
Fig. 4.
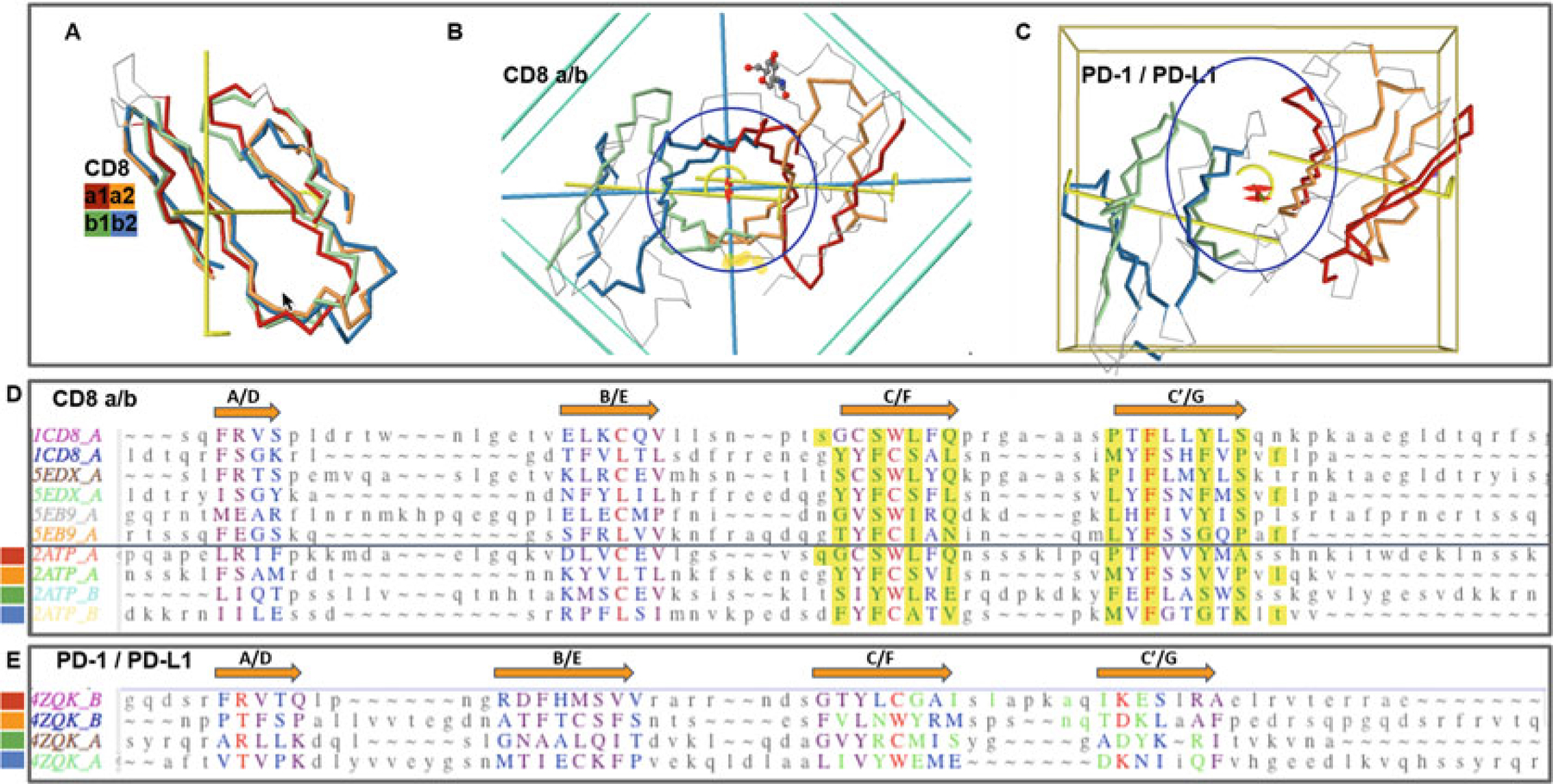
CD8ab and PD1/PDL1 heterodimers. Protodomains and quaternary symmetric arrangements. (a) CD8ab (structure of mouse CD8ab: 2ATP). Four protodomains aligned for CD8a and CD8b colored red/orange and green/blue, respectively. Automatic symmetry detection and protodomain alignment performed with CE-symm and displayed with JMol. Average RMS on protodomains as computed by CE-symm is 2.71. (b) CD8ab dimer with two orthogonal axes of symmetry: Two C2 levels of symmetry detected as overall D2 symmetry, meaning the two axes domain level and dimer level intersect in the center of symmetry, as for a CD8aa homodimer (see schematic representation in Fig. 2d). A small departure from perfect symmetry is observed between the actual domain-level yellow axes of symmetry vs. perfect orthogonality to the dimer axis, perpendicular to the plane of the paper. One can see a pseudosymmetric eight-stranded central barrel formed by the two faces of each monomer, from both sheets G|F||C|C′ facing each other (the symmetric homodimer CD8aa—structure 1CD8 is presented in Fig. S1 with an iCn3D Link). (c) PD-1/PD-L1 receptor ligand interface (structure of human PD1-PDL1: 4ZQK). Here we still have a pseudosymmetry for each domain, and for the heterodimer, the two external faces of the respective Sheet B of PD1 and PDL1 are shifted laterally relative to each other, to form the interface. We still have two C2 levels of symmetry but the domain-level axes to not cross with the dimer axis on the center of symmetry. There is still a C2 domain level of symmetry for each domain, and a dimer center and C2 axis of (pseudo) symmetry, but not a D2 symmetry. The average on automatic detection RMS is 3.38A. (d) Optimized structural alignment of protodomains of CD8a in the homodimer CD8aa (structures of CD8aa: Human 1CD8; Swine 5EDX; Chicken 5EB9) and the heterodimer CD8ab (structure of mouse CD8ab: 2ATP chains A and B, respectively). The RMSD for the optimized multiple domains/protodomains alignment for each first and second protodomain vs. the first Human CD8a protodomain are 1.61, 0.436, 1.71, 0.852, 1.57, 0.522, 1.95, 0.895, and 1.54 A, respectively. The computer-generated alignment is higher by 1–2A (this is usually the case). In this case it does a good job to match key symmetry equivalent residues, especially C/L and W/C. However accurate delineation and multiple structure alignment is only possible through interactive software Cn3D currently. Noticeable is the absolutely conserved F residue in strands C′ and G. Interface residues are contributed pseudosymmetrically as can be seen in the alignment for residues colored in green and highlighted in yellow, except for F colored red. (e) Optimized protodomain alignment of PD-1 and PD-L1 (structure 4ZQK chains B and A, respectively). In this case the automatic alignment is not as good as for CD8ab but is good enough to detect two levels of symmetry. The structural alignment optimized interactively gives a very good RMSD for the four protodomains with 1.73, 1.53, and 1.84 A, respectively, for the second PD-1 and the first and second PDL1 protodomains relative to the first PD-1 protodomain. Noticeable is a C/M match vs. a C/L match between protodomains of PD-1 vs. PD-L1. On the PD-1/PD-L1 interface, it is clearly not as symmetric as for CD8 as the barrel opens on one side vs. the other, with the relative shift of the domains external faces of Sheets B (G|F|C|C′) observed (see c vs. b)