeres in cancer cells by interfering with proper recruitment of the telomerase enzyme complex, which is elicited through m6A-mediated down-regulation of HMBOX1.
Critically short telomeres can eventually induce the DNA damage responses at the chromosome ends (35). These telomere-associated DNA damage signals are referred to as telomere dysfunction–induced foci (TIF) (36). Now, we proceeded to investigate whether METTL3-promoted telomere attrition can cause TIF formation and what role HMBOX1 plays in this process. Thus, we costained γH2AX, a marker for the locations of DNA breaks (37), and telomeric DNA in A549 cells overexpressing METTL3 only or in combination with HMBOX1 (Fig. 4K). In cells of passages 3 to 5, we could barely obtain any signals from γH2AX-specific antibody. On the contrary, late-passage cells with overexpression of METTL3 alone displayed strong immunofluorescence staining of γH2AX, which significantly overlapped with the signals from the telomeric FISH probes. However, such colocalization of the DNA damage sensor protein and telomeres was not observed in control cells or cells coexpressing METTL3 and HMBOX1. Quantification of the overlapped immunofluorescence signals clearly confirmed that TIFs are formed in METTL3-overexpressing cells, which are markedly abolished by reintroduction of HMBOX1 (Fig. 4L). Similar images (fig. S6H) and quantification (fig. S6I) were obtained in LNCaP cells, where TIFs are accumulated only in cells with overabundant METTL3. It is worth noting that TIFs are seen in METTL3-overexpressing cells cultured for extended periods of time, indicating a cumulative effect of METTL3 on induction of telomere dysfunction. In summary, we demonstrated that the METTL3-HMBOX1 axis regulates telomerase recruitment and subsequently telomere length in cancer cells, which leads to the engagement of DNA damage responses.
Overexpression of METTL3 inactivates p53 pathway and generates chromosomal instability in cancer cells via methylation of HMBOX1
DNA damages induced by dysfunctional telomeres can cause genomic instability, a hallmark of cancer, or can be targeted by DNA repair apparatus leading to permanent cell cycle arrest or cell death (38). These two distinct outcomes are dictated by the status of p53 signaling. Therefore, we first examined the activity of p53 in LNCaP cells overexpressing METTL3 or METTL3 plus HMBOX1 (Fig. 5A). To our surprise, p53 itself and p21, the major target of p53 transcriptional activity, are both robustly reduced upon overexpression of METTL3, even in the late-passage cells where γH2AX was significantly increased. This suggests that excessive expression of METTL3 inactivates the p53 pathway, although DNA lesions accumulated in this circumstance, which are recognized by the DNA damage sensor protein γH2AX. Reintroduction of HMBOX1 completely nullified the inhibitory effect of METTL3 on the p53 pathway. Similar observations were obtained in the lung cancer cells A549 (Fig. 5B). However, when we checked the mRNA levels of TP53 and CDKN1A, the gene encoding p21, only CDKN1A showed the same changing pattern with its protein levels, whereas TP53 was hardly altered. This prompted us to examine the expression of MDM2, the primary negative regulator of p53 protein by promoting ubiquitylation and proteasomal degradation (39). We found that MDM2 was significantly up-regulated by METTL3 and restored to the basal level when HMBOX1 was simultaneously overexpressed in the cells (Fig. 5C).
Fig. 5. The METTL3-HMBOX1 axis regulates the status of p53 signaling and genomic stability in cancer cells.
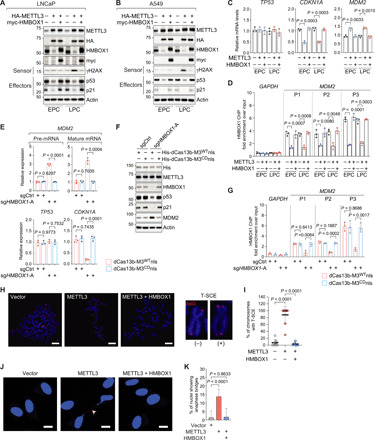
(A and B) Immunoblotting analysis in LNCaP (A) and A549 (B) expressing the indicated plasmid DNAs. EPC (LPC), early (late) passage cells. (C and D) Expression of TP53, CDKN1A, and MDM2 by RT-qPCR (C) and recruitment of HMBOX1 to the promoter region of MDM2 (P1, P2, and P3) by ChIP-qPCR (D) in LNCaP cells as described in (A) and (B). (E to G) Expression of the indicated mRNA molecules (E), immunoblotting (F), and HMBOX1 binding at the indicated chromatin regions (G) in the dCas13b-based, m6A-editing system of LNCaP cells. (H and I) Representative images (H) and quantitative analysis (I) of CO-FISH in A549 cells at high passage numbers, which stably express the indicated plasmid DNAs. Telomere was detected by TelG-Cy3 (red), and DNA was stained with DAPI (blue). Scale bars, 10 μm. (J and K) Representative images (J) and quantification (K) of anaphase bridge (arrowhead) formation in late-passage A549 cells as described in (H) and (I). DNA was stained with DAPI (blue). Scale bars, 10 μm. Eight random microscopic views were examined in each biological replicate for quantification purpose.
We next asked how the METTL3-HMBOX1 axis regulates the expression of MDM2. HMBOX1 was originally considered as a member of the homeobox transcription factors because of the possession of an atypical homeodomain and was shown to suppress the luciferase activity in a luciferase reporter assay (40). More than that, in the published chromatin immunoprecipitation–sequencing (ChIP-seq) data of HMBOX1 that were done in the chronic myeloid leukemia cells K562 (41), we found prominent peaks within 5 kb downstream of the transcription start site of MDM2 (fig. S7A). On the basis of these facts, we surmised that MDM2 is a direct target gene that is transcriptionally repressed by HMBOX1 in cancer cells. Consistent with this hypothesis, both mRNA (fig. S7B) and protein (fig. S7C) levels of MDM2 were significantly augmented when HMBOX1 was silenced by two independent shRNAs in LNCaP and A549. Under this condition, TP53 transcripts were unaffected, whereas p53 protein was markedly decreased. Thus, expression of p21 was consequently suppressed. HMBOX1 robustly binds at the cis-regulatory elements close to MDM2 in these two cancer cell lines, which was almost completely abolished by HMBOX1-specific shRNAs (fig. S7D). Congruent with these results, recruitment of HMBOX1 to the promoter of MDM2 was reduced in METTL3-overexpressing cells, which was recovered upon reintroduction of HMBOX1 into the cells (Fig. 5D).
To further verify that the m6A mark on HMBOX1 is involved in regulation of MDM2 expression, we examined the levels of pre-mRNA and mature mRNA of the gene in dCas13b-based, m6A-editing system and found that both mRNA molecules are significantly elevated in cells expressing dCas13b-M3WTnls and effective sgHMBOX1-A (Fig. 5E). We saw an increase in MDM2 protein under the same condition (Fig. 5F). Upon coexpression of dCas13b-M3WTnls and sgHMBOX1-A, there is no change in the expression of TP53 gene but a significant decrease in the protein product, whereas p21 is markedly diminished at both mRNA and protein levels. Concordantly, chromatin binding of HMBOX1 at the promoter region of MDM2 is reduced (Fig. 5G). Expression of control sgRNA or dCas13b-M3CDnls did not exhibit any effects. Together, we demonstrated strong evidence that METTL3-catalyzed methylation on HMBOX1 impairs the transcriptional suppression of MDM2, which ultimately leads to inactivation of p53 signaling.
It is worth noting that the transcript of MDM2 is also m6A-modified at two consistent sites in several different types of cancer cells, as revealed by MeRIP/m6A-seq data that were generated in previous studies (42) and in ours as well (fig. S7E). Methylation intensities at both locations were enriched by m6A-specific antibody, which were markedly decreased upon METTL3 knockdown, implying that MDM2 is likely to be an authentic target mRNA of m6A machinery (fig. S7F). However, in cells expressing dCas13b-M3WTnls and sgHMBOX1-A, m6A status on MDM2 was not different compared to those expressing control sgRNA or dCas13b-M3CDnls, suggesting that the changes in MDM2 expression we observed in these engineered cell lineages are not due to modulation of m6A signals on MDM2 mRNA itself (fig. S7G). Together, we concluded that HMBOX1 directly suppresses the transcription of MDM2 gene, which is derepressed by METTL3-catalyzed m6A modification on HMBOX1.
