

Abstract
Background & Aims:
The mechanisms of hepatitis C virus (HCV) infection, liver disease progression, and hepatocarcinogenesis are only partially understood. We performed genomic, proteomic, and metabolomic analyses of HCV-infected cells and chimeric mice to learn more about these processes.
Methods:
Huh7.5.1dif (hepatocyte-like cells) were infected with culture-derived HCV and used in RNA-Seq, proteomic, metabolomic, and integrative genomic analyses. uPA/SCID mice were injected with serum from HCV-infected patients; 8 weeks later, liver tissues were collected and analyzed by RNA-seq and proteomics. Using differential expression, gene set enrichment analyses, and protein interaction mapping, we identified pathways that changed in response to HCV infection. We validated our findings in studies of liver tissues from 216 patients with HCV infection and early-stage cirrhosis and paired biopsies from 99 patients with hepatocellular carcinoma, including 17 patients with histologic features of steatohepatitis. Cirrhotic liver tissues from patients with HCV infection were classified into 2 groups based on relative peroxisome function; outcomes assessed included Child-Pugh class, development of hepatocellular carcinoma, survival and steatohepatitis. Hepatocellular carcinomas were classified according to steatohepatitis; the outcome was relative peroxisomal function.
Results:
We quantified 21,950 mRNAs and 8297 proteins in HCV-infected cells. Upon HCV infection of hepatocyte-like cells and chimeric mice, we observed significant changes in levels of mRNAs and proteins involved in metabolism and hepatocarcinogenesis. HCV infection of hepatocyte-like cells significantly increased levels of mRNAs, but not proteins, that regulate the innate immune response—we believe this was due to the inhibition of translation in these cells. HCV infection of hepatocyte-like cells increased glucose consumption and metabolism and the STAT3 signaling pathway and reduced peroxisome function. Peroxisomes mediate beta-oxidation of very long-chain fatty acids (VLCFAs); we found intracellular accumulation of VLCFAs in HCV-infected cells, which is also observed in patients with fatty liver disease. Cells in livers from HCV-infected mice had significant reductions in levels of mRNAs and proteins associated with peroxisome function, indication perturbation of peroxisomes. We associated defects in peroxisome function with outcomes and features of HCV-associated cirrhosis, fatty liver disease, and hepatocellular carcinoma in patients.
Conclusions:
We performed combined transcriptome, proteome, and metabolome analyses of liver tissues from HCV-infected hepatocyte-like cells and HCV-infected mice. We found that HCV infection increases glucose metabolism and the STAT3 signaling pathway and thereby reduces peroxisome function; alterations in expression of peroxisome genes were associated with outcomes of patients with liver diseases. These findings provide insights into liver disease pathogenesis and might be used to identify new therapeutic targets.
Keywords: HCC, signal transduction, metabolic disease, immune regulation
INTRODUCTION
Viruses have developed sophisticated strategies to promote their life cycle, evade the antiviral defense systems and cause disease. As a result, some viruses can persist beyond the stage of acute infection and develop a state of co-existence with the host through either chronic or latent infection. Viral and cellular gene expression are adjusted over time to meet the requirements of persistence. Considering chronic infection as merely an enduring acute phase is thus fundamentally inaccurate. In fact, the modulations of host biology in the long run are profound and continually damaging the host cell and its microenvironment1. This contributes to pathogenic phenotypes including chronic inflammation, tissue injury and cancer. Viral reprogramming of host cells can be investigated by systematic genome-wide profiling of gene-products. Such analysis does not only increase our understanding of the rearrangements of cellular architecture and functions, but also provides mechanistic insight into disease development.
Chronic hepatitis C virus (HCV) infection is an intriguing prototype to study general mechanisms of immune evasion and disease pathogenesis because of the refined strategies to evade antiviral responses, and alteration of metabolic pathways as well as regulatory cell circuits including signaling pathways, translation machinery and RNAi2–5. This may have profound impact on the host cell’s proteome and transcriptome resembling patterns induced by other pathologies such as alcohol or obesity. HCV establishes acute and chronic infection of the liver, which is a leading cause of liver disease progressing from chronic inflammation and metabolic disease to fibrosis, cirrhosis and ultimately hepatocellular carcinoma (HCC). It is assumed that HCV contributes to liver disease directly by viral factors and indirectly through signaling. Indeed, tumor expression profiling of cirrhotic livers revealed similar genetic profiles between HCV-associated HCCs and other etiologies6. Novel direct antivirals achieve very high cure rates, but despite HCV elimination patients with advanced liver disease remain at high risk for HCC development7. Treatment is costly and is currently only available for a fraction of all HCV-infected patients. Furthermore, resolved infection does not provide protection against reinfection, emphasizing the need for increased understanding of immune evasion for effective vaccine development8.
During recent years thematically focused studies have revealed that virus-host interactions and microbial immune evasion involve the manipulation of both host gene transcription and RNA translation9, 10. However, a global and integrated view on the multiple biological layers is required to understand the host antiviral response and mechanisms leading to disease. Using HCV infection as a model we assessed perturbations of the cellular homeostasis contributing to chronic inflammatory disease and virus-induced cancer. We combined a state-of-the art HCV infection model with cutting edge screening technologies including proteomics, RNA sequencing (RNA-Seq), metabolomics and mathematical modeling to gain a multilayered insight into virus-host interaction and its impact for liver disease biology at the systems level. This approach sheds new light on how viruses evade innate immune responses, reprogram host cell metabolism and trigger chronic inflammatory and metabolic disease as well as cancer.
MATERIALS AND METHODS
HCV infection of human hepatocyte chimeric mice.
Mouse uPA+/+/SCID+/+ (uPA/SCID) liver samples used including ethical approval and informed consent have been described11. Mice with human albumin levels >10mg/mL (~80–90 % human hepatocytes repopulation) were used for this study. Briefly, mice were intravenously inoculated with HCV+ patient serum (105 HCV particles, genotype 1b). Five viremic mice (>107 HCV copies/mL) and five control mice were sacrificed at week eight. RNA-seq data on a subset of these mice (three HCV-infected and three non-infected mice) have been described11. All liver samples were snap frozen and stored at −80 °C prior to analysis.
Immunofluorescence microscopy.
Cells were fixed with 4 % paraformaldehyde for 20 min prior permeabilization (15 min) with 0.1 % Triton X-I00, 0.5 % BSA. Antibody incubation was performed as recommended. Slides were mounted with Fluoroshield including DAPI (Sigma). Fluorescence dots (puncta) of 25 uninfected and 25 HCV-infected cells were quantified as described12.
Differentiation and infection of liver cells.
Huh7.5.113 and HepG2-NTCP14 cells have been described. For proliferation arrest and differentiation (Huh7.5.1dif cells), Huh7.5.1 cells were cultured in DMEM containing 1 % DMSO15 for 10 days before infection. 2.5 × 104 Huh7.5.1dif cells (RNA-Seq and metabolomics) or 1.5 × 106 Huh7.5.1dif cells (proteomics) were infected with a MOI of 8 using affinity purified HCVcc (TCID50 6.7 × 105 / mL) or mock-inoculated with FLAG-peptide elution buffer. After 6 h incubation, the inoculum was replaced by fresh medium supplemented with 1 % DMSO. This MOI is >10× higher than the minimal MOI (0.12) required to infect 100 % of Huh7.5.1dif cells after 7 days (Supplementary Fig. S1C).
Proteome analysis.
Protein lysates were harvested (duplicates) at day 0, 3, 7 and 10 post-infection (pi) for both HCV-infected and mock-infected Huh7.5.1dif cells. Cells were lysed for 15 min at room temperature in 8.0 M urea buffer containing 75 mM NaCl, 50 mM Tris pH 8, 1 mM EDTA, aprotinin 2 μg/mL (Sigma-Aldrich), leupeptin 10 μg/mL (Roche), PMSF 1 mM (Sigma-Aldrich), phosphatase inhibitor cocktail 2 and phosphatase inhibitor cocktail 3 (Sigma-Aldrich). Snap-frozen mouse liver tissues were crushed in liquid nitrogen and lysed, accordingly. Lysates were cleared at full speed in a bench top centrifuge for 5 min, and stored at −80 °C. Proteomic analyses of Huh7.5.1dif lysates were performed at the Broad Institute of MIT and Harvard (Cambridge, MA, USA) and the tissue lysates were analyzed at the Max Delbruck Center for Molecular Medicine/Berlin Institute of Health (Berlin, Germany) using 10-plex tandem mass tag (TMT) labeling as described16.
Transcriptome profiling.
RNA samples were collected daily between days 0 and 10 pi (triplicates). Cells were lysed in TCL buffer (Qiagen) supplemented with 1 % 2-mercaptoethanol. Cellular mRNA was isolated and analyzed as described17. RNA-Seq was performed at the Broad Institute of MIT and Harvard (Cambridge, MA, USA). Therefore, HCV RNA was co-amplified with cellular mRNA using a SMART-compatible primer (sequence: 5’-Biotin-AAG CAG TGG TAT CAA CGC AGA GTA CTC TGC GGA ACC GGT GAG TA-3’)18. Cellular mRNA was isolated and analyzed according to the SmartSeq2 protocol as described17, 19. RNA sequencing paired-end reads were aligned to the human hg19 UCSC reference using TopHat software (v2.0.14). HCV RNA sequencing paired-end reads were aligned to the HCV Jc1E2FLAG genome using Bowtie2 (v2.2.5) software. Gene expression levels for 21,950 human genes were estimated using Cufflinks (v2.2.1) with the fragments per kilobase of transcript per million mapped reads method.
Metabolomics and lipid analysis.
