

Abstract
IKZF1 encodes Ikaros, a zinc finger–containing transcription factor crucial to the development of the hematopoietic system. Germline pathogenic variants in IKZF1 have been reported in patients with acute lymphocytic leukemia and immunodeficiency syndromes. Diamond–Blackfan anemia (DBA) is a rare inherited bone marrow failure syndrome characterized by erythroid hypoplasia, associated with a spectrum of congenital anomalies and an elevated risk of certain cancers. DBA is usually caused by heterozygous pathogenic variants in genes that function in ribosomal biogenesis; however, in many cases the genetic etiology is unknown. We identified a germline IKZF1 variant, rs757907717 C > T, in identical twins with DBA-like features and autoimmune gastrointestinal disease. rs757907717 C > T results in a p.R381C amino acid change in the IKZF1 Ik-x isoform (p.R423C on isoform Ik-1), which we show is associated with altered global gene expression and perturbation of transcriptional networks involved in hematopoietic system development. These data suggest that this missense substitution caused a DBA-like syndrome in this family because of alterations in hematopoiesis, including dysregulation of networks essential for normal erythropoiesis and the immune system.
Keywords: congenital hypoplastic anemia
INTRODUCTION
IKZF1 is a member of the essential IKAROS family of hematopoietic transcription factors encoded by IKZF1 on Chromosome 7p12.2 (OMIM: 603023). Germline and somatic genetic variants in IKZF1 are associated with aberrant hematopoiesis. For example, somatic IKZF1 deletions or, less commonly, point mutations occur in ∼15% of pediatric acute lymphoblastic leukemia and are of prognostic importance (Iacobucci and Mullighan 2017; Stanulla et al. 2020). Common IKZF1 single-nucleotide polymorphisms (SNPs) have been associated with germline susceptibility to B-cell precursor acute lymphoblastic leukemia (ALL) (Speedy et al. 2014; Churchman et al. 2018). Rare pathogenic germline variants in IKZF1 have been reported in patients with ALL, as well as those with pancytopenia and immunodeficiency, and in common variable immunodeficiency (CVID) (Goldman et al. 2012; Kuehn et al. 2016; Hoshino et al. 2017; Bogaert et al. 2018; Boutboul et al. 2018; Churchman et al. 2018).
Diamond–Blackfan anemia (DBA; OMIM 105650) is a rare inherited bone marrow failure syndrome characterized by congenital erythroid hypoplasia, typically presenting in infancy with macrocytic anemia, usually with normal white blood cell and platelets (Shimamura and Alter 2010). It is also associated with a spectrum of congenital anomalies and a higher-than-expected risk of certain cancers, including myelodysplastic syndrome, acute myeloid leukemia, and colon adenocarcinoma (Alter et al. 2010; Ball 2011; Gazda et al. 2012; Vlachos et al. 2018). Phenotypic heterogeneity is often observed, even within affected members of the same family (Campagnoli et al. 2004; Farrar and Dahl 2011; Alter et al. 2018; Vlachos et al. 2018). The majority of patients with DBA have autosomal dominant pathogenic germline variants in genes encoding ribosomal biogenesis proteins (small and large subunit components), with two X-linked recessive genes (GATA1 and TSR2) also causative of DBA (Draptchinskaia et al. 1999; Campagnoli et al. 2008; Boria et al. 2010; Narla and Ebert 2010; Farrar et al. 2011; Gazda et al. 2012; Sankaran et al. 2012; Landowski et al. 2013; Gripp et al. 2014; Mirabello et al. 2014; Wang et al. 2015; Bhar et al. 2020).
Here we report on identical twins with a DBA-like phenotype and immune-mediated gastrointestinal disease, found to have a heterozygous germline pathogenic variant in IKZF1 associated with increased protein expression and aberrations in hematopoietic gene expression.
RESULTS
Clinical Presentation and Family History
Individuals NCI-40-1 and NCI-40-2 were the product of an uncomplicated monozygotic twin pregnancy, born at term to nonconsanguineous parents of Northern European ancestry (Fig. 1A). At 14 mo of age, NCI-40-1 had symptomatic anemia requiring packed red blood cell (RBC) transfusion and was presumptively diagnosed with DBA (diagnostic clinical features are shown in Table 1). He did not respond to oral corticosteroids and became transfusion-dependent. At 2 yr of age, he was started on low-dose cyclosporine with improvement of the anemia as reported in 2003 (Bobey et al. 2003). He has been off cyclosporine since age 4 yr, and his hemoglobin remains at the lower end of normal range for age and sex. At 8 yr of age, he underwent a comprehensive evaluation at the National Institutes of Health (NIH) Clinical Center (Table 1) and was found to have microcytic anemia consistent with iron deficiency. Adenotonsillar hypertrophy, allergic rhinitis, asthma, and eczema were also reported in this patient. NCI-40-1 has a history of long-standing gastrointestinal (GI) problems with inflammatory small bowel strictures of unknown etiology, and he had multiple duodenal and gastric ulcers at age 9 yr. At age 11 yr, he underwent endoscopic dilatation of his pylorus and has had multiple such procedures since then. Methotrexate was tried for possible Crohn's disease at age 13 yr of age. His last follow-up was at 22 yr of age at which time he was receiving the JAK inhibitor tofacitinib every 6 wk for atypical Crohn's disease and was feeling well.