In the context of p53 activity deficiency, telomere attrition facilitates the acquisition of complex chromosomal rearrangements, which may produce sister chromatid fusion/exchange, end-to-end chromosome fusions, dicentric chromosomes, and amplification or translocation of terminal telomeric sequences (38). Considering the effects of the METTL3-HMBOX1 axis on the telomere length and the status of p53 activity, we sought to investigate whether there exist any types of telomere-associated chromosomal aberrations when we overexpress METTL3 with or without HMBOX1 in cancer cells. To this end, we performed chromosome orientation–FISH (CO-FISH) that can deduce the strand-specific orientation of telomere sequences (43). Unexpectedly, in METTL3-overexpressing cells at high passage numbers, we observed a marked increase of sister chromatid exchange at the telomeres (T-SCE) (Fig. 5H), which is diminished upon reintroduction of HMBOX1 in the quantitative analysis (Fig. 5I). This cytogenetic anomaly has been found in some types of invasive carcinomas and is reported to accompany the generation of cancer-driving chromosomal instability (44, 45). We observed increased numbers of anaphase bridges in A549 (Fig. 5, J and K) and LNCaP (fig. S7H) cells that overexpress METTL3 alone, but not in control cells or cells coexpressing HMBOX1. Together, our data demonstrate that the METTL3-HMBOX1 axis is a determinant of genomic integrity in cancer cells and that dysregulation of this particular epitranscriptomic program will promote chromosomal instability that fuels tumor progression.
METTL3-induced genomic instability renders the cancer cells to malignant progression, which can be alleviated by HMBOX1
Telomere-driven genomic alterations have been associated with cancer development and metastasis (38). On the basis of the role of the METTL3-HMBOX1 axis in regulation of genomic stability, we investigated whether HMBOX1 antagonizes the tumor-promoting function of METTL3. Overexpression of METTL3 enhanced the tumorigenic potential of LNCaP, Huh-7, and A549 cells, which is blocked by coexpression of HMBOX1 (Fig. 6A). In addition, migratory (Fig. 6B) and invasive (Fig. 6C) capacities of LNCaP cells were significantly boosted by the abundantly expressed METTL3. The tumor-promoting effect of METTL3 is continuously accumulative, as it is more prominent in late-passage cells, which is concordant with the occurrence of METTL3-induced chromosomal instability. However, when HMBOX1 was reintroduced into the cells, the aggressive cancer phenotypes driven by overexpression of METTL3 were substantially suppressed. We obtained similar results in a second cancer cell line, A549 (Fig. 6, D and E). Furthermore, in two independent genome-wide CRISPR-Cas9 knockout screen datasets (fig. S8, A and B), we confirmed the contradictory effects of METTL3 and HMBOX1 on cancer cell proliferation: METTL3 is essential for almost all the cancer cells being tested, whereas HMBOX1 knockout predominantly accelerates the growth of cancer cells. In summary, our results indicate that HMBOX1 protects against METTL3-mediated malignant progression.
Fig. 6. Overexpression of METTL3 drives malignant progression of cancer cells, which can be mitigated by reintroduction of HMBOX1 or removal of m6A mark on HMBOX1.
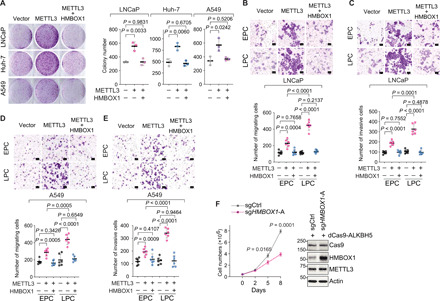
(A) Representative images and quantitative analysis of colony formation assays in LNCaP, Huh-7, and A549, which stably express the indicated plasmid DNAs. (B to E) Representative images and quantification of in vitro transwell migration assays (B and D) and in vitro transwell invasion assays (C and E) in the stable clones of LNCaP (B and C) and A549 (D and E) expressing the specified plasmid DNAs. EPC (LPC), early (late) passage cells. Scale bars, 100 μm. Six random microscopic fields were viewed for quantification. (F) Cell proliferation and Western blot in LNCaP cells expressing the dCas9-based, m6A-editing system.
To further confirm that the m6A modification on HMBOX1 contributes to the anticancer effect of HMBOX1, we measured the proliferative rates of the engineered cells in dCas9-based, m6A-editing system (fig. 6F). Delivery of functional sgHMBOX1 into LNCaP cells expressing the wild-type, dCas9-fused ALKBH5 significantly augmented the levels of HMBOX1 protein and meanwhile abrogated the proliferation of cancer cells. This result directly proves that it is the m6A mark that determines the tumor-suppressive function of HMBOX1.
Dysregulation of the METTL3-HMBOX1 axis associates with telomere shortening and genomic instability in human cancer
Considering the critical role of HMBOX1 in tumor suppression, we wondered whether this gene is misregulated in cancer cells. In the matched panels of tumor-normal cell lines of various types (prostate, liver, and lung), we observed a constant reduction in the expression of HMBOX1 in cancer cells compared to the normal counterparts (fig. S8C). We further sought to confirm what we attained in human cancer cells in tissue samples from cancer patients. In contrast to METTL3, HMBOX1 is significantly down-regulated in most of the cancer types in the TCGA datasets (Fig. 7A). This is further confirmed in independent gene expression profiling in patients with different cancer types, such as prostate (Fig. 7B and fig. S8D) (15, 16), liver (Fig. 7C and fig. S8E) (17, 18), and lung (Fig. 7D and fig. S8F) (19, 20) cancer. All these data suggested that HMBOX1 is aberrantly down-regulated in human cancer.
Fig. 7. METTL3 and HMBOX1 are both misregulated in human cancer, which are associated with aberrant signals of m6A modification on HMBOX1, telomere shortening, and altered cancer genome.
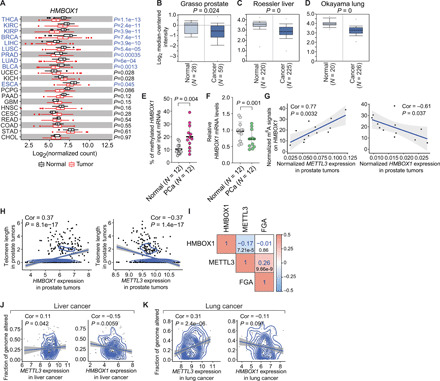
(A) HMBOX1 expression in a panel of human tumors and the corresponding normal tissues in TCGA. Cancers that show significant down-regulation of HMBOX1 were highlighted in blue. (B to D) Comparison of HMBOX1 expression between tumors and the corresponding normal tissue counterparts in prostate (B) (15), liver (C) (18), and lung (D) (19) cancer. N, case numbers. (E and F) MeRIP-qPCR analysis of m6A signals on HMBOX1 (E) or RT-qPCR detecting the expression of HMBOX1 (F) in 12 pairs of prostate cancer (PCa) and adjacent normal tissues (Normal). (G) Correlation of m6A signals on HMBOX1 with expression of METTL3 or HMBOX1 in the tumor tissues mentioned in (E) and (F). (H) Correlation between telomere length and expression of HMBOX1 or METTL3 in prostate tumors. (I) Correlation matrix showing correlation among the mRNA levels of METTL3 and HMBOX1 and fractions of altered genome in prostate cancer. Numbers in blue, correlation coefficients. Numbers in black, P values. (J and K) Correlation between the fractions of cancer genome with alterations and expression of METTL3 or HMBOX1 in liver (J) and lung (K) cancer. Correlation coefficients (Cor) were determined by Pearson correlation, and P values in (E) and (F) were calculated by two-tailed paired t test.
Next, we asked whether the m6A pattern on HMBOX1, which we characterized in cultured cells, could be repeated in human tissues and how it correlates with the expression of METTL3 and HMBOX1. In 12 pairs of primary prostate tumors and normal adjacent tissues, we consistently found stronger m6A intensities (Fig. 7E) and lower mRNA levels (Fig. 7F) of HMBOX1 in prostate tumors than in the normal counterparts. The m6A signals are positively correlated with the expression of METTL3, but negatively with that of HMBOX1 in human tumor tissues (Fig. 7G). All these results together support that HMBOX1 is highly methylated, which possibly causes its down-regulation in human cancer.
Last, we investigated whether the METTL3-HMBOX1 axis is associated with telomere shortening and genomic instability in tissue samples from cancer patients. To this end, we linked the telomere length in prostate tumors with the expression of either METTL3 or HMBOX1 (Fig. 7H) (46). While HMBOX1 is favorably correlated with the length of telomeres, which is in agreement with its role as a positive regulator of telomerase activity and telomere homeostasis, there is an inverse relationship between the telomere length and METTL3 expression. These results inspired us to perform similar correlation analysis with the genomic alterations in human cancer. In the TCGA datasets, levels of METTL3 mRNA are negatively correlated with the expression of HMBOX1 but positively with the fractions of altered genome, both of which are statistically significant (Fig. 7I). Notably, this positive association between METTL3 level and abnormalities of cancer genome could be constantly observed in other cancer types, such as liver (Fig. 7J) and lung (Fig. 7K) carcinoma (46). As for HMBOX1, although not significant in some cases, its expression is always negatively correlated with the portions of genome alterations in all the cancer types that we examined. Together, we concluded that METTL3-catalyzed m6A modification on certain transcripts, such as HMBOX1, plays a profound role in establishing genomic instability that is a characteristic of most cancer cells.