Analysis of polar metabolites was performed in Huh7.5.1dif cells infected with HCV Jc1E2FLAG (MOI=8). Intra/extra-cellular metabolites were analyzed by mass spectrometry as described20 by the Barteesy lab at the Massachusetts General Hospital (Boston, MA, USA). Methyl derivatives of very long-chain fatty acids (VLCFA) were extracted in the presence of internal standards by the Ricci and Dali-Youcef labs and analyzed by gas chromatography-mass spectrometry (GC-MS) at the Hôpitaux Universitaires de Strasbourg (France) as described21.
Bioinformatical analyses
Proteins and transcripts were mapped despite possible many-to-many relationships (i.e. isoforms) by constructing “analysis groups”22 Host responses to HCV infection were assessed over a time course on a temporal population level. Pre-ranked gene set enrichment analysis (GSEA)23 was performed on a temporal population level, taking time points as continuous class label, and taking the control time course as day 0 samples. To compare the two different sources, an indirect comparison was applied. Each time point for both sources were mapped independently to a functional metaspace by performing GSEA, implemented in GenePattern genomic analysis toolkits24 for each gene set in the collection. For each time point, we compared the HCV-infected samples with the mock samples using the Signal-To-Noise ratio and the default setting for GSEA. Correlation to time point for the HCV-infected samples was used to obtain a general enrichment over the entire time course. Next, we compared the significant enrichment scores (ES) (p-value <0.005) for the transcriptional and proteomic data sets in this metaspace. The following gene sets from the Molecular Signature Database25 (MSigDB, ver.4.0) were included for the analysis: BIOCARTA, BIOPLEX, KEGG, NABA, PID, REACTOME, SA, SIG, ST, CORUM, HCVpro, HALLMARK, E1, E2, E3, DUB. “Leading-edge” genes were extracted from all previous GSEA analyses, followed by overlap analyses using the hypergeometric test. The data set was compared to HCVpro database signatures identifying significant overlaps with the current knowledge in HCV-host interactions. Raw reads of patient’s samples with low ISG26 were trimmed using cutadapt27 and mapped to the human genome hg19 using HISAT2. Reads mapping to GENCODE v19 genes were counted with htseq-count. Differentially expressed genes were analyzed using DESeq2. Common transcription factors as potential regulators of HCV-impaired peroxisome expression were identified using Enrichr28. The top 30 transcription factors were retrieved from a combined score ranking (calculated by multiplying p-value (Fisher extract test) and z-score) of the protein-protein interaction (PPI) database and those profiled by ChIP-Seq in mammalian cells (ChEA). These were then successively compared to transcription factors retrieved manually using GeneCards (www.genecards.org), that potentially regulate more than 2 peroxisomal genes. Inference of the transcriptional regulatory networks underlying HCV infection using AMARETTO is described in supplementary information.
RESULTS
An integrated proteogenomic approach reveals the spatiotemporal map of HCV-hepatocyte interactions during infection
We used an hepatocyte-like cell culture model consisting of DMSO-differentiated Huh7-derived liver cells (Huh7.5.1dif) because it is suitable for robust, long-term culture and has been shown to have a similar phenotype than primary human hepatocytes in cell culture15, 29. Indeed, Huh7.5.1dif are quiescent and display enhanced hepatocyte-specific marker expression compared to undifferentiated Huh7.5.1 cells (Supplementary Fig. S1A–B). Huh7.5.1dif were infected with HCVcc (Jc1E2FLAG) prior sampling during a 10 days culture period (Fig. 1A). HCV protein expression was visualized by a HCV peptide time course analysis (Fig. 1B) and quantified 59 peptides from 8 viral proteins (Fig. 1C). During infection all HCV peptides increased in abundance until day 7 pi (Fig. 1C) simultaneously (p<0.0001, Chi-Square Test), with indifferent expression levels or kinetics (p>0.05, U-Test). These results suggest comparable half-lives of the viral proteins indicating that the HCV polyprotein abundance defines the amount of individual cleavage products. We therefore focused our investigations on these first 7 days. In total we quantified 21,950 mRNAs and 8,297 proteins providing a multidimensional atlas of persistent HCV infection. The atlas reveals a time-resolved proteogenomic state of HCV-infected cells. 7,416 proteins (90.8 %) were uniquely mapped to overlapping mRNAs using integrative functional genomic analyses (Fig. 2A, Supplementary table S1). The replicates (n=2) demonstrated excellent sensitivity and technical quality of the approach superior to recent studies for other viral infections22 (Supplementary Fig. S2).
Fig. 1. Mapping of HCV protein expression by time-resolved proteogenomics.
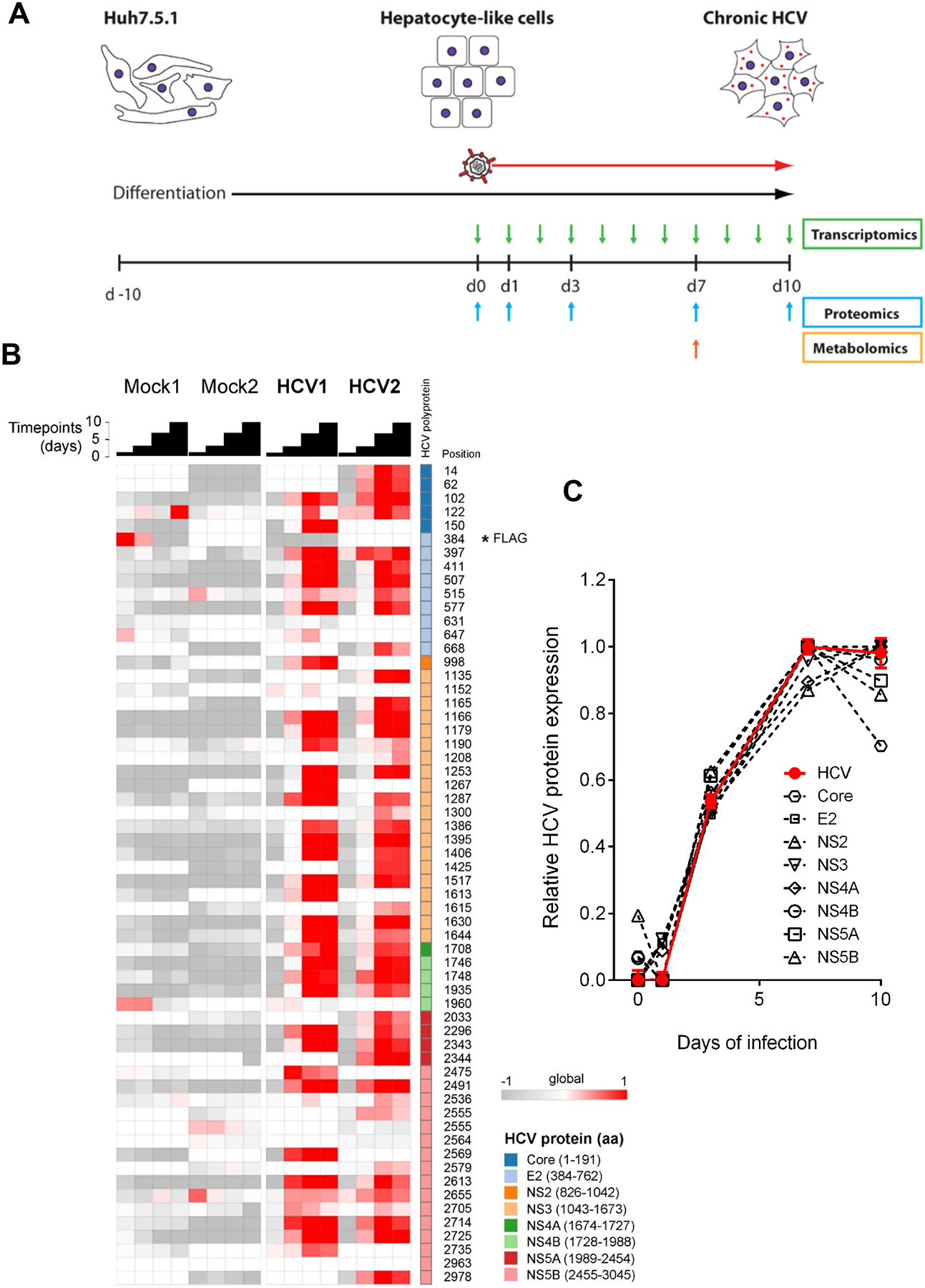
(A) Hepatocyte-like Huh7.5.1dif cells were infected with HCVcc (Jc1E2FLAG) over 10 days. Sampling intervals for transcriptomics, metabolomics and proteomics are indicated. (B) Quantification of HCV-specific peptides over the infection time course relative to non-infected and mock-infected (Mock) cells. The data represent TMT ratios with the 131 channels in the denominator (internal reference = mix of all samples). All ratios were normalized for the median of all 120,000 distinct peptides for each time point. The FLAG peptide is indicated by asterisk. (C) Increase of HCV protein abundance between 3–7 days pi. Data are displayed as relative expression (mean) of all peptides corresponding to a given HCV protein over the time course. HCV replication is depicted in red as median of total HCV peptide abundance ± standard error of the median.
Fig. 2. Persistent HCV infection manipulates host pathways and triggers an attenuated innate immune response.