Figure 1.

Pedigree of NCI-40 and bone marrow characteristics of the proband. (A) Pedigree illustrating clinical features and carrier status of tested family members. Clinical features of NCI-40-1 (proband) and NCI-40-2 are described in detail in the text. Their mother, NCI-40-4, had a history of anemia and is heterozygous for the p.R381C variant. Her sister, the children's maternal aunt, NCI-40-6, had a history of aplastic anemia and thyroid cancer but did not have genetic testing. (WT) Wild-type, p.R381C (IKZF1 isoform Ik-x rs757907717 C > T) heterozygote, (6) number of siblings. (B–F) NCI-40-1 evaluation at 8 yr of age. (B) Hematoxylin and eosin (H&E) stain of bone marrow core biopsy at 40× magnification shows maturing erythroid islands, adequate myeloid:erythroid (M:E) ratio, and mild eosinophilia. (C) Bone marrow core biopsy at 100×. (D) Bone marrow aspirate smear at 50× magnification with progressively maturing trilineage hematopoiesis and no dysplastic changes or increase in blasts. (E) Stainable histiocytic iron (blue) in bone marrow. (F) Normal peripheral blood smear. (G–K) NCI-40-2 evaluation at 8 yr of age. (G) Core biopsy H&E stain at 40× magnification shows higher cellularity than his twin brother, has focal increase in megakaryocytes, maturing erythroid islands, adequate M:E ratio, and mild eosinophilia. (H) Bone marrow core biopsy at 100×. (I) Bone marrow aspirate smear at 50× magnification has progressively maturing trilineage hematopoiesis and no dysplastic changes or increase in blasts. (J) Reduced iron staining. (K) Normal peripheral blood smear with some platelet clumps (50× magnification).
Table 1.
Clinical characteristics of participants
| Clinical parameter (unit) | NCI 40-1 | NCI 40-2 | ||
|---|---|---|---|---|
| Age 14 mo (normal range) | Age 8 yr (normal range) | Age 22 mo (normal range) | Age 8 yr (normal range) | |
| White blood count (×109/L) | 14.8 (6–17) | 7.88 (3.3–9.6) | 7.5 (6–17) | 7.5 (3.3–9.6) |
| Hemoglobin (g/L) | 46 (110–140) | 111 (115–155) | 79 (110–140) | 112 (115–155) |
| Mean corpuscular volume (fL) | 75 (71–89) | 70 (79–98) | 74 (71–89) | 75 (79–98) |
| Platelet count (×109/L) | 349 (150–400) | 326 (154–345) | 356 (150–400) | 356 (154–345) |
| Reticulocyte count (%) | 0 (1–2) | 0.66 (0.66–2.47) | 0 (1–2) | 0.72 (0.66–2.47) |
| HbF (%) | 1.7 (1–2) | 0 (0–2) | 1.2 (1–2) | 0 (0–2) |
| eADA (IU/g of hemoglobin) | 1.23 (≤0.96) | 1.05 (≤0.96) | 1.07 (≤0.96) | |
| Bone marrow evaluation | Pure red blood cell aplasia | 46, XY Hypocellular at 50%–60% (normal 90%) M:E ratio 3.4:1 Erythroid precursors 29% Granulocytes 67% Lymphocytes 14% Occasional megaloblastic features |
Pure red blood cell aplasia | 46, XY 70% cellularity, focal hypercellularity M:E ratio 3:1 Erythroid precursors 24% Granulocytes 59% Lymphocytes 16% Occasional megaloblastic precursors |
| Erythrocyte sedimentation rate (mm/h) | 23 (<25) | 25 (<25) | ||
| C-reactive protein (mg/dL) | <0.4 (<0.8) | <0.4 (0.8) | ||
| IgG (mg/dL) | 1170 (613–1295) | 994 (613–1295) | ||
| IgA (mg/dL) | 161 (69–309) | 96 (69–309) | ||
| IgM (mg/dL) | 62 (53–335) | 53 (53–335) | ||
| Evidence of hemolysis | No | No | ||
| Anti-thyroglobulin antibody (IU/mL) | 64 (0–59) | 18 (0–59) | ||
| Response to childhood vaccinations | Normal | Normal | ||
| Dysmorphic features | None | None | None | None |
| EKG and echocardiogram | Normal | Normal | ||
| Abdominal ultrasound | Normal | Normal | ||
Evaluations reported at age 8 yr were done at the NIH Clinical Center. The values at the other ages were obtained from medical record review. Empty cells indicate study not done.
(HbF) Hemoglobin F, (eADA) erythrocyte adenosine deaminase, (M:E) myeloid:erythroid, (EKG) electrocardiogram.
His identical twin brother, NCI-40-2, was diagnosed with pure RBC aplasia at age 22 mo, confirmed by bone marrow biopsy also leading to a presumptive diagnosis of DBA (Table 1). Similar to his twin brother, he did not respond to oral corticosteroid therapy but did respond to low-dose cyclosporine after being RBC transfusion–dependent for 5 mo (Bobey et al. 2003). He has not taken cyclosporine since age 4 yr and was evaluated with his brother at the NIH Clinical Center at 8 yr of age (Table 1). NCI 40-2 also had a long-standing history of fibrostenotic duodenal disease of unknown etiology, needing surgical dilatation, and received infliximab for the chronic gut inflammation.