DISCUSSION
Genomic instability driven by telomere dysfunction is closely associated with full malignant progression (38). Here, we elucidated a novel role of the epitranscriptomic mark m6A in control of the telomere homeostasis and chromosomal reorganization in cancer cells (Fig. 8). In multiple types of human cancer, we constantly observed an increase in METTL3 expression, which is concordant with the elevated m6A levels in total. HMBOX1 was identified as a de novo target of m6A modification in cancer cells. Methylation of HMBOX1 is commonly detected in several lines of cancer cells, and the signals are much stronger in prostate cancer compared to the normal counterparts. Attachment of the methyl groups facilitates the degradation of HMBOX1 mRNA, which is mediated by the m6A reader protein YTHDF2. Therefore, in both cultured cancer cells and human tumor tissues, HMBOX1 expression is significantly reduced. In line with its function in recruitment of telomerase holoenzyme complex to telomeres, lowly expressed HMBOX1 is inadequate to maintain the telomere length with cumulative rounds of cell division. We also found that HMBOX1, as a transcriptional repressor, suppresses the expression of MDM2 and is essential for competency of p53 signaling. Therefore, overexpression of METTL3 in human cancer leads to telomere dysfunction and inactivation of p53-dependent DNA damage response pathway, a coordinative condition that causes various types of telomere-associated chromosomal aberrations, enhancing the tumorigenicity and aggressiveness of cancer cells. All these effects can be reversed by either reintroduction of HMBOX1 protein or removal of the targeted m6A mark on the transcript. Together, we revealed an unexpected regulatory role of m6A modification in telomere biology and genome maintenance.
Fig. 8. Schematic illustration of the oncogenic function of the METTL3-HMBOX1 axis.
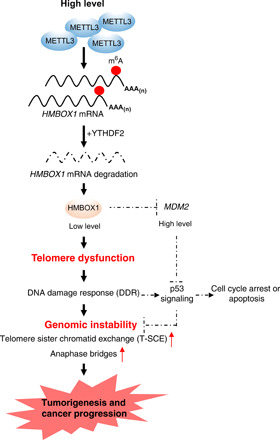
Overexpression of METTL3 in human cancer leads to augmented m6A signals on HMBOX1. Alteration to this particular m6A epitranscriptomic program facilitates the degradation of HMBOX1 mRNAs, causes progressive telomere shortening, inactivates p53 signaling, and eventually generates genomic instability in cancer cells, which drives the full malignant progression.
To identify the genuine m6A target mRNAs that account for the oncogenic function of the epitranscriptomic mark, we focused on the methylated transcripts whose m6A signals are stronger in prostate adenocarcinoma cells than in the normal epithelial cells and expression is dependent on the methyltransferase activity of METTL3. In compliance with both criteria, we pinned down HMBOX1. We found in the published MeRIP/m6A-seq datasets that the same area at the 3′ UTR of HMBOX1 transcript is highly methylated in several other cancer cell lines. m6A modification of HMBOX1 can be validated in tissue samples from cancer patients, which is more prominent in prostate tumors than in the normal counterparts. Such particular methylation signals are positively correlated with METTL3 expression but negatively with HMBOX1 mRNA levels. Together, our work has uncovered HMBOX1 as an important and general downstream target of m6A signaling in human cancer. We also provided the original line of evidence that mRNA methylation can be detected in human tissue samples, which offers the prospects of characterizing m6A pattern in a real clinical context and potential application of m6A mark on certain mRNAs as new tumor biomarkers.
We demonstrated that telomere shortening in cancer may be attributed to the dysregulation of m6A machinery. Telomeres in most solid tumors, for example, colorectal cancer, prostate cancer, and hepatocellular carcinoma, are often shorter than those found in the normal counterparts, which appear in early-stage tumors and persist through the deterioration of the disease (47). This is coincident with overexpression of METTL3 and down-regulation of HMBOX1 in human cancer, which fits our original model of action by which the METTL3-HMBOX1 axis regulates telomere homeostasis. Extremely short telomeres can either be protective against carcinogenesis or enhance both tumorigenic and metastatic ability of cancer cells via induction of genomic instability, which is determined by the status of p53-centered DNA damage response (12, 13). We found that the METTL3-HMBOX1 axis plays a very important role in control of p53 activity. This is due to HMBOX1-mediated transcriptional suppression of MDM2, the principal E3 ubiquitin ligase for p53 protein (39). MDM2 up-regulation has been found in various types of cancer (48), and our findings suggest that METTL3-catalyzed methylation on HMBOX1 may be the reason for MDM2 overexpression and genomic instability in cancer with the wild-type but insufficient p53. Excessively overexpressed METTL3 exacerbates chromosomal abnormalities in cancer cells and enhances the malignant phenotypes, which can be restored by coexpression of HMBOX1. Analysis of the clinical data is in accordance with our findings in human cancer cell lines, corroborating the importance of the METTL3-HMBOX1 axis in regulation of cancer genome. Our study explicitly explains how an aberration in the epitranscriptomic mark m6A generates telomere dysfunction, causes defective p53, and eventually induces genomic instability to support cancer progression.
Many cancer cells need the reactivated telomerase complex to maintain the telomere length within a certain range so that the infinite proliferation is possible (47). Here, we demonstrated that up-regulation of METTL3 in cancer leads to telomere shortening by hindering HMBOX1-facilitated recruitment of telomerase to telomeric chromatin, which seems contradictory to the essential role of telomerase activity in promoting tumorigenesis. Our data can possibly explain the incompatible results. First, compared to the normal cells that have no functional telomerase complex at all, a significant proportion of the holoenzyme can still associate with telomeres even in the METTL3-overexpressing cancer cells, although much lower than in the control cells or in cells coexpressing HMBOX1. This suggests that METTL3 does not completely block the action of the telomerase complex. On the other hand, METTL3-induced telomere attrition increases the tendency of cancer genome to undergo alterations, creating a permissive environment that drives full malignant transformation. In addition, in METTL3-overexpressing scenarios, we observed prevalent presence of T-SCEs encompassing large proportions of metaphase chromosomes. T-SCEs are usually seen in the telomerase-negative backgrounds (49) and has been considered as a characteristic of homologous recombination–based ALTs, another way to maintain telomere length in cancer cells (50). Reactivation of the telomerase complex or deployment of the ALT mechanism is necessary for cancer initiation and progression, and coexistence of both mechanisms has also been found in vivo in some tumors (51). In our case, elevation of METTL3 down-regulates HMBOX1, which is demonstrated to hinder proper telomerase recruitment. Occurrence of T-SCE may thereby reflect an ALT-like phenotype that the cancer cells adapt to overcome the limitation of shortened telomeres on continuous growth. Together, our data reconcile the conflict between action of METTL3 on telomere attrition and cancer evolution.
In this study, we demonstrated two types of chromosomal abnormalities that are regulated by the METTL3-HMBOX1 axis in cancer cells. Unequal T-SCE has been demonstrated to render high proliferative rate, immortalization potential, and avoidance of senescence in cancer cells (45). T-SCE represents a source of chromosomal rearrangements that are often seen in human malignant tumors, such as complex karyotypes and anaphase bridges (52). We found that, in METTL3-overexpressing cells where T-SCE is prevalent, chromatin bridges during anaphase become robustly noticeable. It has been suggested that alterations, like mutation, loss, or amplification of chromosome materials may happen during bridging events and subsequently results in altered functions of key genes that drive cancer initiation and progression (53). Further investigation is warranted to define such genetic changes that are conferred by METTL3-induced anaphase bridges, as it will definitely help elucidate the mechanism of action of the epitranscriptomic mark m6A in human cancer.
Our work highlights several profound clinical implications of m6A signaling. First, it implies that genomic aberrations in cancer cells can potentially be fixed or hindered by pharmacological manipulation of protein enzymes involved in m6A modification, such as METTL3. Second, the methyl group on HMBOX1 is proved to be the functional entity that mediates the mRNA decomposition and controls the proliferative rates of cancer cells. Therefore, implementation of CRISPR-based, m6A-editing tools may represent an effective and specific way to change the expression of particular genes for the purpose of cancer treatment. Last, the critical role of telomere length homeostasis in aging is well known since a long time (54). Our discovery of the close association between m6A modification on HMBOX1 and telomere shortening makes it appealing to investigate whether the same mechanism underlies the process of aging and the etiology of other aging-associated diseases including cancer.