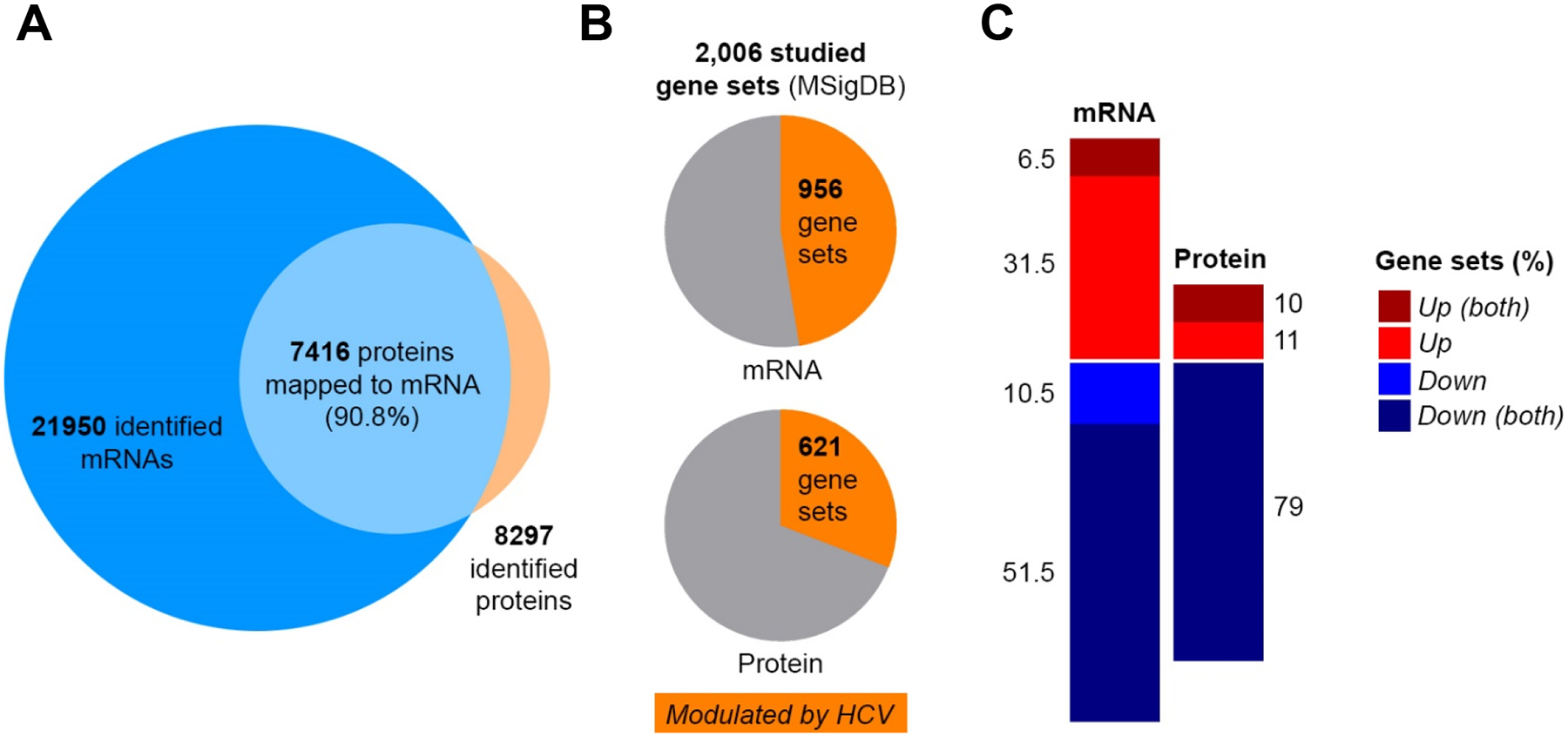
HCV-infected Huh7.5.1dif cells relative to mock-infected cells until 7 days pi. (A) Proteogenomic mapping of HCV infection. (B) Gene sets obtained from the MSigDB that are modulated by HCV infection on the RNA and protein level. (C) Stimulation (red) and suppression (blue) of host pathways (gene sets) by HCV infection.
Disparate dynamics between host cell mRNA and proteins upon HCV infection
We then analyzed 2,006 predefined gene sets representing specific host pathways, cellular functions (MSigDB database) and protein complexes (Bioplex) with our proteogenomic data set using GSEA23. This approach classifies differentially expressed genes or proteins according to their representation within a pre-defined gene set associated to a phenotype. 47.4 % (956 gene sets, transcriptomic data set) and 31 % (621 gene sets, proteomic data set) of these gene sets were significantly altered by HCV over 7 days pi (Fig. 2B). Most of these gene sets were generally downregulated (negatively enriched). In gene set collections with >50 gene sets (BIOCARTA, BIOPLEX, KEGG, PID, REACTOME, CORUM, HALLMARK) a median of 62 % and 79 % of RNA and protein, were negatively enriched, respectively. Moreover, 83 % of these downregulated gene sets on the RNA level were also significantly impaired on the protein level. In contrast, only 17 % of the upregulated pathways on the RNA level matched to the corresponding protein trends (Fig. 2C). This suggests that HCV infection shuts down most of the non-vital processes on the transcriptional and/or at the posttranscriptional level to divert the resources toward viral replication and persistence. Upregulated pathways include the antiviral response and inflammation as well as pro-viral signaling pathways as discussed in the supplementary information (Supplementary Fig. S3, Supplementary table S2).
Persistent HCV infection impairs peroxisome function, lipid metabolism, fatty acid and bile acid metabolism
A striking observation from our proteogenomic atlas of HCV infection is a strongly impaired peroxisomal function as suggested by the GSEA at the RNA and protein levels (Fig. 3A). Peroxisomes are involved in lipid synthesis, signaling, ß-oxidation of VLCFAs and the detoxification of hydrogen peroxide30. Accordingly, we observed an impaired expression of genes involved in peroxisomal biogenesis, bile acid metabolism, fatty acid metabolism and cholesterol biosynthesis (Fig. 3A). We confirmed these findings in a transcriptomic database comprising liver biopsies of 25 patients with chronic HCV infection and 6 non-infected individuals26 and in livers of HCV-infected chimeric mice (n=3) (Fig. 3A). Comparing the GSEA results of HCV-infected patients with Huh7.5.1dif we identified 46 leading-edges of the HALLMARK_PEROXISOME gene signature that are impaired in infected Huh7.5.1dif cells and in liver tissue of HCV-infected patients. The expression of 11 of these leading-edge genes changed significantly (p<0.05, Wald test) in HCV-infected Huh7.5.1dif cells and in the livers of HCV-infected patients (Fig. 3B, Supplementary Fig. S4). We further validated this finding by demonstrating impaired catalase expression in infected cells (Fig. 3C, Supplementary Fig. S5), which is a peroxisome-specific enzyme. Consistently, quantification of catalase-stained peroxisomes revealed a significantly (p<0.005, T-Test) lower number of catalase puncta formed in HCV-infected cells compared to uninfected hepatocytes (Fig. 3C) suggesting an impaired metabolic function of these organelles and an accumulation of fatty acids in infected cells. Whether less peroxisomes are formed during HCV infection cannot be concluded for sure, however a positive correlation between catalase expression and peroxisome abundance has been recently suggested31. Moreover, we demonstrate that HCV infection of Huh7.5.1dif increases intracellular concentrations of very long-chain fatty acids with a chain length of 20–26 carbons, while shorter fatty acids (C16–C18) are less accumulated (Fig. 4A). This is consistent with the formation of intrahepatic lipid droplets during infection, which are important during viral assembly32 and the accumulation of hepatic lipids during steatosis. At the same time, impaired peroxisomes will increase oxidative stress imposed by HCV infection increasing the oncogenic pressure on infected cells. In contrast to HCV, persistent HBV infection increased peroxisomal function and its associated metabolic processes (Fig. 3D), which was confirmed by GSEA of gene expression in HBV-infected primary human hepatocytes (data set GSE69590)33. This is consistent with the clinical pathology of chronic HBV infection where steatosis is much less common34.
Fig. 3. HCV but not HBV infection impairs expression of peroxisomal genes in Huh7.5.1dif cells and liver tissue of patients.