Their family history was notable for anemia at 30 yr of age in their mother, and a history of aplastic anemia in their maternal aunt, who also developed thyroid cancer at age 38 yr. Their maternal grandmother died of breast cancer at age 60 yr and maternal grandfather died of lung cancer at age 78 yr. Their father and brother were healthy.
Genomic Analyses
Insertions or deletions were not identified in blood-derived DNA from the proband or his brother. Filtering of exome sequencing for rare variants identified 74 heterozygous variants shared by both twins and a parent (Supplemental Tables S1 and S2). No homozygous variants were identified. Notably, variants in ribosome assembly genes typically associated with DBA were not identified. Ribosome assembly assays of pre-rRNA processing did not identify an assembly defect in the proband's lymphocytes (Supplemental Fig. S1). A nonsynonymous variant (Chr 7:50468032 in GRCh37, rs757907717 C > T) in IKZF1 was the strongest candidate for disease association in this family because prior studies identified germline mutations in IKZF1 in individuals with ALL, CVID, and pancytopenia (Table 2; Goldman et al. 2012; Kuehn et al. 2016; Hoshino et al. 2017; Boutboul et al. 2018; Churchman et al. 2018). This variant was predicted deleterious or disease-causing by multiple in silico prediction models and occurred in a well-conserved region of the gene (Fig. 2). Specifically, rs757907717 C > T is predicted disease-causing by MutationTaster, deleterious by Provean, SIFT, PolyPhen, and FATHHM but predicted tolerated by METASVM. Its CADD PHRED score is 25.4. The IKZF1 protein has numerous isoforms. We used NM_001220765, exon7, c.C1141T, p.R381C for all experiments here because it is the predominant form found in normal hemopoietic cells (Payne et al. 2001).
Table 2.
Variant table
Figure 2.

Protein sequence conservation and structural prediction suggests IKZF1 p.R381C is pathogenic. (A) The four functional isoforms of IKZF1 depicted with predicted Zn finger, DNA binding domains, and location of the variant p.R423C in isoform Ik-1. The variant location is p.R381C in isoform Ik-x. Isoform Ik-x is the isoform used in the described biochemical experiments in this manuscript. Note that the figure is not drawn to scale. (B) M-Coffee alignment reveals strict conservation of p.R381 (blue arrow and box) across orthologs from evolutionarily divergent species. (C) “OpenPredictProtein-effect of point mutation” algorithm predicts any changes at the p.R381C position (blue arrow) to be highly deleterious. (D) Jpred server prediction of secondary structure indicates that a R → C mutation (top vs. bottom panel, blue arrow heads) at p.R423 (equivalent to p.R381C in isoform Ik-x) would result in a loss of a B-sheet and potentially alter secondary structures up to 18 amino acid residues upstream (broad green arrows represent predicted B-sheets; red box indicates potential helical structure). (E) Phosphosite Plus analysis of IKZF1 Ik-1 isoform indicates post-translational modification sites near the p.R423 residue. Of note are p.S427-phos, p.K429-Ub, and p.K429-SUMO.
Functional Consequences of IKZF1 p.R381C
IKZF1 p.R381C Protein Is Increased but Stable Compared with Wild Type
Upon initial review of the predicted secondary structure and known and post-translational modifications of IKZF1 using UNIPROT and PhosphositePlus databases, we hypothesized that the p.R381C variant could be altering protein stability, as it lies nearby several post-translational modifications in a highly conserved region of the protein (Fig. 2). Epstein–Barr virus (EBV)-transformed lymphoblasts with the p.R381C variant showed increased IKZF1 protein levels by immunoblotting compared to controls (Fig. 3). K562 cell lines stably transduced with IKZF1 Ik-x isoform wild type (WT) or p.R381C were treated with 60 µg/mL cycloheximide for up to 12 h (Supplemental Fig. S2). The results did not show significant differences between WT or p.R381C forms of IKZF1. Addition of MG132, a potent proteasome blocking peptide, to the assay also had little effect (Supplemental Fig. S3). Because of the proximity of the p.R381C residue to reported post-translational modification sites (Fig. 2E), we hypothesized that the variant could alter protein stability by disrupting ubiquitination at residue p.K387 (p.K429 in the full-length isoform). We therefore evaluated whether ubiquitination was affected by the p.R381C variant. HEK cells were stably transduced with IKZF1 WT or p.R381C containing a carboxy-terminal V5 tag. Cells treated with 2.5 µM MG132 were immunoprecipitated for the V5 epitope tag and subsequently immunoblotted for ubiquitin, revealing little difference between the WT and mutant forms of IKZF1 (Supplemental Fig. S4). This result is unsurprising as IKZF1 is reported to be degraded in multiple myeloma cell lines by kinase inhibition in a proteasome- and lysosome-independent mechanism (Liu et al. 2015) as well as through the Cul4CRBN (CUL4–RBX1–DDB1–CRBN) ubiquitin ligase complex (Fischer et al. 2014).
Figure 3.