MATERIALS AND METHODS
Cell culture
Human hepatocellular carcinoma cell line Huh-7, human non–small cell lung cancer line A549, human prostatic carcinoma cell line 22Rv1, and human embryonic kidney cell line HEK293T were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (P/S). The human hepatocellular carcinoma cell line HepG2 was cultured in MEM supplemented with 10% FBS and 1% P/S. Human prostate adenocarcinoma cell lines LNCaP and C81 were cultured in RPMI 1640 supplemented with 10% FBS and 1% P/S. The human normal adult liver epithelial cell line THLE-2 and human nontumorigenic lung epithelial cell line BEAS-2B were cultured in a BEGM Bronchial Epithelial Cell Growth Medium kit supplemented with 10% FBS and 1% P/S. The human prostate epithelial cell line RWPE-1 was cultured with Keratinocyte Serum Free Medium (SFM) supplemented with bovine pituitary extract (0.05 mg/ml), human recombinant epidermal growth factor (EGF) (5 ng/ml), and 1% P/S. All cell lines were grown in a 5% CO2 cell culture incubator at 37°C. A549, 22Rv1, HEK293T, HepG2, LNCaP, THLE-2, BEAS-2B, and RWPE-1 were originally purchased from the American Type Culture Collection. All cell lines were routinely tested for mycoplasma contamination using the Mycoplasma Detection Kit–DigitalTest v2.0 (BioTool, catalog no. B39132). These cell lines were not listed in the database of commonly misidentified cell lines maintained by the International Cell Line Authentication Committee.
Animal models
All the animal work was performed with the approval of the University of Texas Health Science Center at San Antonio (UTHSCSA) Institutional Animal Care Committee, and the animals were handled in accordance with institutional and national guidelines for animal experiments.
Human tissue specimens
The use of the human specimens was reviewed and approved by the Institutional Review Board committees of University of Washington, Seattle. Samples in the human TMAs were deidentified, and only those with complete clinical information, follow-up data, and good tissue quality were included. The primary prostate cancer cohort comprised 127 patients with duplicated 1-mm cores, containing matched benign prostate and prostate carcinoma taken at prostatectomy. Eighty-two of these 127 pairs of normal and tumor tissues were shown reliable staining signals and were therefore scored. For confirmation of m6A signals on HMBOX1 in human tissue samples, optimum cutting temperature (OCT)–embedded frozen tissues from 12 patients with matched prostate cancer and adjacent normal tissues were shaved (10 μm), followed by RNA extraction.
Expression plasmids and siRNAs
The lentiviral vectors expressing shRNAs against nonspecific control sequences (shCtrl), METTL3 (shM3#1 and #2), or HMBOX1 (shHM1#1 and #2) were purchased from Sigma-Aldrich (St. Louis, Missouri). The coding sequences of METTL3 were amplified from HeLa complementary DNA (cDNA) using primers that are listed in table S1. PCR products were digested with Xba I and Sal I and then ligated into pLenti–CMV (cytomegalovirus)–GFP (green fluorescent protein)–hygro (654-4) (Addgene, #17446). Point mutations on METTL3 (the catalytically dead mutations and the synonymous mutations conferring resistance to METTL3-specific shRNA) were generated by mutagenesis PCR using the Q5 Site-Directed Mutagenesis Kit (New England Biolabs Inc., Beverly, MA) using primers that are listed in table S1. The coding sequences of HMBOX1 were also amplified from HeLa cDNA using primers listed in table S1. PCR products were digested with Bam HI and Xho I and then ligated into pLenti-CMV-GFP Puro (658-5) (Addgene, #17448) digested with Bam HI and Sal I.
HMBOX1-targeting sgRNAs for dCas9- and dCas13b-based, m6A-editing systems were designed on the basis of the principles described previously (29, 34) and cloned into Cas9 (Addgene, #68463) and Cas13 (Addgene, #103854) sgRNA vector, respectively. PAMers in dCas9-based system were designed accordingly (29), and the high-performance liquid chromatography (HPLC)–purified PAMer sequences were commercially synthesized by Integrated DNA Technologies. The sequences of sgRNAs and PAMers used in this study are listed in table S2. Control siRNA (D-001206-14) and the SMARTpool siRNAs targeting METTL3 (M-005170-01-0005), METTL14 (M-014169-00-0005), ALKBH5 (M-004281-01-0005), YTHDF2 (M-021009-01-0005), YTHDC2 (M-014220-00-0005), or TERT (M-003547-02-0005) were purchased from GE Dharmacon.
Antibodies and reagents
Antibodies used in this study include αm6A (Millipore, ABE572 or MABE1006) for MeRIP/m6A-seq and MeRIP-qPCR, αHMBOX1 (Novus Biologicals, NBP1-31316) for ChIP-qPCR and immunoblotting, αγH2AX (JBW301, Millipore, 05-636) for immunofluorescence-FISH and Western blot, αTPP1 (Proteintech, 25849-1-AP) for immunoprecipitation (IP) and immunoblotting, and αTRF2 (Proteintech, 66893-1-Ig) for immunofluorescence-FISH. Other antibodies that were applied in Western blot analysis include αMETTL3 (Proteintech, 15073-1-AP), αMETTL14 (Sigma-Aldrich, HPA038002), αALKBH5 (Sigma-Aldrich, HPA007196), αHA (16B12) (BioLegend, #901501), αmyc (9E10) (Santa Cruz Biotechnology, sc-40), αHis (Proteintech, #66005-1), αCRISPR-Cas9 (Abcam, ab191468), αAR (H-280) (Santa Cruz Biotechnology, sc-13062), αYTHDF2 (Proteintech, 24744-1-AP), αYTHDC2 (Proteintech, 27779-1-AP), αp53 (DO-1) (Santa Cruz Biotechnology, sc-126), αp21 (Cell Signaling Technology, #2947), αMDM2 (Proteintech, 19058-1-AP), αTelomerase reverse transcriptase (TERT) (Abcam, ab32020), and αβ-actin (Sigma-Aldrich, A5441). Reagents that were used in IP, ChIP, and MeRIP are Protein A/G Plus Agarose (Santa Cruz Biotechnology, sc-2003) and Dynabeads Protein A (Thermo Fisher Scientific, #10006D) or Protein G (Thermo Fisher Scientific, #10007D) immunoprecipitation kit.
Lentivirus production and generation of stable clones
Lentivirus-mediated establishment of stable clones was performed as previously described (55). Briefly, lentiviral vectors for gene overexpression or shRNA knockdown were cotransfected into HEK293T cells together with packaging plasmid (pCMV delta R8.9 or psPAX2) and envelope-expressing plasmid (VSV-G). The lentiviral particles were harvested 48 and 72 hours after transfection, clarified through filters with a 0.45-μm pore size, concentrated with a Lenti-X concentrator (TAKARA), and finally added into the host cells in the presence of polybrene (5 μg/ml; Sigma-Aldrich). Twenty-four hours after infection, puromycin (1 μg/ml; Sigma-Aldrich) or hygromycin B (100 to 200 μg/ml; Sigma-Aldrich) was added into the culture medium and incubated with the cells for at least two to three passages.
RNA isolation and reverse transcription qPCR
RNA isolation and reverse transcription qPCR (RT-qPCR) assays were performed as previously described (55). Briefly, total RNAs were extracted using the TRIzol Reagent (Thermo Fisher Scientific) and quantified by Cytation5 (BioTek). One microgram of RNAs was reverse-transcribed into cDNA using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). qPCR was performed using SYBR Green qPCR Master Mix (Bio-Rad). The primers used in RT-qPCR were listed in table S3.
For mRNA stability assays, specified siRNAs or plasmids were transiently transfected to LNCaP cells using Lipofectamine 3000. Seventy-two hours after transfection, cells were treated with ActD (5 μg/ml; Sigma-Aldrich) and total RNAs were collected either immediately (0 hour) or 2, 4, 6, and 8 hours after the treatment. Last, mRNAs were subjected to RT-qPCR analysis as described above. All the RT-qPCR data are normalized to the mRNA levels of GAPDH and presented as the means of three biological replicates ± SD.
Immunoprecipitation
Co-IP was performed as previously described (55). Briefly, cells were collected and lysed in the lysis buffer [0.5% IGEPAL CA-630, 0.5% Triton X-100, 150 mM NaCl, 2 mM EDTA (pH 8.0), 50 mM tris-HCl (pH 7.5), 10 mM sodium fluoride, 2 mM sodium orthovanadate, 1 mM phenylmethanesulfonylfluoride, and 1× protease inhibitor (Roche)]. Lysates were precleared with Protein A/G Plus Agarose (Santa Cruz Biotechnology) and then incubated with indicated primary antibody and Dynabeads Protein A or Protein G magnetic beads (Thermo Fisher Scientific) at 4°C overnight. The immunoprecipitates were washed three times using the lysis buffer, subjected to SDS–polyacrylamide gel electrophoresis, and finally analyzed using immunoblotting with specified antibodies.