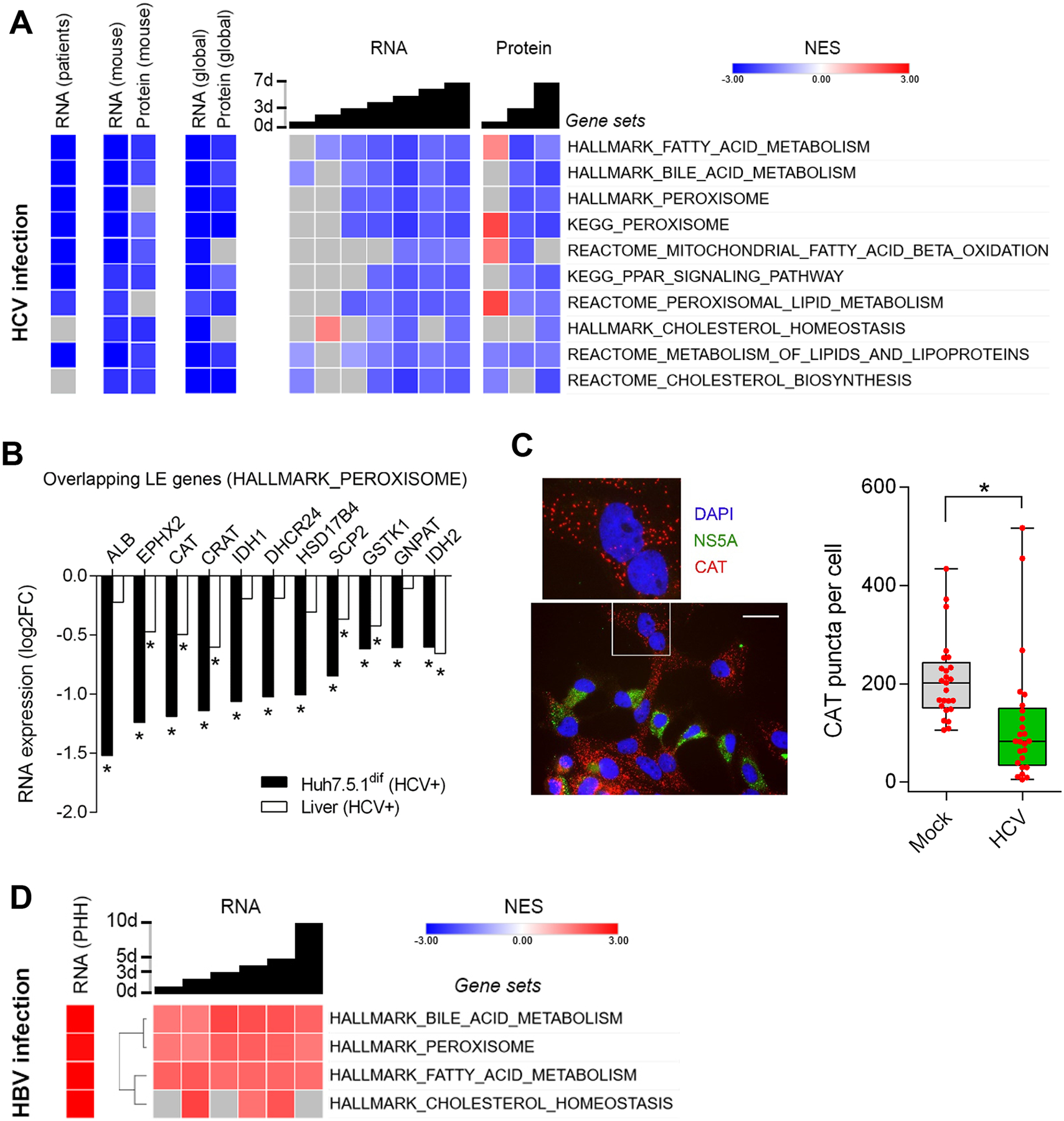
(A) HCV infection impairs metabolic pathways associated to peroxisomal function and lipid homeostasis. GSEA of RNA-Seq data from liver tissue of 25 chronic HCV-infected patients vs. 6 non-infected individuals26, and transcriptomics and proteomics of HCV-infection time-course of Huh7.5.1dif relative to mock-infected cells (n=2). (B) Expression of 11 peroxisomal genes is significantly (p<0.05, Wald test) suppressed by HCV in Huh7.5.1dif and in liver tissue of patients with chronic HCV infection26. Log fold change of leading-edge gene expression of the HALLMARK_PEROXISOME gene set (A). (C) Peroxisome marker expression is significantly (p=0.0048, T-Test) perturbed in HCV-infected hepatocyte-like cells. Immunofluorescence microscopy of Huh7.5.1dif cells infected for 3 days with HCVcc. The peroxisomal marker catalase (CAT) is stained in red, nuclear DNA (DAPI) in blue and HCV (NS5A) in green (see also supplementary Fig. S5). Quantification of catalase-stained peroxisomes in 25 random HCV-infected cells and 25 mock cells is shown in box/whiskers; bar represents 200 μm. (D) HBV infection promotes peroxisome function. GSEA of transcriptomics of HBV-infection of HepG2-NTCP for 10 days (n=3), and primary human hepatocytes (n=3) infected for 40 days with HBV (genotype D) (GSE69590). NES are displayed in red (increased), blue (decreased), and gray (no significant change). Temporal analysis of infected Huh7.5.1dif are presented as global trend (global) and individual timepoints. Statistical cut-off for GSEA of liver tissues was FDR q<0.05 and for Huh7.5.1dif p<0.005.
Fig. 4. Persistent HCV infection enhances metabolism of intracellular very long-chain fatty acids and glucose and creates a Warburg-like shift of the lactate flux.
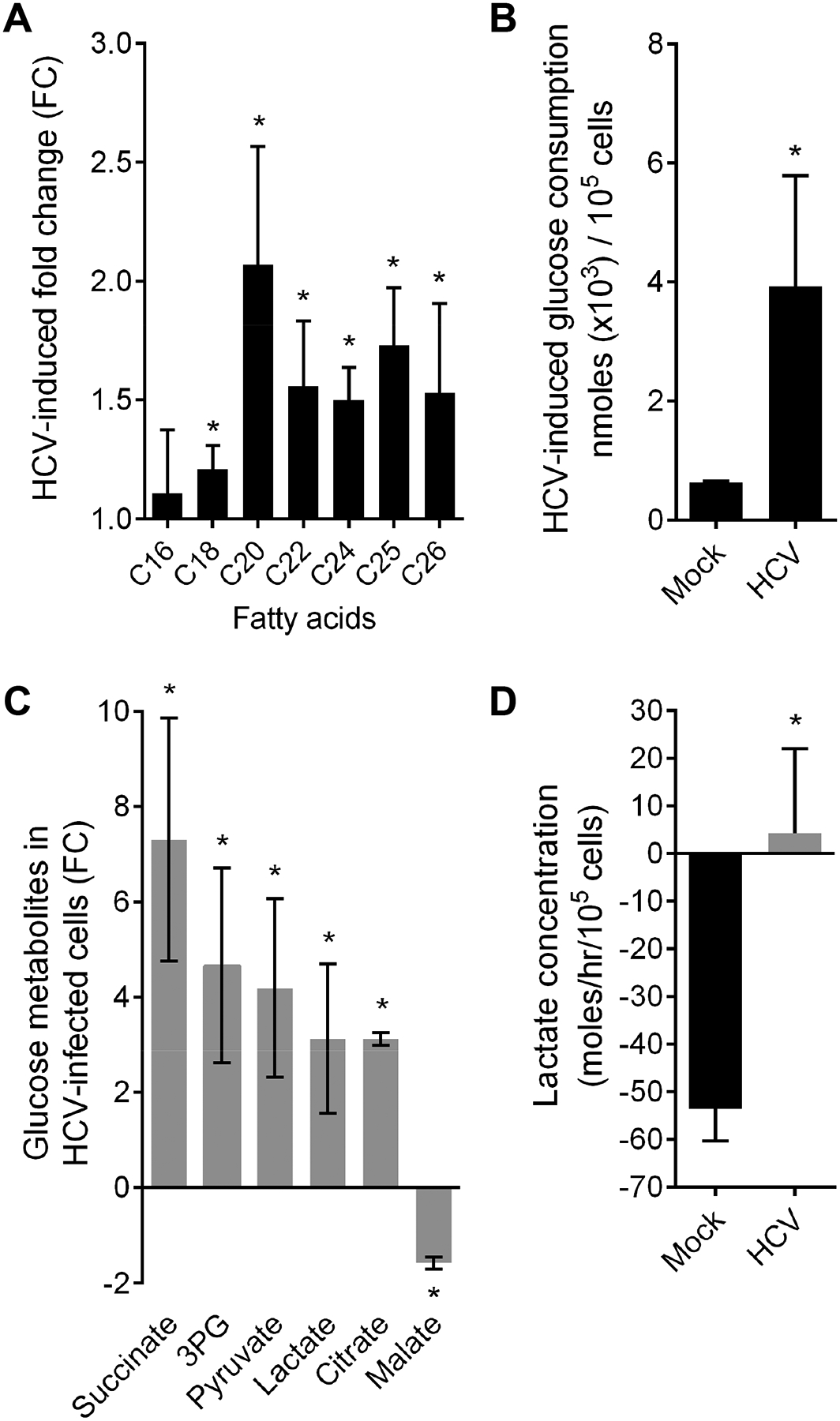
(A) HCV infection induces intracellular accumulation of very long-chain fatty acids (VLCFA). Data expressed as mean fold change of intracellular C16 (palmitic acid), C18 (oleic acid), C20 (arachidic acid), C22 (behenic acid), C24 (lignoceric acid), C25 (pentacosanoic acid), C26 (cerotic acid) fatty acids relative to mock-infected cells ±SEM (2–3 independent experiments). (B) HCV-infection increases the concentration of glucose metabolites in infected Huh7.5.1dif except for malate. Mean fold change of intracellular polar metabolites per 100,000 cells ±SD (4 independent experiments in triplicates). (C) HCV infection of Huh7.5.1dif increases glucose consumption ~6 fold. Glucose consumption of HCV- and mock-infected (CTRL) cells was measured at day 7 pi in supernatants. Mean glucose consumption per 100,000 cells ±SD (2 independent experiments in triplicates). (D) HCV creates a Warburg-like shift in infected cells. Lactate uptake (negative value) from culture supernatant inverses to lactate secretion (positive value) in infected Huh7.5.1dif cells. Mean lactate flux ±SEM (mole/h/100,000 cells; 4 independent experiments in triplicates). * p<0.05, T-Test.