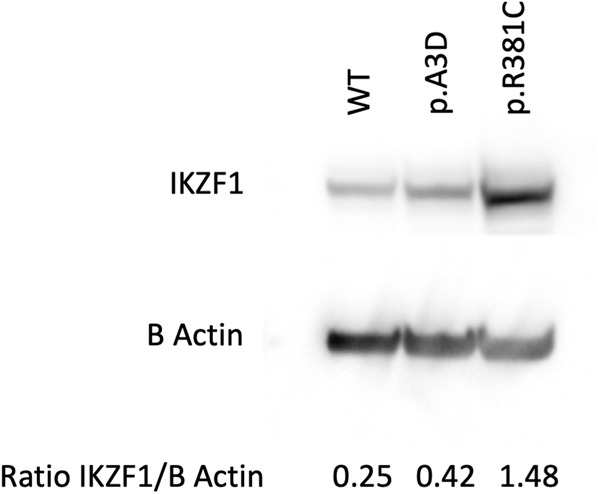
Increased IKZF1 protein levels are associated with the p.R381C variant. Epstein–Barr virus (EBV)-transformed lymphoblasts with the IKZF1 p.R381C variant show increased IKZF1 protein levels by immunoblotting compared with wild type (WT) control and an unrelated individual with IKZF1 p.A3D variant. Signal densities quantified by ImageJ.
IKZF1 p.R381C Has Little Detectable Effect on Interactions with Known Protein Complexes
To determine whether IKZF1 p.R381C had any effect on known protein–protein interactions, we conducted a series of coimmunoprecipitations. IKZF1 and GATA1 are known to interact to control immunoglobulin gene expression (Bottardi et al. 2009). Therefore, we started with coimmunoprecipitations of these proteins with WT or p.R381C IKZF1 (Supplemental Fig. S5). There was no difference between these interactions in the WT and p.R381C background cell lines. IKZF1 is also reported to be involved in the recruitment of the NURD complex to heterochromatin (Kim et al. 1999). We tested whether the IKZF1 mutant form was impaired for interactions with NURD complex members by coimmunoprecipitation (Supplemental Fig. S6). No discernible difference was detected between IKZF1 WT or p.R381C mutant–expressing cells with NURD members CHD4, MTA1, or MBD2.
IKZF1 p.R381C Alters Hematopoietic Gene Expression Profiles
Whole-transcriptome RNA sequencing was conducted to determine whether overexpression of the WT or p.R381C IKZF1 functionally altered gene expression profiles. Differential expression analysis revealed that overexpression of the p.R381C mutant compared with WT resulted in significant changes in gene networks involved in cancer, cell signaling, apoptosis, and hematopoiesis (Fig. 4; Supplemental Table S3). Notably, the gene expression differences observed in the K562 background were more distinct than in the HEK cell background as observed by PCA (Supplemental Fig. S7). HEK cells do not express high levels of endogenous IKZF1, whereas the K562 erythroleukemia cells express moderate levels of IKZF1, suggesting that networks and pathways involved in IKZF1-mediated signaling exist in the K562 cells but not in HEK cells.
Figure 4.

IKZF1 p.R381C alters hematopoietic gene expression. RNA from stable cell lines expressing wild-type or p.R381C IKZF1 was extracted and sequenced on Illumina Novaseq S2 platform. Differentially expressed gene lists identified by RNA-seq analysis were queried in Ingenuity Pathway Analysis (QIAGEN) software. (A) Differentially expressed genes identified by RNA-seq form a network containing factors with functions critical to differentiation, proliferation, and development of the hematological system. (B) IKZF1 plays a central role in hematopoiesis, lymphoid, myeloid, and megakaryocytic proliferation and differentiation, leukemia, and erythroid biology. (C) Top functions of network presented in A. The full table is presented in Supplemental Table S3.
Gene expression was validated by quantitative polymerase chain reaction (qPCR) for selected genes integral to the networks identified by pathway analyses (Fig. 5). This list was further validated by selection of targets with large ΔΔCT between WT and p.R381C by immunoblot. Interestingly, IKZF3/Aiolos, a protein that directly interacts with IKZF1 (Kim et al. 1999; Hung et al. 2016) and is integral to hematopoiesis, was up-regulated many-fold in the presence of IKZF1 p.R381C but largely undetectable in cells expressing WT IKZF1. The perturbation of IKZF3 levels by IKZF1 mutation could be directly responsible for the pathogenic effect observed in patients with this germline variant. Of note, IKZF1 expression in transformed cell lines is comparable by both whole-transcriptome and qPCR (Fig. 5).
Figure 5.

Differentially expressed genes validated by quantitative polymerase chain reaction (qPCR) and immunoblot. (A) RNA-seq log2 fold change, standard error, and adjusted P-value for selected genes present as nodes in network analysis. (B) Targets identified as critical nodes in network analysis were selected for validation by qPCR. TaqMan probes were utilized to perform qPCR. Relative quantity values by ΔΔ−CT between wild-type (WT) and mutant (MUT) samples (n = 3 biological replicates, n = 3 technical replicates) normalized to β-actin and GAPDH are reported. Genes highlighted by the rectangle are included in immunoblot analysis. IKZF3/Aiolos signal was undetected after 40 cycles in WT samples but displayed a value of ∼32 CT in MUT samples. SNAI2 amplified in WT samples but was undetected in MUT samples after 40 cycles. (C) Targets were further validated by immunoblot. Immunoblots were quantified using density analysis in ImageJ software.