MeRIP/m6A-seq and targeted MeRIP-qPCR
MeRIP was carried out using the published protocol with some adjustments (56). Basically, total RNAs were extracted using the TRIzol reagent (Invitrogen) and chemically fragmented in the RNA fragmentation buffer [100 mM tris-HCl (pH 7.4) and 100 mM ZnCl2] at 70°C for 8 min. Fragmented RNAs were incubated with 5 μg of anti-m6A antibody (Millipore, ABE572) and a mixture of Dynabeads Protein A and Protein G magnetic beads (Thermo Fisher Scientific) in IP buffer [150 mM NaCl, 10 mM tris-HCl (pH 7.5), 0.1% IGEPAL CA-630, and 0.4 U SUPERase•In RNase Inhibitor (Invitrogen)] at 4°C for 4 hours. The immunoprecipitates were then washed twice in IP buffer, twice in low-salt IP buffer (IP buffer with 50 mM NaCl instead of 150), and twice in high-salt IP buffer (IP buffer with 500 mM NaCl instead of 150) at 4°C for 10 min each time. After washing and purification, the m6A-enriched RNAs were finally eluted into nuclease-free water for 2 min at room temperature. The purified RNAs were used for MeRIP/m6A-seq library construction using the SMARTer Stranded Total RNA-Seq Kit v2-Pico Input Mammalian (TAKARA, 634413) and loaded onto an Illumina NextSeq 75PE sequencer. Alternatively, they were subjected to qPCR analysis using primers that were listed in table S3.
Setup of m6A-editing systems based on dCas9 or dCas13b
LNCaP cells were cotransfected with HMBOX1-targeting sgRNA and plasmid expressing dCas9 or dCas13b fusion protein at a mass ratio of 1:3 using Lipofectamine 3000. In dCas9-based platform, PAMers were transiently delivered into cells using Lipofectamine 3000 on the next day or once every 2 to 3 days in the cell proliferation assay. Seventy-two hours after transfection, cells were subjected to RT-qPCR, mRNA stability assay, MeRIP-qPCR, ChIP-qPCR, or IP. Sequences of control sgRNA, HMBOX1-targeting sgRNAs, and PAMers were listed in table S2.
Chromatin immunoprecipitation–quantitative polymerase chain reaction
ChIP was performed as previously described with some modifications (55). Briefly, cells were double cross-linked by 6 mM disuccinimidyl glutarate (CovaChem) for 30 min and then 1% formaldehyde (Sigma-Aldrich) for 10 min at room temperature. Nuclei were extracted sequentially using buffers LB1 [50 mM Hepes-KOH (pH 7.5), 140 mM NaCl, 1 mM EDTA (pH 8.0), 10% (v/v) glycerol, 0.5% NP-40, 0.25% Triton X-100, and 1× cOmplete protease inhibitor], LB2 [10 mM tris-HCl (pH 8.0), 200 mM NaCl, 1 mM EDTA (pH 8.0), 0.5 mM EGTA (pH 8.0), and 1× cOmplete protease inhibitor], and LB3 [10 mM tris-HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA (pH 8.0), 0.5 mM EGTA (pH 8.0), 0.1% Na-deoxycholate, 0.5% N-lauroyl sarcosine, and 1× cOmplete protease inhibitor]. Extracted chromatin was sheared with a Q800R sonicator (Qsonica) and then incubated with indicated antibody conjugated to a mixture of Dynabeads Protein A and Protein G magnetic beads (Thermo Fisher Scientific) at 4°C overnight. The immunoprecipitates were washed three times in radioimmunoprecipitation assay buffer [50 mM Hepes-KOH (pH 7.5), 500 mM LiCl, 1 mM EDTA, 1% NP-40, 0.7% sodium deoxycholate, and 1× cOmplete protease inhibitor], twice in 1× TE buffer [10 mM tris-HCl (pH 8.0) and 1 mM EDTA (pH 8.0)], and finally eluted in the elution buffer [1% SDS, 10 mM tris-HCl (pH 8.0), and 10 mM EDTA (pH 8.0)]. ChIP’d DNA was reverse cross-linked at 65°C overnight, purified by the treatment with RNase A and proteinase K, and finally subjected to qPCR with primers listed in table S4.
Cell proliferation assay and PD calculation
Cells (0.4 × 106 to 0.6 × 106) were initially plated in a 60-mm culture dish and counted on the indicated day using a hemocytometer and Cytation5. PD was calculated on the basis of the following formula: 3.32*log10(Efinal/Pinitial), where Pinitial is the number of cells seeded in a dish at the beginning of each passage and Efinal is the final number of cells grown on the same dish after certain days.
Soft agar colony formation assays
Live cells were first mixed with 0.3% agarose (Sigma-Aldrich, A0701) and then plated into six-well plates that were coated with 0.6% agar (Sigma-Aldrich, A1296) at low seeding density (5000 per well for LNCaP, 3000 per well for Huh-7, and 3000 per well for A549). Eighteen to 24 days afterward, colonies were stained with crystal violet (Sigma-Aldrich, C0775) and counted using ImageJ.
In vitro cell migration and invasion assays
The transwell migration/invasion assays were carried out using the Falcon permeable chamber with 8.0-μm transparent PET (polyethylene terephthalate) membrane (Corning, 353097) according to the manufacturer’s instruction. Briefly, chambers coated with (invasion assay) or without (migration assay) Corning Matrigel Basement Membrane Matrix (Corning, 354234) were first placed into the wells of companion plates, which were covered by the cOmplete cell culture medium containing 10% FBS. LNCaP cells (5 × 104) or A549 cells (5 × 103) were suspended in medium with no FBS and added onto the top of the chambers. Sixteen hours later, cells at the bottom of the chamber were fixed with 3.7% formaldehyde, permeabilized with 100% methanol, and stained with crystal violet (Sigma-Aldrich, C0775). Representative images of migrated and invaded cells were taken using Cytation5 and quantified using ImageJ.
In vivo tumor growth in xenograft mouse model of prostate cancer
Five 6-week-old male BALB/c nude mice (Charles River Laboratories) were castrated. After all the animals were completely recovered, 2 × 106 viable 22Rv1 cells were suspended in serum-free medium containing 50% Corning Matrigel Membrane Matrix HC (Corning, 08-774-391) and injected subcutaneously into the lower flanks of each mouse. To exclude the possibility of bias against distinct proliferative rates in the engineered cell lineages, we designed the injection pattern as follows. Control cells (“Vec. + shCtrl”) and METTL3 knockdown cells (“shM3#1 + Vec.”) were implanted in the left and right flank of the same mouse, respectively. Cells substituted with the wild-type METTL3 (“shM3#1 + M3-WTR”) or catalytically dead mutant (“shM3#1 + M3-CDR”) were injected into the left and right flank of the same animal, respectively. The xenografts tumor volume was measured using calipers twice a week and calculated using the formula length × width2/2.
Telomere repeat amplification protocol assay
Telomerase activities were assessed using the TRAPeze Telomerase Detection Kit (Millipore, S7700) according to the manufacturer’s instructions. Briefly, LNCaP cells were lysed in the lysis buffer supplied by the manufacturer for 30 min at 4°C. After centrifugation at 12,000g for 20 min at 4°C, telomerase substrate oligonucleotide (TS primer) and telomere repeat amplification protocol (TRAP) primer mix were added into the cleared lysates and then underwent PCR amplification using Titanium Taq DNA Polymerase (TAKARA, 639208). The PCR conditions were denaturation for 30 min at 30°C, followed by 32 cycles of annealing for 15 s at 94°C, 30 s at 59°C, and 1 min at 72°C. TRAP products were resolved using nondenaturing polyacrylamide gel electrophoresis and stained with SYBR Green I Nucleic Acid Gel Stain (Thermo Fisher Scientific, S7563) for 30 min before the gel images were scanned using the ChemiDoc Imaging System (Bio-Rad).
Telomere length measurement by qPCR
Relative telomere length was determined using a modified qPCR assay (57). Genomic DNA was prepared using the QIAamp Genomic DNA Kit (51304) (QIAGEN). Equal amounts of genomic DNA were added into two different qPCRs, one with telomere primers (qTelomere-hTEL) and the other with primers targeting the reference single-copy gene HBG (qTelomere-hBG). Primer information was provided in table S3.