Impaired PPAR signaling is a regulator of peroxisomal function during HCV infection
We hypothesized that impaired peroxisomal function was the result of direct, virus-related and indirect effects linked to host cell metabolism. The nuclear receptor peroxisomal proliferator-activated receptor (PPAR) is associated with peroxisome function and highly expressed in the liver. VLCFAs activate PPAR-alpha35 and high glucose levels impair its activity36. Indeed, we observed an impaired PPAR signaling expression signature in HCV-infected Huh7.5.1dif cells and in the liver of patients on both RNA and the protein level (Fig. 3A), suggesting a perturbed switch between fatty acid and glucose metabolism in HCV-infected hepatocytes. This is consistent with suppressed PPAR-alpha expression by HCV infection37. Therefore, we studied the metabolic status of HCV-infected Huh7.5.1dif cells using mass spectrometry-based metabolomic profiling20. Analysis of polar metabolites revealed elevated concentrations of the glucose metabolites succinate, pyruvate, 3-phosphoglycerate and citrate in HCV-infected Huh7.5.1dif cells (Fig. 4B, Supplementary table S4), consistent with the high glucose-dependence of the HCV replication38. Indeed, our analyses revealed an increased glucose consumption (Fig. 4C) and an accumulation of lactate and decrease of malate arguing for impaired gluconeogenesis in infected cells (Fig. 4B). In healthy individuals, lactate is produced and secreted by glycolysis-dependent tissues including skeletal muscle, bone marrow and hypoxic tissue. Lactate is then rapidly metabolized in the liver to glucose and energy. As shown in Supplementary Fig. S6 HCV infection induces hypoxia presumably contributing to lactate production. When measuring the hepatocellular lactate flux, a significant accumulation inside the cells was observed (Fig. 4D). Collectively, these data demonstrate that persistent HCV infection causes elevated glucose levels in infected cells contributing to impaired peroxisomal functions. Moreover, infection creates a Warburg-like metabolic shift of the host metabolism which is a hallmark of cancer or cells undergoing carcinogensis39. Next, we aimed to identify common transcription factors of peroxisomal genes that are potentially interacting with HCV proteins. Thereto, we identified common transcription factor binding sites among the combined 85 leading edge genes of the HALLMARK_PEROXISOME gene set in livers of HCV-infected patients and Huh7.5.1dif cells (Supplementary table S5) by using Enrichr28. Furthermore, we added to the 11 leading edge genes described in Fig. 3B common transcription factors potentially binding to more than 2 leading edge genes. As a result, we predicted 15 transcription factors potentially involved in the transcription of peroxisomal genes and suppressed by HCV infection (Supplementary table S5). Among these HCV-sensitive regulators were PPAR-alpha (PPARA) and its functional partner Retinoid X Receptor Beta (RXRB). Moreover, a comparison of the identified transcription factors with the HCVpro protein interaction database of HCV-host interactions40, revealed that HCV core protein associates to PPAR-alpha and RXR-beta. In line with these computational analyses, we observed that HCV infection strongly inhibits PPAR-alpha expression in Huh7.5.1dif (Fig. 5A). Finally, we analyzed the RNA-Seq profiles of the HCV infection and non-infected control time courses using the AMARETTO algorithm41. This regulatory network inference tool learns regulatory modules by connecting known regulatory driver genes with co-expressed target genes that they control. Interestingly, AMARETTO highlighted interleukin 6 receptor (IL-6R) as a driver candidate of a module comprising a significant (FDR<0.001) functional negative enrichment of pathways associated to peroxisome function (7 genes), fatty acid (12 genes) and lipid metabolism (20 genes) (Fig. S7, Supplementary Table S6–7). This suggests a functional role for IL-6/STAT3 signaling in the regulation of peroxisome function in HCV infection. Indeed, infection induces a STAT3 transcriptional signature5 (Supplementary Fig. S6) and STAT3 activation cause a rapid inhibition of IL-6R transcripts (Fig. 5B) as part of a negative feedback regulation. Moreover, inhibition of STAT3 activity by niclosamide (Fig. 5C) rescued the virus-induced inhibition of peroxisomal genes (Fig. 5D) suggesting a regulatory link between STAT3 signaling and peroxisomes. Indeed, inflammatory pathways regulate PPAR family members42.
Fig. 5. HCV inhibits peroxisomal gene expression by suppressing PPAR-alpha function via STAT3 signaling.
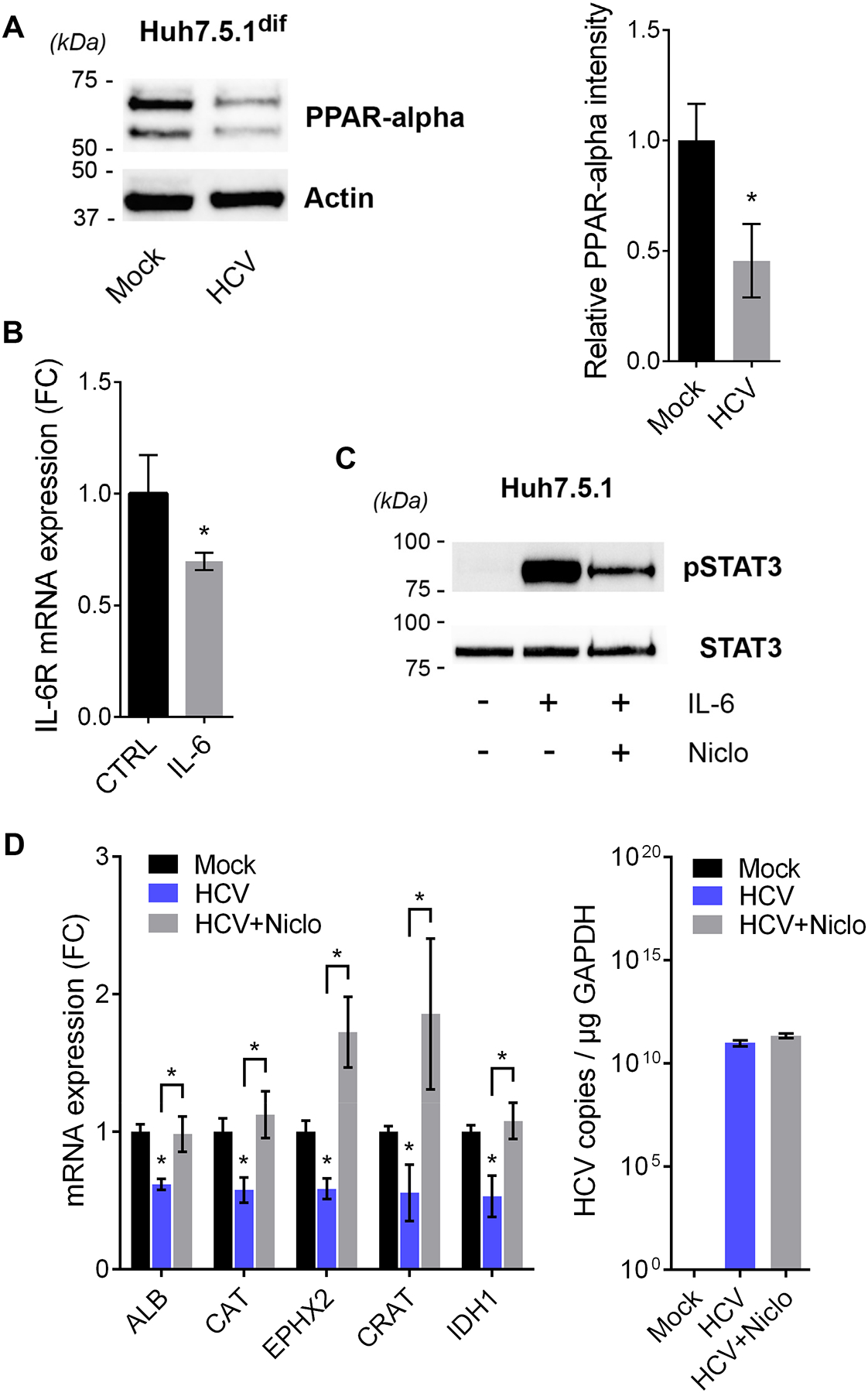
(A) HCV infection of Huh7.5.1dif significantly (p<0.05, U-Test) inhibits PPAR-alpha expression. Western blotting for PPAR-alpha and actin after 7 days of infection with HCVcc (Jc1). Quantification of band intensities of PPAR-alpha and actin. Mean relative PPAR-alpha intensity ±SD (2 independent experiments in triplicates). (B) Activation of the IL-6/STAT3 pathway rapidly downregulates IL-6 receptor (IL-6R). Huh7.5.1 cells were incubated 6 h with 10 ng/mL IL-6 prior RNA extraction and qPCR analysis. Mean fold change (FC) ±SEM triplicates. * p<0.05 (T-Test). (C) Niclosamide inhibits IL-6-induced STAT3 phosphorylation. Incubation of Huh7.5.1 cells with 2 μM niclosamide for 24 h and/or 10 ng IL-6 (30 min). Western blotting for pSTAT3 (Y705) and total STAT3. (D) HCV-induced inhibition of peroxisomal genes is rescued by the STAT3-inhibitor niclosamide. Huh7.5.1dif cells were infected with HCVcc (Jc1) for 7 days. At day 6 pi cells were treated with solvent control or 2 μM niclosamide. Niclosamide reversed the HCV-induced inhibition of 5 top leading-edge genes of the HALLMARK_PEROXISOME gene set (Fig. 3B, Supplementary Fig. S4), while it had no effect on HCV replication. Mean fold change of copy number normalized to GAPDH from triplicates ±SD. *p<0.05 T-Test.
Taken together, our data suggest that HCV infection suppresses peroxisomal function at different levels by interfering with PPAR-alpha expression and function. This impacts the metabolic switch between fatty acid and glucose metabolism and thus contributes to glucose-dependence and lipid accumulations in infected hepatocytes that is a hallmark of chronic liver disease and HCC development.
Impaired peroxisomal function is associated with HCV clinical liver disease including cirrhosis and HCC
As perturbed peroxisome function might be relevant for outcome of HCV-associated liver disease, we analyzed the gene expression in liver biopsies from 216 patients with HCV-related early-stage liver cirrhosis43 as well as in paired liver biopsies of patients with HCV-associated HCC44 (Supplementary table S8–9). Comparing the enrichment of the HALLMARK_PEROXISOME gene set in each individual sample to the clinical outcome of the patients, we revealed a significant association of peroxisomal gene expression with liver cirrhosis (p=0.001, log-rank test), HCC development (p=0.03, log-rank test) and patient survival from HCC (p=0.006, log-rank test) (Fig. 6A). This suggests that HCV patients with low peroxisomal function have high risk for poor clinical outcomes. Moreover, peroxisomal gene expression significantly correlates with collagen, type I, alpha 1 expression (COL1A1) (R=−0.16, p=0.02, Spearman correlation test) and with α-smooth muscle actin (ACTA2) (R= −0.19, p=0.004, Spearman correlation test) (Fig. 6B), suggesting elevated extracellular matrix deposition and liver injury in patient livers with low peroxisomal activity6. This also reflects the observed negative association of the peroxisomal gene expression with the Child-Pugh liver disease score in Fig. 6A. Interestingly, peroxisomal gene expression was reduced in tumor (T) and adjacent tissue (A) of HCC patients with steatohepatitis (SH-HCC) (Fig. 6C) suggesting that an impaired peroxisome is not only associated to fatty liver development but also a hallmark of tumors reflecting their glucose dependence and the shutting down of beta-oxidation45, 46. We validated key findings in Fig. 6 using CAT expression (Supplementary Fig. S8) thus, confirming the reliability of CAT as functional peroxisomal marker in patients. Reactivation of peroxisomal function during liver disease may therefore reduce oxidative stress and the risk of disease progression toward liver cirrhosis and HCC.