DISCUSSION
IKZF1 is an important hematopoietic transcription factor with numerous isoforms; its complex role in hematopoiesis is still being elucidated. An array of phenotypes have been reported associated with germline IKZF1 variation and range from common SNPs associated with risk of pediatric leukemia to rare pathogenic variants associated with CVID, immune dysregulation, and, now, possibly DBA-like features.
The siblings presented here had RBC aplasia phenotypes as infants consistent with a presumed diagnosis DBA but were also quite different than a typical child with DBA because their anemia responded to cyclosporine (Bobey et al. 2003; Ball 2011; Li et al. 2018). They later had autoimmune GI manifestations that are not typical of DBA but have been reported. Notably, chronic colitis of unclear etiology has recently been described in two individuals with DBA found to have germline pathogenic variants in RPS20 (Bhar et al. 2020).
The autoimmune GI manifestations in the twins reported here are consistent with those of autoimmune disease associated with both rare and common IKZF1 variants. Several studies have reported associations between heterozygous pathogenic IKZF1 variants and immune deficiency, impaired B-cell development, and/or autoimmune diseases, including immune thrombocytopenic purpura, IgA vasculitis, and systemic lupus erythematosus (SLE), CVID, anti-phospholipid syndrome, and immune thrombocytopenia (Wojcik et al. 2007; Hoshino et al. 2017; Van Nieuwenhove et al. 2018; Dieudonne et al. 2019; Eskandarian et al. 2019). Common variants in IKZF1 have also been associated with risk of SLE and inflammatory bowel disease (He et al. 2010; Wang et al. 2013; Tajuddin et al. 2016).
Our studies show that the IKZF1 p.R381C amino acid change due to rs757907717 C > T most significantly appears to change hematopoietic gene expression profiles. Increased IKZF1 protein was noted in the p.R381C form compared with the WT, although our studies did not identify increased protein stability in the presence of CHX or MG132, changes in ubiquitination, or alterations in interactions with GATA1 and NURD complex proteins. It remains possible that the p.R381C variant alters the half-life of the IKZF1-containing complexes in ways we were unable to detect, presumably skewing the dynamics and kinetics of these complexes and their interactions with chromatin, functionally altering timing and reprogramming of transcriptional networks.
The rs757907717 C > T variant resulting in p.R381C in IKZF1 Ik-x protein was reported as p.R423C in isoform Ik-1 (Fig. 2) in a study of germline pediatric ALL (Churchman et al. 2018). The authors reported alterations in IKZF1 protein localization, increased cell–cell adhesion, and a modest response to dasatinib and dexamethasone associated with this variant. Our finding of increased IKZF1 with the p.R381C variant suggest that dysregulation of protein turnover or altered protein levels could cause mislocalization of IKZF1. In fact, protein phosphatase 1 (PP1) has been shown to regulate IKZF1 turnover and localization by binding to a site approximately 40 amino acids carboxy-terminal to p.R423/p.R381 at amino acids 459–470 (Popescu et al. 2009). Although p.R381C is not in this PP1 binding domain, it could potentially alter the affinity of this or other post-translational modifiers controlling IKZF1 stability and localization.
We identified several dysregulated transcriptional networks attributable to IKZF1 p.R381C by comparing transcriptomes from K562 cells harboring IKZF1 WT or p.R381C constructs. These networks include functions critical in hematopoiesis; differentiation of lymphoid, myeloid, erythroid, and megakaryocytic lineages; and the development of leukemia and anemia. As these experiments were performed in model cell lines containing constructs overexpressing IKZF1, these data should be interpreted with caution. Regardless, by comparing the overexpression of WT and p.R381C, functional consequences of the variant in question may be deduced. IKZF1 is reported to interact directly with SMARCA4 (BRG1) in a SWI/SNF type complex (Kim et al. 1999), and we found in the OregAnno tracks on the UCSC genome browser potential SMARCA4 binding sites in enhancers proximal to both IKZF1 and IKZF3 genes (data not shown). This suggests a feedback loop by which ectopic IKZF1 expression or protein dysregulation increases levels of IKZF1 and IKZF3 (Aiolos), potentially driving alternative hematopoietic developmental programs. IKZF3 is normally expressed solely in B and T lineage cells and is up-regulated upon terminal differentiation (Morgan et al. 1997). Therefore, altering the developmental timing of IKZF3 expression could drive differentiation programs toward lymphoid lineages and away from erythroid lineages.
Both IKZF1 and IKZF3 are known substrates of the Cul4CRBN E3 ubiquitin ligase complex, which is activated by lenalidomide (Krönke et al. 2017). The interaction between lenalidomide, Cul4CRBN, and IKAROS family members is hypothesized to be a mechanism of action driving its anticancer activity in multiple myeloma (Krönke et al. 2017). Lenalidomide has also been reported to induce IKZF1 degradation in low-risk del5q MDS (MDSL) cells, resulting in activation of a calcium/calpain proapoptotic pathway (Fang et al. 2016). Additionally, lenalidomide was recently reported to up-regulate Regulator of Calcineurin 1 (RCAN1), which, in turn, suppressed calcineurin (CaN) (He et al. 2020). Depletion of IKZF1 in MDSL with siRNA increased RCAN1 mRNA (He et al. 2020). Future studies are required to understand whether variants in IKZF1 are associated with changes in this pathway.