Telomere TIF assay
TIF assay was carried out on the basis of the established immunofluorescence-FISH protocol with some adjustments (36). Briefly, LNCaP or A549 cells grown on coverslips were fixed with 3.7% formaldehyde for 10 min, permeabilized by 0.5% Triton X-100 for 10 min, and blocked with 1× phosphate-buffered saline (PBS) containing 0.5% bovine serum albumin (BSA) and 0.2% cold fish gelatin for 10 min. Cells were then incubated with anti-γH2AX antibody at 4°C for 16 hours. After washing with 1× PBS three times, cells were incubated with Alexa Fluor 594 anti-mouse secondary antibody (Invitrogen) at room temperature for 1 hour. After washing six times with 1× PBS, telomere FISH was performed with a telomere-specific peptide nucleic acid (PNA) conjugate (TelC-FITC, F1009) (PNA Bio) according to the manufacturer’s instructions. Cells were counterstained and mounted with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, H-1200). Immunofluorescence images were captured using a confocal laser scanning microscope (LSM 710, Carl Zeiss) and analyzed using ImageJ.
Telomere FISH
Telomere FISH was performed on metaphase spreads using a reported protocol (58). A549 cells were synchronized in G2-M phase with demecolcine (100 ng/ml; Gibco) for 4 hours, detached from the culture vessel, washed in 1× PBS, and allowed to swell in 75 mM KCl at 37°C for 40 to 60 min. Hypotonically swollen cells were fixed in a solution containing methanol and acetic acid in proportion of 3:1. Chromosome spreads were obtained according to standard cytogenetic methods, washed with fresh fixative, and dried overnight. Slides were rehydrated in 1× PBS for 5 min, fixed in 4% paraformaldehyde (PFA) for 2 min at room temperature, and washed three times in 1× PBS for 5 min each. Next, spreads were treated with pepsin (1 mg/ml) for 10 min at 37°C, washed twice in 1× PBS for 2 min, and fixed one more time in 4% PFA. Slides were subjected to a graded ethanol series (70, 90, and 100% for 5 min each) and allowed to air dry. Dehydrated slides were overlaid with 6 nM telomeric PNA probe (TelG-Cy3, PNA Bio) in hybridization solution [10 mM tris-HCl (pH 7.2), 70% formamide, and 0.5% blocking reagent (Roche)] and denatured for 5 min at 80°C, followed by hybridization for 2 hours at room temperature. Slides were washed twice in hybridization wash buffer #1 [10 mM tris-HCl (pH 7.2), 70% formamide, 0.1% BSA] for 15 min each and three times in hybridization wash buffer #2 [0.1 M tris-HCl (pH 7.2), 0.15 M NaCl, 0.08% Tween 20] for 5 min each, with 300 nM DAPI added to the second wash. Slides were subjected to a graded ethanol series as described above, air-dried, and finally mounted with VECTASHIELD HardSet mounting medium (Vector Laboratories, H140010).
Chromosome orientation–fluorescence in situ hybridization
CO-FISH was performed on metaphase spreads as described previously (43). Basically, A549 cells were cultured in fresh medium supplemented with 7.5 mM 5-bromo-2′-deoxyuridine (BrdU; MP Biomedicals) and 2.5 mM 5-bromo-2′-deoxycytidine (BrdC; Sigma-Aldrich) for around one cell cycle (16 to 18 hours). Chromosome spreads were prepared as aforementioned. Slides were rehydrated in 1× PBS for 5 min and treated with deoxyribonuclease (DNase)–free RNase A (0.5 mg/ml; Sigma-Aldrich) for 10 min at 37°C. Metaphase spreads were stained with Hoechst 33258 (0.5 μg/ml; Sigma-Aldrich) in 2× SSC buffer for 15 min at room temperature. Slides were then flooded with 2× SSC and exposed to long-wave (~365-nm) ultraviolet (UV) light (Stratalinker 1800 UV irradiator) for 5.4 × 103 J/m2 at room temperature. Afterward, the BrdU/BrdC-substituted DNA strands were digested in exonuclease III solution (10 U/μl; New England Biolabs Inc.) for 30 min at 37°C. Slides were washed in 1× PBS for 5 min, subjected to a graded ethanol series (70, 90, and 100% for 5 min each), and allowed to air dry. Dehydrated slides were overlaid with 6 nM telomeric PNA probe (TelG-Cy3, PNA Bio) in hybridization solution [10 mM tris-HCl (pH 7.2), 70% formamide, and 0.5% blocking reagent (Roche)], denatured for 5 min at 80°C, and hybridized for 2 hours at room temperature. Slides were washed twice in hybridization wash buffer #1 for 15 min each and three times in hybridization wash buffer #2 for 5 min each, with 300 nM DAPI added to the second wash. Slides were subjected to a graded ethanol series as described above, air-dried, and finally mounted with VECTASHIELD HardSet mounting medium (Vector Laboratories, H140010).
TERC RNA fluorescence in situ hybridization
TERC RNA FISH was performed as previously described with some adjustments (59). After carrying out the immunostaining with anti-TRF2 antibody, cells were refixed with 3.7% formaldehyde in PBS for 10 min, permeabilized by 70% ethanol for 1 hour at 4°C, washed, and rehydrated in Wash buffer A (Biosearch) for 5 min at room temperature. Cells were then hybridized in the hybridization solution containing a mixture of Quasar 570–labeled oligonucleotide probes that are complementary to different regions of TERC RNA (Biosearch) for 16 hours at 37°C. After hybridization, cells were washed with Wash buffer A and B (Biosearch) and twice with 1× PBS. Cells were counterstained and mounted with DAPI (Vector Laboratories, H-1200). Immunofluorescence images were captured using a confocal laser scanning microscope (LSM 710, Carl Zeiss) and analyzed using ImageJ. The TERC-specific probes used in RNA FISH were listed in table S5.
Immunohistochemical staining
TMA sections (5 μm) were deparaffinized and rehydrated in xylene and graded ethanol series. Heat-induced antigen retrieval was performed in 10 mM citrate buffer (pH 6.0). Endogenous peroxidase and avidin/biotin were blocked (Vector Laboratories Inc., Burlingame, CA), and TMAs were incubated with 5% normal goat-horse-chicken serum at 4°C overnight. Sections were incubated with the primary antibody recognizing METTL3 (Proteintech, #15073-1-AP, 1:500), followed by the biotinylated secondary antibody (Vector Laboratories Inc.) and ABC reagent (Vector Laboratories Inc.). Staining was detected with stable DAB (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions. All sections were counterstained with hematoxylin and mounted with Cytoseal XYL (Richard Allan Scientific, San Diego, CA). Rabbit IgG was used as negative controls.
Quantification of IHC
METTL3 IHC staining was assessed using a combination of both intensity (score 0, no staining; score 1, weak; score 2, moderate; and score 3, strong) and proportion of stained cells (score 0, 0 to 50%; score 1, 51 to 60%; score 2, 61 to 70%; score 3, 71 to 80%; score 4, 81 to 90%; and score 5, 91 to 100%). The final score was obtained by multiplying the intensity and the proportion scores, resulting in a value ranging from 0 to 15. The samples with scores of 0 to 9, 10 to 12, and 15 were respectively classified as low, moderate, and high expression. All histological evaluations were carried out in a double-blind manner.
MeRIP/m6A-seq data analysis
MeRIP/m6A-seq data were analyzed using computational pipelines that were originally described (60). Cutadapt was used to remove the first nine bases and N’s on both ends of reads. Reads with a minimum of 25 bases were kept and aligned to human genome (hg19) using STAR. Duplicated reads were marked and removed by Picard. The bam files were configured in bed12 format by bedtools, shifted to 200 nucleotides (nt) using a custom script, and finally transformed back to bed6 format. Each sample was normalized to 10 million reads in total using Macs2 bdgopt subcommand. MeTDiff with default parameters was used to call m6A peaks by combining signals from both biological replicates. All the peaks in LNCaP and RWPE-1 cells were merged using a custom script, and thereby, a reference peak set was generated, based on which the differential m6A intensities were derived. Guitar package was used to annotate and plot the distribution of m6A peaks across the whole transcriptome. The motif enrichment analysis was performed with Homer in RNA mode, which considered a window size of 400 bp around the center of m6A peaks and allowed one mismatch. The Gene Ontology (GO) analysis of the transcripts with common or differential m6A peak signals was conducted by clusterProfiler.
RNA-seq data analysis
Classical analysis strategy was implemented to analyze the RNA-seq data (61). The sequencing reads were aligned to human genome (hg19) using STAR 2.5.2b. After removing reads that were mapped to rRNAs, read counting for each gene was conducted by featureCounts package with default parameter. Genes with less than one read in at least two samples were discarded. DESeq2 1.14.1 was used to call differentially expressed genes with fold change ≥ 1.3 and false discovery rate ≤ 0.05 as the cutoff (62). The gene expression data from the cohorts of patients with prostate, liver, and lung cancer were retrieved from Firehose by RTCGAToolbox.
ChIP-seq data analysis
Reads were aligned to human genome (hg19) using bowtie (63). Peaks were then called using MACS with a cutoff of q value at 1 × 10−5 (64). Peaks overlapped with the UCSC blacklist regions were removed.