Fig. 6. Significant association of hepatic peroxisome expression with clinical outcomes and phenotypes in viral and metabolic liver disease.
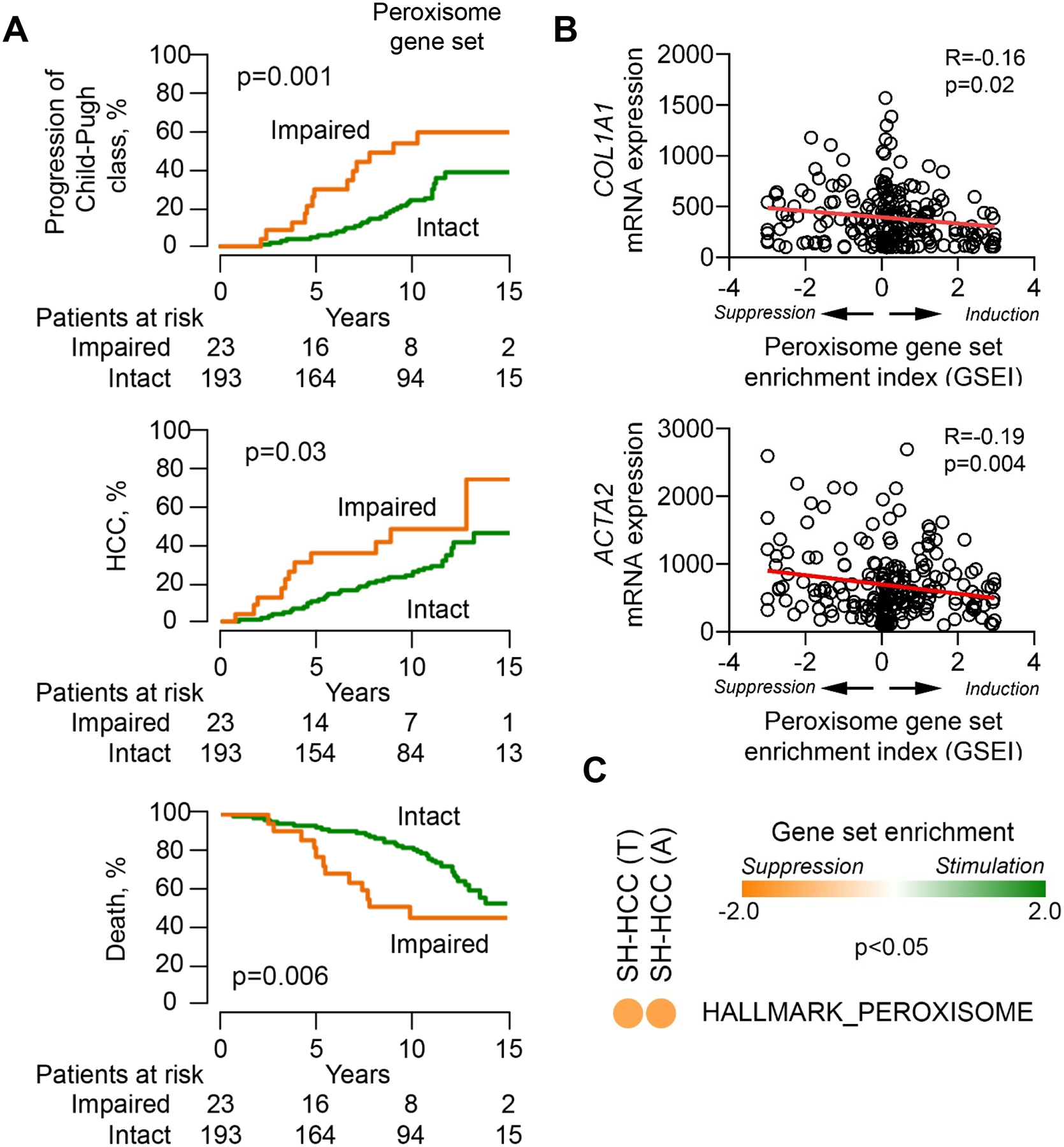
(A) Patients with impaired peroxisomal function showed worse outcome compared to those with intact function (log-rank test). Early-stage HCV cirrhosis patients (n=216)43 are classified into 2 groups based on relative peroxisomal function. Therefore, modulation of the HALLMARK_PEROXISOME gene set25 in each individual sample was used to infer peroxisomal function in the liver tissues. Impaired peroxisomal function was defined by coordinated suppression of the gene set determined by modified gene set enrichment analysis with statistical significance (false discovery rate <0.10)6. (B) Expression of fibrosis-related genes, COL1A1 and ACTA2, tend to be higher in the livers with a suppressed peroxisome pathway (Spearman correlation test). Induction or suppression of the HALLMARK_PEROXISOME gene set were measured by gene set enrichment index (GSEI)6. GSEI was calculated from gene set enrichment p value based on iterative random gene permutations (1,000 times). GSEI of +3 indicates induction at enrichment p= 0.001, GSEI of −3 indicates suppression at enrichment p=0.001, and GSEI of 0 indicates no modulation at enrichment p=1.0. (C) The peroxisome is significantly (p<0.05) suppressed in histological steatohepatitis HCC (SH-HCC) (n=17) compared to other histological types (n=82) in tumor samples (T) (NES=−1.38) and in tissue adjacent to tumors (A) (NES=−1.43) of paired liver biopsies44. GSEA using the HALLMARK_PEROXISOME gene set.
DISCUSSION
Chronic liver disease generally progresses from steatosis, inflammation to fibrosis, cirrhosis and ultimately to HCC. The striking similarities in liver disease progression independent of the underlying etiology, suggests the presence of common drivers and deregulated pathways that promote liver pathogenesis. In this study, we have unraveled the temporal proteogenomic atlas of persistent HCV infection that sheds light to open questions in HCV-host interactions and uncovers virus-induced perturbations of host cell circuits driving viral pathogenesis and disease progression. Using a highly efficient and reproducible infection model, we mapped and quantified for the first time the entire viral proteome in the early and steady-state phase of infection. Surprisingly, infection of the quiescent hepatocyte-like cells did mount a robust interferon response on the RNA level including expression signatures from dsRNA sensors RIG-I, MDA5, TLR3 and their downstream effector IRF3. This is consistent with reports demonstrating MDA5 being more critical for HCV sensing than RIG-I47, 48. HCV replication intermediates are also partially recognized by the TLR3 protein, however not sufficient enough to mount a fully effective host response against HCV49. However, all these antiviral gene expression patterns do not translate into protein in Huh7.5.1dif. These findings support a model where HCV infection overcomes the antiviral defense in hepatocytes mainly by inhibiting the translation of ISG mRNAs via the induction of PKR activity as previously suggested50. Indeed, HCV-infection of our model strongly induced PKR (EIF2AK2) expression at the RNA level which led to a self-limiting expression of PKR protein that spiked at day 1 pi (Supplementary table S1). Why in HCV-infected chimeric mice this evasion strategy seems to be distinctly different from HCV-infected patients and Huh7.5.1dif is an interesting observation and may be useful to refine our understanding of viral immune evasion. This shows that the Huh7.5.1dif is a useful model to study and understand the innate immune responses to HCV infection and serves as an example of how this data set can serve as a resource file to understand temporal changes and putative discrepancies in the proteogenomic landscape of HCV-host interactions. Using this atlas, we validated cancer relevant pro-oncogenic pathways and gene signatures in a thus far unparalleled depth and comprehension (Supplementary Fig. S6) and correlated it with metabolic features resembling cancer, i.e. a Warburg-like shift of the lactate flux in infected cells. Of note, the transcriptomic patterns identified in HCV-infected Huh7.5.1dif were similar to those in liver tissue of patients and HCV-infected human liver chimeric mice (Figs. 3A, Supplementary Figs. S3 and S6) further emphasizing the suitability of this atlas to identify previously unrecognized disease-relevant processes in vivo.