In summary, the p.R381C IKZF1 variant was associated with DBA-like features with immune-related GI phenotype and an alteration in hematopoietic gene expression networks. Additional studies are required to understand the specific IKZF1-containing complexes affected and the developmental timing of hematopoiesis affected by IKZF1 dysregulation, as well as the mechanisms by which IKZF1 and RCAN1 may interact, and the role of calcineurin inhibition on normal and mutant IKZF1 function. The recognition of other cases like these with DBA-like phenotypes associated with immune-related findings as possibly being associated with IKZF1 will aid in further characterizing what the other manifestations may be, and how to appropriately manage these individuals.
METHODS
Study Population
Members of family NCI-40 were participants in an institutional review board approved study at the National Cancer Institute (NCI) titled “Cancer in Inherited Bone Marrow Failure Syndromes” (https://marrowfailure.cancer.gov ClinicalTrials.gov Identifier: NCT00027274) (Alter et al. 2010, 2018). Study participants or their guardians signed informed consent, and completed detailed family and medical history questionnaires. Medical records were reviewed in detail and participants underwent comprehensive clinical evaluations and biospecimen collection at the National Institutes of Health (NIH) Clinical Center. The patient samples and cells used in the experiments described below were obtained at the NIH Clinical Center evaluation, at 8 yr of age. All laboratory personnel were blinded to the clinical and genetic status of analyzed samples.
Genomic Characterization
DNA from participants was extracted from whole blood using standard procedures. Whole-exome sequencing data were generated at the NCI Cancer Genomics Research Laboratory as previously described (Supplemental Methods) (Ballew et al. 2013; Shi et al. 2014). A custom CGH array (Agilent) consists of 52,797 probes, with a spacing of ∼150 bp, designed to target 107 genes of interest in DBA was used to identify copy number variants (Chandrasekharappa et al. 2013; Mirabello et al. 2017). Additional details are in the Supplemental Methods.
Functional Studies
Hek293T (human embryonic kidney cells) and K562 (human erythroleukemic cells) were transfected with a lentivirus vector made by site-directed mutagenesis. These cell lines were used for coimmunoprecipitation and immunoblots. The details are reported in the Supplemental Methods. Ribosome assembly assays on patient-derived mononuclear cells were performed as described in the Supplemental Methods.
RNA Sequencing and Gene Expression Analyses
RNA from stable cell lines expressing both WT and p.R381C IKZF1 was extracted and sequenced on Illumina NovaSeq S2 platform. Trimmed reads were aligned to GRCh38 human reference genome (Illumina iGenomes NCBI GRCh38) and gene reads count were quantified according to Illumina iGenomes NCBI GRCh38 annotation file using STAR/2.7.3a. Additional details are reported in the Supplemental Methods.
The analyses of pathways and networks significantly enriched in differentially expressed genes were performed using Ingenuity Pathway Analysis software (IPA; QIAGEN, https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/). Selected targets were validated by qPCR using TaqMan style probes (Supplemental Table S4) (Thermo Fisher Scientific). Briefly, total RNA was extracted from K562 cells harboring stable IKZF1 WT or MUT constructs. cDNA was synthesized using SuperscriptIII kit (Thermo Fisher Scientific) and subjected to qPCR with TaqMan probes on a Quantstudio6 Real-time instrument (Thermo Fisher Scientific). Results were quantified by the ΔΔ−CT method.
ADDITIONAL INFORMATION
Data Deposition and Access
Genomic data are available through controlled access in dbGaP per the National Institutes of Health (NIH) genomic data sharing policy (Study Accession: phs001481.v1.p1). The IKZF1 variant was submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and can be found under accession number VCV000988782.1.
Ethics Statement
This study was approved by the Institutional Review Board of the National Cancer Institute (study ID number 02-C-0052; ClinicalTrials.gov Identifier: NCT00027274). The participants, their parents, or legal guardians provided written, informed consent. Minors provided written, informed assent.
Acknowledgments
We are grateful to the patients and their family members for their valuable contributions to this study. We also thank the referring clinicians and consultants. Mention of any product or service does not constitute endorsement by the National Cancer Institute (NCI), NIH, or the United States Department of Health and Human Services (U.S. DHHS).
Author Contributions
S.A.B. designed and performed experiments; collected, analyzed, and interpreted data; and cowrote the manuscript. P.P.K., N.G., and B.P.A. collected, analyzed, and interpreted data and cowrote the manuscript. A.J.B., L.J., F.X.D., and S.R.E. performed experiments and collected, analyzed, and interpreted data. M.S. and J.D. analyzed and interpreted data and performed statistical analysis. S.C.C. and I.M. collected, analyzed, and interpreted data. K.C.A. designed figures and interpreted data. L.M. designed research and interpreted data. S.A.S. designed research, analyzed and interpreted data, and cowrote the manuscript. All authors reviewed, edited, and approved the final manuscript.
Funding
This work was supported in part by the intramural research program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health, by the intramural research program of the National Human Genome Research Institute, National Institutes of Health, and by contract HHSN261201700004C with Westat, Inc.