Data collection and visualization
All MeRIP/m6A-seq and ChIP-seq data were visualized in the Integrative Genomics Viewer (IGV) (65). All the genomic datasets were normalized to 10 million reads per sample.
Statistical analysis
All the experiments were repeated using at least three biological and technical replicates, and the results were shown as the average ± SD, unless otherwise stated. P values were calculated using two-tailed unpaired t test or the appropriate statistical methods as mentioned in the figure legends.
Acknowledgments
We thank H. H. He at Princess Margaret Cancer Centre and Bryce Lakely from the University of Washington for technical support. We are also very grateful to M. Liss at University of Texas Health Science Center at San Antonio (UTHSCSA) for advice and discussion. Last, we really appreciate the generosity of Y. Xiang at UTHSCSA, S.-B. Qian at Cornell University, and D. Liu at Harvard University for sharing reagents with us. Some of the results presented in this study are based on data generated by the TCGA Research Network (https://cancer.gov/tcga). Funding: This work was supported by grants from CPRIT awards (RR140072 to K.X. and RP170345 to R.D.L.R.), Voelcker Fund Young Investigator award (to K.X.), and Early Investigator Research Award from DoD (W81XWH-17-1-0255 to M.Y.) and partially by the funding from the Mays Cancer Center (P30 CA054174 to T.L.J.-P. and R.J.L. at the Institutional Biospecimen and Translational Genomics Core Laboratory of UTHSCSA). Author contributions: J.H.L. and J.H. performed all the biochemical, biological, and molecular biology assays in this study, except the chromosomal instability analyses. Z.Z. analyzed all the next-generation sequencing data and published data in patient samples under the instructions of S.Z. and K.X. B.d.l.P.A. and E.D. carried out all the experiments examining telomere dysfunction and chromosomal abnormalities in cancer cells. H.-M.L. and J.G. did the IHC staining in prostate tissue microarrays. C.J.P., A.R.D., P.G.G., P.V.E., M.P.R., and A.E.S. carried out the pathological analyses of IHC staining. R.D.L.R., K.V., M.Y., Y.L., J.S., and D.P. assisted J.H.L. and J.H. in their work. P.Y. and M.K.R. helped generate the transcriptome-wide MeRIP/m6A-seq data. H.-M.L., T.L.J.-P., and R.J.L. dissected and supplied proper samples from prostate cancer patients. This study was conceptually monitored by K.X. with the advice from S.Z., R.J.L., C.J.P., P.V.E., and E.D. All the authors helped design the study and write the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All the genome-wide datasets generated in this study, including RNA-seq and MeRIP/m6A-seq, have been deposited at the Gene Expression Omnibus database (http://ncbi.nlm.nih.gov/geo/) with an accession number GSE147891. The HMBOX1 ChIP-seq data are from GEO: GSE96356, and the MeRIP/m6A-seq data done in Huh-7, HepG2, and A549 cells are from GEO: GSE130891, GEO: GSE37003, and GEO: GSE76367, respectively. The cohort studies of patients with prostate, liver, and lung cancer are all from Oncomine with GEO accession numbers available as GSE35988, GSE6764, GSE14520, GSE31210, and GSE7670. The CRISPR-Cas9 knockout screening data are retrieved from DepMap. Pan-cancer analysis of gene expression, data on telomere length in prostate cancer, and information about genome alterations in prostate, liver, and lung cancer are all from the TCGA Program. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/31/eabg7073/DC1
REFERENCES AND NOTES
- 1.Wei C. M., Gershowitz A., Moss B., Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell 4, 379–386 (1975). [DOI] [PubMed] [Google Scholar]
- 2.Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob-Hirsch J., Amariglio N., Kupiec M., Sorek R., Rechavi G., Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Wang X., Feng J., Xue Y., Guan Z., Zhang D., Liu Z., Gong Z., Wang Q., Huang J., Tang C., Zou T., Yin P., Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature 534, 575–578 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Ping X. L., Sun B. F., Wang L., Xiao W., Yang X., Wang W. J., Adhikari S., Shi Y., Lv Y., Chen Y. S., Zhao X., Li A., Yang Y., Dahal U., Lou X. M., Liu X., Huang J., Yuan W. P., Zhu X. F., Cheng T., Zhao Y. L., Wang X., Danielsen J. M. R., Liu F., Yang Y. G., Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 24, 177–189 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jia G., Fu Y., Zhao X., Dai Q., Zheng G., Yang Y., Yi C., Lindahl T., Pan T., Yang Y. G., He C., N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng G., Dahl J. A., Niu Y., Fedorcsak P., Huang C. M., Li C. J., Vågbø C. B., Shi Y., Wang W. L., Song S. H., Lu Z., Bosmans R. P. G., Dai Q., Hao Y. J., Yang X., Zhao W. M., Tong W. M., Wang X. J., Bogdan F., Furu K., Fu Y., Jia G., Zhao X., Liu J., Krokan H. E., Klungland A., Yang Y. G., He C., ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 49, 18–29 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi H., Wei J., He C., Where, when, and how: Context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell 74, 640–650 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X., He C., Reading RNA methylation codes through methyl-specific binding proteins. RNA Biol. 11, 669–672 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peer E., Rechavi G., Dominissini D., Epitranscriptomics: Regulation of mRNA metabolism through modifications. Curr. Opin. Chem. Biol. 41, 93–98 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Chen X. Y., Zhang J., Zhu J. S., The role of m6A RNA methylation in human cancer. Mol. Cancer 18, 103 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cong Y.-S., Wright W. E., Shay J. W., Human telomerase and its regulation. Microbiol. Mol. Biol. Rev. 66, 407–425 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cosme-Blanco W., Shen M. F., Lazar A. J. F., Pathak S., Lozano G., Multani A. S., Chang S., Telomere dysfunction suppresses spontaneous tumorigenesis in vivo by initiating p53-dependent cellular senescence. EMBO Rep. 8, 497–503 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chin L., Artandi S. E., Shen Q., Tam A., Lee S. L., Gottlieb G. J., Greider C. W., DePinho R. A., p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 97, 527–538 (1999). [DOI] [PubMed] [Google Scholar]
- 14.Yates L. R., Campbell P. J., Evolution of the cancer genome. Nat. Rev. Genet. 13, 795–806 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grasso C. S., Wu Y. M., Robinson D. R., Cao X., Dhanasekaran S. M., Khan A. P., Quist M. J., Jing X., Lonigro R. J., Brenner J. C., Asangani I. A., Ateeq B., Chun S. Y., Siddiqui J., Sam L., Anstett M., Mehra R., Prensner J. R., Palanisamy N., Ryslik G. A., Vandin F., Raphael B. J., Kunju L. P., Rhodes D. R., Pienta K. J., Chinnaiyan A. M., Tomlins S. A., The mutational landscape of lethal castration-resistant prostate cancer. Nature 487, 239–243 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arredouani M. S., Lu B., Bhasin M., Eljanne M., Yue W., Mosquera J. M., Bubley G. J., Li V., Rubin M. A., Libermann T. A., Sanda M. G., Identification of the transcription factor single-minded homologue 2 as a potential biomarker and immunotherapy target in prostate cancer. Clin. Cancer Res. 15, 5794–5802 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wurmbach E., Chen Y. B., Khitrov G., Zhang W., Roayaie S., Schwartz M., Fiel I., Thung S., Mazzaferro V., Bruix J., Bottinger E., Friedman S., Waxman S., Llovet J. M., Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology 45, 938–947 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Roessler S., Jia H. L., Budhu A., Forgues M., Ye Q. H., Lee J. S., Thorgeirsson S. S., Sun Z., Tang Z. Y., Qin L. X., Wang X. W., A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res. 70, 10202–10212 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okayama H., Kohno T., Ishii Y., Shimada Y., Shiraishi K., Iwakawa R., Furuta K., Tsuta K., Shibata T., Yamamoto S., Watanabe S. I., Sakamoto H., Kumamoto K., Takenoshita S., Gotoh N., Mizuno H., Sarai A., Kawano S., Yamaguchi R., Miyano S., Yokota J., Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 72, 100–111 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Su L. J., Chang C. W., Wu Y. C., Chen K. C., Lin C. J., Liang S. C., Lin C. H., Whang-Peng J., Hsu S. L., Chen C. H., Huang C. Y. F., Selection of DDX5 as a novel internal control for Q-RT-PCR from microarray data using a block bootstrap re-sampling scheme. BMC Genomics 8, 140 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X., Huang J., Zou T., Yin P., Human m6A writers: Two subunits, 2 roles. RNA Biol. 14, 300–304 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dejardin J., Kingston R. E., Purification of proteins associated with specific genomic loci. Cell 136, 175–186 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kappei D., Butter F., Benda C., Scheibe M., Draškovič I., Stevense M., Novo C. L., Basquin C., Araki M., Araki K., Krastev D. B., Kittler R., Jessberger R., Londoño-Vallejo J. A., Mann M., Buchholz F., HOT1 is a mammalian direct telomere repeat-binding protein contributing to telomerase recruitment. EMBO J. 32, 1681–1701 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gokhale N. S., McIntyre A. B. R., Mattocks M. D., Holley C. L., Lazear H. M., Mason C. E., Horner S. M., Altered m6A modification of specific cellular transcripts affects flaviviridae infection. Mol. Cell 77, 542–555.