Peroxisomes are key organelles for VLCF metabolism and detoxification of membrane-permeable peroxides and oxidative stress. We observed a marked decrease of the peroxisomal function in HCV-infected hepatocytes, in the livers of HCV patients and in infected chimeric mice, which is distinctly different from patients with chronic hepatitis B where peroxisomal function is increased (Fig. 3). Consistently, HCV but rarely HBV infection is associated with fat accumulations in the liver leading to NAFLD and NASH51. Both HBV and HCV induce NF-kB signaling52, contributing to chronic liver inflammation, injury and disease progression. Inflammation is a regulator of host metabolism53. However, only HCV but not HBV infection impairs peroxisomal function and predominantly induces fat inclusions in hepatocytes arguing for an additional HCV-specific molecular mechanism suppressing fatty acid metabolism. Complementary to previous lipidomic analysis that suggested an HCV-induced accumulation of phospholipids and sphingomyelins38 our data revealed that HCV cause a phenotype of VLCFA accumulation, which corresponds to impaired peroxisomal beta-oxidation. Consequently, HCV infection of differentiated cells as used here mimics a phenotype resembling fatty liver disease and its complication NASH, which is characterized by fatty acid accumulations and chronic inflammation54. HCV infection requires lipid droplets for particle assembly55. The suppressed peroxisomal activity in HCV-infected cells is thus of potential advantage to the virus since it contributes to accumulation of long-chain fatty acids in infected hepatocytes with potential impact on liver pathogenesis. Indeed, HCV-infected cells display a higher abundance of phosphatidylcholines and triglycerides with longer chain fatty acids56, which are preferentially metabolized in the peroxisome. This resembles the situation in NASH patients, where increased systemic phosphatidylcholine levels are observed57 and suggests that HCV infection is a model to study the molecular mechanisms of fatty liver disease independent of the underlying etiology. Moreover, high levels of VLCFA in serum provoked hepatic steatosis, NASH, and HCC in an animal model58. Our data suggest that re-activation of HCV-impaired nuclear receptors like PPAR-alpha has a potential clinical relevance for HCC prevention. Furthermore, targeting of PPAR-alpha is in development for NASH, where treatment options are currently limited. Indeed, PPAR stimulation improves steatosis, inflammation and fibrosis in pre-clinical models of NAFLD59, 60, albeit with limited clinical efficacy. Alternatively, our data highlight a potential new strategy for a restoration of peroxisomal function using clinical STAT3 inhibitors. Collectively, these data indicate that HCV and NASH share similar pathways driving the pathogenesis of liver disease, which are distinctly different from HBV-associated liver disease. The highly similar results obtained in livers of HCV-infected chimeric mice and livers and HCV patients suggest the hepatocyte-like HCV-Huh7.5.1dif as suitable model for the discovery of targets and compounds of metabolic liver disease. Our atlas validated previous transcriptomic studies demonstrating an association of HCV with genes involved in lipid metabolism and reactive oxygen species61, 62 in an unprecedented temporal resolution, which allowed the prediction of regulatory networks (Supplementary data S7). The multi-omics approach in this study integrated transcriptome, proteome, polar metabolites and lipid analysis revealed novel mechanistic insights in the regulation of peroxisomal function by an interplay of glucose levels and HCV-induced cytokine signaling.
Finally, the temporal proteogenomic atlas of HCV infection is a useful and unique resource data set for researchers to validate individual hypotheses in virus-host interactions and liver disease biology. Moreover, the convenient upscaling, the high reproducibility, and the high similarity with gene expression profiles in livers of HCV patients emphasizes the potential of this model for screening approaches targeting drivers of liver disease pathobiology and cancer-risk to identify therapeutics for liver disease in general.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Francis V. Chisari (The Scripps Research Institute) for providing Huh7.5.1 cells, Charles M. Rice (Rockefeller University) for providing JFH1-based sequences and Arvind Patel (MRC Virology Unit, Glasgow) for mAb AP33. Jim Boccachio (Broad Institute) for performing RNA-Seq analyses. We thank Drs. Aviv Regev and Orit Rozenblatt-Rosen for helpful discussions and all members of the AMARETTO/Community-AMARETTO algorithm and software development team, including Drs. Vincent Carey (BWH, HMS, Broad), Jill Mesirov, Michael Reich, Ted Liefeld, Thorin Tabor (UCSD), Jayendra Ravindra Shinde and Shaimaa Hesham Bakr (Stanford).
Grant support:
This work was supported by the European Union (ERC-AdG-2014 HEPCIR to T.F.B. and Y. H., EU H2020 HEPCAR 667273 to T.F.B. and J.L.), the French Cancer Agency (ARC IHU201301187 to T.F.B.), the US Department of Defense (W81XWH-16-1-0363 to T.F.B. and Y. H.), the National Institutes (NIAID R03AI131066 to N.P. and T.F.B, NCI 1R21CA209940 to N.P. and T.F.B. and O.G., NIAID 5U19AI123862-02 to T.F.B, NCI/ITCR U01 CA214846 to N.P. and O.G.), the Fondation de l’Université de Strasbourg (HEPKIN) (TBA-D0N-0002) and the Inserm Plan Cancer 2019–2023 to T.F.B. This work has benefitted from support by the Initiative of excellence IDEX-Unistra (ANR-10-IDEX-0002-02 to J.L.) and has been published under the framework of the LABEX ANR-10-LAB-28 (HEPSYS). Inserm Plan Cancer, IDEX and LABEX are initiatives from the French program “Investments for the future”. Work of R.B. was supported by the Deutsche Forschungsgemeinschaft (TRR179, TP9). K.C. was supported by Research Program on Hepatitis from Japanese Agency for Medical Research and development (AMED) Japan (JP18fk0210020h0002).The work of R.R. and G.M was supported by a European Research Council (ERC) starting grant (ERC-2011-StG, 281271-STRESS METABOL), by the European Foundation for the Study of Diabetes (EFSD)/Lilly European Diabetes Research Program grant and by the ANR-10-LABX-0030-INRT grant, a French State fund managed by the ANR under the frame program Investissements d’Avenir ANR-10-IDEX-0002-02.
Abbreviations:
- ATCC
American Type Culture Collection
- ChEA
DNA sequencing after chromatin immunoprecipitation in mammalian cells
- ChIP-Seq
DNA sequencing after chromatin immunoprecipitation
- CTRL
control
- DAPI
4’,6-diamidino-2-phenylindole
- DMSO
dimethyl sulfoxide
- dsRNA
double-stranded RNA
- EDTA
ethylenediaminetetra-acetic acid
- EMT
epithelial-mesenchymal transition
- ES
enrichment score
- GC-MS
gas chromatography-mass spectrometry
- GEO
Gene expression Omnibus
- GSEA
gene set enrichment analysis
- HBsAg
hepatitis B virus surface antigen
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HCVcc
cell culture-derived hepatitis C virus
- IL-6
Interleukin 6
- ISGs
interferon-stimulated genes
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- MAPK
mitogen-activated protein kinase
- MOI
Multiplicity of Infection
- MSigDB
The Molecular Signatures Database
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- NES
normalized enrichment score
- NF-kB
Nuclear factor kappa B
- pi
post-infection
- PMSF
phenylmethylsulfonyl fluoride
- PPARA
Peroxisome proliferator-activated receptor alpha
- PPI
protein-protein interaction
- RNAi
RNA interference
- RNA-Seq
RNA sequencing
- SD
standard deviation
- SH
steatohepatitis
- SH-HCC
steatohepatitis-associated hepatocellular carcinoma
- SRA
Sequence Read Archive
- STAT3
Signal transducer and activator of transcription 3
- TCID50
50 % tissue culture infective dose
- TMT
tandem mass tag
- uPA/SCID
urokinase-type plasminogen activator/severe combined immunodeficiency
- VLCFA
very long-chain fatty acid.
Footnotes
Disclosures: The authors have nothing to disclose, no conflict of interest.
Proteogenomic profiling:
The complete RNA-seq time-course profiling of HCV-infected Huh7.5.1dif including baseline expression levels are accessible in GEO (GSE126831). Transcriptomic profiling of HCV-infected chimeric mice can be retrieved from SRA data set SRP170244 (biosamples: SAMN10465389, SAMN10465390, SAMN10465391, SAMN10465395, SAMN10465396, SAMN10465397). Proteomic profiling of HCV-infected Huh7.5.1dif and chimeric mice are accessible at MassIVE (MSV000083382). RNA-Seq from infected vs. control patients were obtained from the GEO data set GSE84346 (low ISG samples).