Competing Interest Statement
The authors have declared no competing interest.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Alter BP, Giri N, Savage SA, Peters JA, Loud JT, Leathwood L, Carr AG, Greene MH, Rosenberg PS. 2010. Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br J Haematol 150: 179–188. 10.1111/j.1365-2141.2010.08212.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alter BP, Giri N, Savage SA, Rosenberg PS. 2018. Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 103: 30–39. 10.3324/haematol.2017.178111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball S. 2011. Diamond Blackfan anemia. Hematol Am Soc Hematol Educ Program 2011: 487–491. 10.1182/asheducation-2011.1.487 [DOI] [PubMed] [Google Scholar]
- Ballew BJ, Yeager M, Jacobs K, Giri N, Boland J, Burdett L, Alter BP, Savage SA. 2013. Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in dyskeratosis congenita. Hum Genet 132: 473–480. 10.1007/s00439-013-1265-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhar S, Zhou F, Reineke LC, Morris DK, Khincha PP, Giri N, Mirabello L, Bergstrom K, Lemon LD, Williams CL, et al. 2020. Expansion of germline RPS20 mutation phenotype to include Diamond–Blackfan anemia. Hum Mutat 41: 1918–1930. 10.1002/humu.24092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobey NA, Carcao M, Dror Y, Freedman MH, Dahl N, Woodman RC. 2003. Sustained cyclosporine-induced erythropoietic response in identical male twins with Diamond–Blackfan anemia. J Pediatr Hematol Oncol 25: 914–918. 10.1097/00043426-200311000-00018 [DOI] [PubMed] [Google Scholar]
- Bogaert DJ, Kuehn HS, Bonroy C, Calvo KR, Dehoorne J, Vanlander AV, De Bruyne M, Cytlak U, Bigley V, De Baets F, et al. 2018. A novel IKAROS haploinsufficiency kindred with unexpectedly late and variable B-cell maturation defects. J Allergy Clin Immunol 141: 432–435 e437. 10.1016/j.jaci.2017.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boria I, Garelli E, Gazda HT, Aspesi A, Quarello P, Pavesi E, Ferrante D, Meerpohl JJ, Kartal M, Da Costa L, et al. 2010. The ribosomal basis of Diamond–Blackfan anemia: mutation and database update. Hum Mutat 31: 1269–1279. 10.1002/humu.21383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottardi S, Ross J, Bourgoin V, Fotouhi-Ardakani N, Affar El B, Trudel M, Milot E. 2009. Ikaros and GATA-1 combinatorial effect is required for silencing of human γ-globin genes. Mol Cell Biol 29: 1526–1537. 10.1128/MCB.01523-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutboul D, Kuehn HS, Van de Wyngaert Z, Niemela JE, Callebaut I, Stoddard J, Lenoir C, Barlogis V, Farnarier C, Vely F, et al. 2018. Dominant-negative IKZF1 mutations cause a T, B, and myeloid cell combined immunodeficiency. J Clin Invest 128: 3071–3087. 10.1172/JCI98164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagnoli M, Garelli E, Quarello P, Carando A, Varotto S, Nobili B, Longoni D, Pecile V, Zecca M, Dufour C, et al. 2004. Molecular basis of Diamond–Blackfan anemia: new findings from the Italian registry and a review of the literature. Haematologica 89: 480–489. [PubMed] [Google Scholar]
- Campagnoli M, Ramenghi U, Armiraglio M, Quarello P, Garelli E, Carando A, Avondo F, Pavesi E, Fribourg S, Gleizes P, et al. 2008. RPS19 mutations in patients with Diamond–Blackfan anemia. Hum Mutat 29: 911–920. 10.1002/humu.20752 [DOI] [PubMed] [Google Scholar]
- Chandrasekharappa SC, Lach FP, Kimble DC, Kamat A, Teer JK, Donovan FX, Flynn E, Sen SK, Thongthip S, Sanborn E, et al. 2013. Massively parallel sequencing, aCGH, and RNA-seq technologies provide a comprehensive molecular diagnosis of Fanconi anemia. Blood 121: e138–e148. 10.1182/blood-2012-12-474585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman ML, Qian M, te Kronnie G, Zhang R, Yang W, Zhang H, Lana T, Tedrick P, Baskin R, Verbist K, et al. 2018. Germline genetic IKZF1 variation and predisposition to childhood acute lymphoblastic leukemia. Cancer Cell 33: 937–948 e938. 10.1016/j.ccell.2018.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieudonne Y, Guffroy A, Vollmer O, Carapito R, Korganow AS. 2019. IKZF1 loss-of-function variant causes autoimmunity and severe familial antiphospholipid syndrome. J Clin Immunol 39: 353–357. 10.1007/s10875-019-00643-2 [DOI] [PubMed] [Google Scholar]
- Draptchinskaia N, Gustavsson P, Andersson B, Pettersson M, Willig T, Dianzani I, Ball S, Tchernia G, Klar J, Matsson H, et al. 1999. The gene encoding ribosomal protein S19 is mutated in Diamond–Blackfan anaemia. Nat Genet 21: 169–175. 10.1038/5951 [DOI] [PubMed] [Google Scholar]