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin S., Choe J., Du P., Triboulet R., Gregory R. I., The m6A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell 62, 335–345 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee Y., Choe J., Park O. H., Kim Y. K., Molecular mechanisms driving mRNA degradation by m6A modification. Trends Genet. 36, 177–188 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Du H., Zhao Y., He J., Zhang Y., Xi H., Liu M., Ma J., Wu L., YTHDF2 destabilizes m6A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat. Commun. 7, 12626 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kretschmer J., Rao H., Hackert P., Sloan K. E., Höbartner C., Bohnsack M. T., The m6A reader protein YTHDC2 interacts with the small ribosomal subunit and the 5′–3′ exoribonuclease XRN1. RNA 24, 1339–1350 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu X. M., Zhou J., Mao Y., Ji Q., Qian S. B., Programmable RNA N6-methyladenosine editing by CRISPR-Cas9 conjugates. Nat. Chem. Biol. 15, 865–871 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu C., Liu K., Tempel W., Demetriades M., Aik W. S., Schofield C. J., Min J., Structures of human ALKBH5 demethylase reveal a unique binding mode for specific single-stranded N6-methyladenosine RNA demethylation. J. Biol. Chem. 289, 17299–17311 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng X., Luo Z., Jiang S., Li F., Han X., Hu Y., Wang D., Zhao Y., Ma W., Liu D., Huang J., Songyang Z., The telomere-associated homeobox-containing protein TAH1/HMBOX1 participates in telomere maintenance in ALT cells. J. Cell Sci. 126, 3982–3989 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou S., Xiao Y., Zhuang Y., Liu Y., Zhao H., Yang H., Xie C., Zhou F., Zhou Y., Knockdown of homeobox containing 1 increases the radiosensitivity of cervical cancer cells through telomere shortening. Oncol. Rep. 38, 515–521 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Xin H., Liu D., Wan M., Safari A., Kim H., Sun W., O’Connor M. S., Songyang Z., TPP1 is a homologue of ciliate TEBP-beta and interacts with POT1 to recruit telomerase. Nature 445, 559–562 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Wilson C., Chen P. J., Miao Z., Liu D. R., Programmable m6A modification of cellular RNAs with a Cas13-directed methyltransferase. Nat. Biotechnol. 38, 1431–1440 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takai H., Smogorzewska A., de Lange T., DNA damage foci at dysfunctional telomeres. Curr. Biol. 13, 1549–1556 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Mender I., Shay J. W., Telomere dysfunction induced foci (TIF) analysis. Bio Protoc. 5, e1656 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mah L.-J., El-Osta A., Karagiannis T. C., γH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 24, 679–686 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Deng Y., Chang S., Role of telomeres and telomerase in genomic instability, senescence and cancer. Lab. Invest. 87, 1071–1076 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Chene P., Inhibiting the p53-MDM2 interaction: An important target for cancer therapy. Nat. Rev. Cancer 3, 102–109 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Chen S., Saiyin H., Zeng X., Xi J., Liu X., Li X., Yu L., Isolation and functional analysis of human HMBOX1, a homeobox containing protein with transcriptional repressor activity. Cytogenet. Genome Res. 114, 131–136 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Davis C. A., Hitz B. C., Sloan C. A., Chan E. T., Davidson J. M., Gabdank I., Hilton J. A., Jain K., Baymuradov U. K., Narayanan A. K., Onate K. C., Graham K., Miyasato S. R., Dreszer T. R., Strattan J. S., Jolanki O., Tanaka F. Y., Cherry J. M., The encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 46, D794–D801 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schwartz S., Mumbach M. R., Jovanovic M., Wang T., Maciag K., Bushkin G. G., Mertins P., ter-Ovanesyan D., Habib N., Cacchiarelli D., Sanjana N. E., Freinkman E., Pacold M. E., Satija R., Mikkelsen T. S., Hacohen N., Zhang F., Carr S. A., Lander E. S., Regev A., Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 8, 284–296 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bailey S. M., Goodwin E. H., Meyne J., Cornforth M. N., CO-FISH reveals inversions associated with isochromosome formation. Mutagenesis 11, 139–144 (1996). [DOI] [PubMed] [Google Scholar]
- 44.Bodvarsdottir S. K., Steinarsdottir M., Bjarnason H., Eyfjord J. E., Dysfunctional telomeres in human BRCA2 mutated breast tumors and cell lines. Mutat. Res. 729, 90–99 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Laud P. R., Multani A. S., Bailey S. M., Wu L., Ma J., Kingsley C., Lebel M., Pathak S., DePinho R., Chang S., Elevated telomere-telomere recombination in WRN-deficient, telomere dysfunctional cells promotes escape from senescence and engagement of the ALT pathway. Genes Dev. 19, 2560–2570 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barthel F. P., Wei W., Tang M., Martinez-Ledesma E., Hu X., Amin S. B., Akdemir K. C., Seth S., Song X., Wang Q., Lichtenberg T., Hu J., Zhang J., Zheng S., Verhaak R. G. W., Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 49, 349–357 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okamoto K., Seimiya H., Revisiting telomere shortening in cancer. Cell 8, 107 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oliner J. D., Saiki A. Y., Caenepeel S., The role of MDM2 amplification and overexpression in tumorigenesis. Cold Spring Harb. Perspect. Med. 6, a026336 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bailey S. M., Brenneman M. A., Goodwin E. H., Frequent recombination in telomeric DNA may extend the proliferative life of telomerase-negative cells. Nucleic Acids Res. 32, 3743–3751 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cesare A. J., Reddel R. R., Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 11, 319–330 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Perrem K., Colgin L. M., Neumann A. A., Yeager T. R., Reddel R. R., Coexistence of alternative lengthening of telomeres and telomerase in hTERT-transfected GM847 cells. Mol. Cell. Biol. 21, 3862–3875 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salawu A., Wright K., Al-Kathiri A., Wyld L., Reed M., Sisley K., Sister chromatid exchange and genomic instability in soft tissue sarcomas: Potential implications for response to DNA-damaging treatments. Sarcoma 2018, 3082526 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weyburne E., Bosco G., Cancer-associated mutations in the condensin II subunit CAPH2 cause genomic instability through telomere dysfunction and anaphase chromosome bridges. J. Cell. Physiol. 236, 3579–3598 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shin J.-S., Hong A., Solomon M. J., Lee C. S., The role of telomeres and telomerase in the pathology of human cancer and aging. Pathology 38, 103–113 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Yang M., Lee J. H., Zhang Z., de la Rosa R., Bi M., Tan Y., Liao Y., Hong J., Du B., Wu Y., Scheirer J., Hong T., Li W., Fei T., Hsieh C. L., Liu Z., Li W., Rosenfeld M. G., Xu K., Enhancer RNAs mediate estrogen-induced decommissioning of selective enhancers by recruiting ERα and its cofactor. Cell Rep. 31, 107803 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zeng Y., Wang S., Gao S., Soares F., Ahmed M., Guo H., Wang M., Hua J. T., Guan J., Moran M. F., Tsao M. S., He H. H., Refined RIP-seq protocol for epitranscriptome analysis with low input materials. PLOS Biol. 16, e2006092 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cawthon R. M., Telomere measurement by quantitative PCR. Nucleic Acids Res. 30, e47 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Doksani Y., de Lange T., Telomere-internal double-strand breaks are repaired by homologous recombination and PARP1/Lig3-dependent end-joining. Cell Rep. 17, 1646–1656 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tang M., Li Y., Zhang Y., Chen Y., Huang W., Wang D., Zaug A. J., Liu D., Zhao Y., Cech T. R., Ma W., Songyang Z., Disease mutant analysis identifies a new function of DAXX in telomerase regulation and telomere maintenance. J. Cell Sci. 128, 331–341 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meyer K. D., Saletore Y., Zumbo P., Elemento O., Mason C. E., Jaffrey S. R., Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149, 1635–1646 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yalamanchili H. K., Wan Y.-W., Liu Z., Data analysis pipeline for RNA-seq experiments: From differential expression to cryptic splicing. Curr. Protoc. Bioinformatics 59, 11.15.1–11.15.21 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Langmead B., Trapnell C., Pop M., Salzberg S. L., Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nussbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robinson J. T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E. S., Getz G., Mesirov J. P., Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/31/eabg7073/DC1