REFERENCES
- 1.Pittman KJ, Aliota MT, Knoll LJ. Dual transcriptional profiling of mice and Toxoplasma gondii during acute and chronic infection. BMC Genomics 2014; 15:806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lupberger J, Casanova C, Fischer B, et al. PI4K-beta and MKNK1 are regulators of hepatitis C virus IRES-dependent translation. Sci Rep 2015;5:13344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lupberger J, Zeisel MB, Xiao F, et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med 2011;17:589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mailly L, Xiao F, Lupberger J, et al. Clearance of persistent hepatitis C virus infection in humanized mice using a claudin-1-targeting monoclonal antibody. Nat Biotechnol 2015;33:549–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Renne N, Roca Suarez AA, Duong FHT, et al. miR-135a-5p-mediated downregulation of protein tyrosine phosphatase receptor delta is a candidate driver of HCV-associated hepatocarcinogenesis. Gut 2018;67:953–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakagawa S, Wei L, Song WM, et al. Molecular Liver Cancer Prevention in Cirrhosis by Organ Transcriptome Analysis and Lysophosphatidic Acid Pathway Inhibition. Cancer Cell 2016;30:879–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Meer AJ, Feld JJ, Hofer H, et al. Risk of cirrhosis-related complications in patients with advanced fibrosis following hepatitis C virus eradication. J Hepatol 2017;66:485–493. [DOI] [PubMed] [Google Scholar]
- 8.Bartenschlager R, Baumert TF, Bukh J, et al. Critical challenges and emerging opportunities in hepatitis C virus research in an era of potent antiviral therapy: Considerations for scientists and funding agencies. Virus Res 2018;248:53–62. [DOI] [PubMed] [Google Scholar]
- 9.Arvey A, Tempera I, Tsai K, et al. An atlas of the Epstein-Barr virus transcriptome and epigenome reveals host-virus regulatory interactions. Cell Host Microbe 2012;12:233–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramage HR, Kumar GR, Verschueren E, et al. A combined proteomics/genomics approach links hepatitis C virus infection with nonsense-mediated mRNA decay. Mol Cell 2015;57:329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamdane N, Jühling F, Crouchet E, et al. HCV induces persistent epigenetic reprogramming post sustained virological response associated with liver cancer risk. Gastroenterology 2019;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Di Cara F, Sheshachalam A, Braverman NE, et al. Peroxisome-Mediated Metabolism Is Required for Immune Response to Microbial Infection. Immunity 2017;47:93–106 e7. [DOI] [PubMed] [Google Scholar]
- 13.Zhong J, Gastaminza P, Cheng G, et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 2005;102:9294–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verrier ER, Yim SA, Heydmann L, et al. Hepatitis B virus evasion from cGAS sensing in human hepatocytes. Hepatology 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bauhofer O, Ruggieri A, Schmid B, et al. Persistence of HCV in quiescent hepatic cells under conditions of an interferon-induced antiviral response. Gastroenterology 2012;143:429–38 e8. [DOI] [PubMed] [Google Scholar]
- 16.Svinkina T, Gu H, Silva JC, et al. Deep, Quantitative Coverage of the Lysine Acetylome Using Novel Anti-acetyl-lysine Antibodies and an Optimized Proteomic Workflow. Molecular & cellular proteomics : MCP 2015;14:2429–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trombetta JJ, Gennert D, Lu D, et al. Preparation of Single-Cell RNA-Seq Libraries for Next Generation Sequencing. Curr Protoc Mol Biol 2014;107:4 22 1–4 22 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiao F, Fofana I, Heydmann L, et al. Hepatitis C virus cell-cell transmission and resistance to direct-acting antiviral agents. PLoS Pathog 2014;10:e1004128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Picelli S, Faridani OR, Bjorklund AK, et al. Full-length RNA-seq from single cells using Smart-seq2. Nature protocols 2014;9:171–81. [DOI] [PubMed] [Google Scholar]
- 20.Nicolay BN, Gameiro PA, Tschop K, et al. Loss of RBF1 changes glutamine catabolism. Genes & development 2013;27:182–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takemoto Y, Suzuki Y, Horibe R, et al. Gas chromatography/mass spectrometry analysis of very long chain fatty acids, docosahexaenoic acid, phytanic acid and plasmalogen for the screening of peroxisomal disorders. Brain Dev 2003;25:481–7. [DOI] [PubMed] [Google Scholar]
- 22.Jovanovic M, Rooney MS, Mertins P, et al. Immunogenetics. Dynamic profiling of the protein life cycle in response to pathogens. Science 2015;347:1259038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reich M, Liefeld T, Gould J, et al. GenePattern 2.0. Nat Genet 2006;38:500–1. [DOI] [PubMed] [Google Scholar]
- 25.Liberzon A, Birger C, Thorvaldsdottir H, et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boldanova T, Suslov A, Heim MH, et al. Transcriptional response to hepatitis C virus infection and interferon-alpha treatment in the human liver. EMBO Mol Med 2017;9:816–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011;17:10–12. [Google Scholar]
- 28.Chen EY, Tan CM, Kou Y, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 2013;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sainz B Jr., Chisari FV. Production of infectious hepatitis C virus by well-differentiated, growth-arrested human hepatoma-derived cells. J Virol 2006;80:10253–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lodhi IJ, Semenkovich CF. Peroxisomes: a nexus for lipid metabolism and cellular signaling. Cell metabolism 2014;19:380–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee JN, Dutta RK, Maharjan Y, et al. Catalase inhibition induces pexophagy through ROS accumulation. Biochem Biophys Res Commun 2018;501:696–702. [DOI] [PubMed] [Google Scholar]
- 32.Paul D, Madan V, Bartenschlager R. Hepatitis C virus RNA replication and assembly: living on the fat of the land. Cell Host Microbe 2014;16:569–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoneda M, Hyun J, Jakubski S, et al. Hepatitis B Virus and DNA Stimulation Trigger a Rapid Innate Immune Response through NF-kappaB. J Immunol 2016;197:630–43. [DOI] [PubMed] [Google Scholar]
- 34.Fan JG, Farrell GC. Epidemiology of non-alcoholic fatty liver disease in China. J Hepatol 2009;50:204–10. [DOI] [PubMed] [Google Scholar]
- 35.Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature 2000;405:421–4. [DOI] [PubMed] [Google Scholar]
- 36.Roduit R, Morin J, Masse F, et al. Glucose down-regulates the expression of the peroxisome proliferator-activated receptor-alpha gene in the pancreatic beta -cell. J Biol Chem 2000;275:35799–806. [DOI] [PubMed] [Google Scholar]
- 37.Dharancy S, Malapel M, Perlemuter G, et al. Impaired expression of the peroxisome proliferator-activated receptor alpha during hepatitis C virus infection. Gastroenterology 2005;128:334–42. [DOI] [PubMed] [Google Scholar]
- 38.Diamond DL, Syder AJ, Jacobs JM, et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog 2010;6:e1000719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.San-Millan I, Brooks GA. Reexamining cancer metabolism: lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2017;38:119–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kwofie SK, Schaefer U, Sundararajan VS, et al. HCVpro: hepatitis C virus protein interaction database. Infect Genet Evol 2011;11:1971–7. [DOI] [PubMed] [Google Scholar]
- 41.Champion M, Brennan K, Croonenborghs T, et al. Module Analysis Captures Pancancer Genetically and Epigenetically Deregulated Cancer Driver Genes for Smoking and Antiviral Response. EBioMedicine 2018;27:156–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mukherji A, Kobiita A, Ye T, et al. Homeostasis in intestinal epithelium is orchestrated by the circadian clock and microbiota cues transduced by TLRs. Cell 2013;153:812–27. [DOI] [PubMed] [Google Scholar]
- 43.Hoshida Y, Villanueva A, Sangiovanni A, et al. Prognostic gene expression signature for patients with hepatitis C-related early-stage cirrhosis. Gastroenterology 2013;144:1024–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoshida Y, Villanueva A, Kobayashi M, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med 2008;359:1995–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buzzai M, Bauer DE, Jones RG, et al. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid β-oxidation. Oncogene 2005;24:4165. [DOI] [PubMed] [Google Scholar]
- 46.Fujiwara N, Nakagawa H, Enooku K, et al. CPT2 downregulation adapts HCC to lipid-rich environment and promotes carcinogenesis via acylcarnitine accumulation in obesity. Gut 2018;67:1493–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cao X, Ding Q, Lu J, et al. MDA5 plays a critical role in interferon response during hepatitis C virus infection. J Hepatol 2015;62:771–8. [DOI] [PubMed] [Google Scholar]
- 48.Hiet MS, Bauhofer O, Zayas M, et al. Control of temporal activation of hepatitis C virus-induced interferon response by domain 2 of nonstructural protein 5A. J Hepatol 2015;63:829–37. [DOI] [PubMed] [Google Scholar]
- 49.Grunvogel O, Colasanti O, Lee JY, et al. Secretion of Hepatitis C Virus Replication Intermediates Reduces Activation of Toll-Like Receptor 3 in Hepatocytes. Gastroenterology 2018;154:2237–2251 e16. [DOI] [PubMed] [Google Scholar]
- 50.Garaigorta U, Chisari FV. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe 2009;6:513–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pais R, Rusu E, Zilisteanu D, et al. Prevalence of steatosis and insulin resistance in patients with chronic hepatitis B compared with chronic hepatitis C and non-alcoholic fatty liver disease. Eur J Intern Med 2015;26:30–6. [DOI] [PubMed] [Google Scholar]
- 52.Hiscott J, Kwon H, Genin P. Hostile takeovers: viral appropriation of the NF-kappaB pathway. J Clin Invest 2001;107:143–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Konner AC, Bruning JC. Toll-like receptors: linking inflammation to metabolism. Trends Endocrinol Metab 2011;22:16–23. [DOI] [PubMed] [Google Scholar]
- 54.Farrell GC, Haczeyni F, Chitturi S. Pathogenesis of NASH: How Metabolic Complications of Overnutrition Favour Lipotoxicity and Pro-Inflammatory Fatty Liver Disease. Adv Exp Med Biol 2018;1061:19–44. [DOI] [PubMed] [Google Scholar]
- 55.Miyanari Y, Atsuzawa K, Usuda N, et al. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol 2007;9:1089–97. [DOI] [PubMed] [Google Scholar]
- 56.Hofmann S, Krajewski M, Scherer C, et al. Complex lipid metabolic remodeling is required for efficient hepatitis C virus replication. Biochim Biophys Acta Mol Cell Biol Lipids 2018;1863:1041–1056. [DOI] [PubMed] [Google Scholar]
- 57.Anjani K, Lhomme M, Sokolovska N, et al. Circulating phospholipid profiling identifies portal contribution to NASH signature in obesity. J Hepatol 2015;62:905–12. [DOI] [PubMed] [Google Scholar]
- 58.Hardwick JP, Osei-Hyiaman D, Wiland H, et al. PPAR/RXR Regulation of Fatty Acid Metabolism and Fatty Acid omega-Hydroxylase (CYP4) Isozymes: Implications for Prevention of Lipotoxicity in Fatty Liver Disease. PPAR Res 2009;2009:952734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol 2015;62:720–33. [DOI] [PubMed] [Google Scholar]
- 60.Friedman SL, Neuschwander-Tetri BA, Rinella M, et al. Mechanisms of NAFLD development and therapeutic strategies. Nat Med 2018;24:908–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blackham S, Baillie A, Al-Hababi F, et al. Gene expression profiling indicates the roles of host oxidative stress, apoptosis, lipid metabolism, and intracellular transport genes in the replication of hepatitis C virus. J Virol 2010;84:5404–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deng Y, Wang J, Huang M, et al. Inhibition of miR-148a-3p resists hepatocellular carcinoma progress of hepatitis C virus infection through suppressing c-Jun and MAPK pathway. J Cell Mol Med 2019;23:1415–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.