- Eskandarian Z, Fliegauf M, Bulashevska A, Proietti M, Hague R, Smulski CR, Schubert D, Warnatz K, Grimbacher B. 2019. Assessing the functional relevance of variants in the IKAROS family zinc finger protein 1 (IKZF1) in a cohort of patients with primary immunodeficiency. Front Immunol 10: 568. 10.3389/fimmu.2019.00568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Liu X, Bolanos L, Barker B, Rigolino C, Cortelezzi A, Oliva EN, Cuzzola M, Grimes HL, Fontanillo C, et al. 2016. A calcium- and calpain-dependent pathway determines the response to lenalidomide in myelodysplastic syndromes. Nat Med 22: 727–734. 10.1038/nm.4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar JE, Dahl N. 2011. Untangling the phenotypic heterogeneity of Diamond Blackfan anemia. Semin Hematol 48: 124–135. 10.1053/j.seminhematol.2011.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar JE, Vlachos A, Atsidaftos E, Carlson-Donohoe H, Markello TC, Arceci RJ, Ellis SR, Lipton JM, Bodine DM. 2011. Ribosomal protein gene deletions in Diamond–Blackfan anemia. Blood 118: 6943–6951. 10.1182/blood-2011-08-375170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer ES, Böhm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, Nagel J, Serluca F, Acker V, Lingaraju GM, et al. 2014. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512: 49–53. 10.1038/nature13527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazda HT, Preti M, Sheen MR, O'Donohue MF, Vlachos A, Davies SM, Kattamis A, Doherty L, Landowski M, Buros C, et al. 2012. Frameshift mutation in p53 regulator RPL26 is associated with multiple physical abnormalities and a specific pre-ribosomal RNA processing defect in Diamond–Blackfan anemia. Hum Mutat 33: 1037–1044. 10.1002/humu.22081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman FD, Gurel Z, Al-Zubeidi D, Fried AJ, Icardi M, Song C, Dovat S. 2012. Congenital pancytopenia and absence of B lymphocytes in a neonate with a mutation in the Ikaros gene. Pediatr Blood Cancer 58: 591–597. 10.1002/pbc.23160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gripp KW, Curry C, Olney AH, Sandoval C, Fisher J, Chong JX, UW Center for Mendelian Genomics; Pilchman L, Sahraoui R, Stabley DL, et al. 2014. Diamond–Blackfan anemia with mandibulofacial dystostosis is heterogeneous, including the novel DBA genes TSR2 and RPS28. Am J Med Genet A 164A: 2240–2249. 10.1002/ajmg.a.36633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He CF, Liu YS, Cheng YL, Gao JP, Pan TM, Han JW, Quan C, Sun LD, Zheng HF, Zuo XB, et al. 2010. TNIP1, SLC15A4, ETS1, RasGRP3 and IKZF1 are associated with clinical features of systemic lupus erythematosus in a Chinese Han population. Lupus 19: 1181–1186. 10.1177/0961203310367918 [DOI] [PubMed] [Google Scholar]
- He X, Dou A, Feng S, Roman-Rivera A, Hawkins C, Lawley L, Zhang J, Wunderlich M, Mizukawa B, Halene S, et al. 2020. Cyclosporine enhances the sensitivity to lenalidomide in MDS/AML in vitro. Exp Hematol 86: 21–27 e22. 10.1016/j.exphem.2020.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino A, Okada S, Yoshida K, Nishida N, Okuno Y, Ueno H, Yamashita M, Okano T, Tsumura M, Nishimura S, et al. 2017. Abnormal hematopoiesis and autoimmunity in human subjects with germline IKZF1 mutations. J Allergy Clin Immunol 140: 223–231. 10.1016/j.jaci.2016.09.029 [DOI] [PubMed] [Google Scholar]
- Hung KH, Su ST, Chen CY, Hsu PH, Huang SY, Wu WJ, Chen MJ, Chen HY, Wu PC, Lin FR, et al. 2016. Aiolos collaborates with Blimp-1 to regulate the survival of multiple myeloma cells. Cell Death Differ 23: 1175–1184. 10.1038/cdd.2015.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobucci I, Mullighan CG. 2017. Genetic basis of acute lymphoblastic leukemia. J Clin Oncol 35: 975–983. 10.1200/JCO.2016.70.7836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Sif S, Jones B, Jackson A, Koipally J, Heller E, Winandy S, Viel A, Sawyer A, Ikeda T, et al. 1999. Ikaros DNA-binding proteins direct formation of chromatin remodeling complexes in lymphocytes. Immunity 10: 345–355. 10.1016/S1074-7613(00)80034-5 [DOI] [PubMed] [Google Scholar]
- Krönke J, Knop S, Langer C. 2017. Prognostic impact of Ikaros expression in lenalidomide-treated multiple myeloma. Oncotarget 8: 106163–106164. 10.18632/oncotarget.22572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn HS, Boisson B, Cunningham-Rundles C, Reichenbach J, Stray-Pedersen A, Gelfand EW, Maffucci P, Pierce KR, Abbott JK, Voelkerding KV, et al. 2016. Loss of B cells in patients with heterozygous mutations in IKAROS. N Engl J Med 374: 1032–1043. 10.1056/NEJMoa1512234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landowski M, O'Donohue MF, Buros C, Ghazvinian R, Montel-Lehry N, Vlachos A, Sieff CA, Newburger PE, Niewiadomska E, Matysiak M, et al. 2013. Novel deletion of RPL15 identified by array-comparative genomic hybridization in Diamond–Blackfan anemia. Hum Genet 132: 1265–1274. 10.1007/s00439-013-1326-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Lodish HF, Sieff CA. 2018. Critical issues in Diamond–Blackfan anemia and prospects for novel treatment. Hematol Oncol Clin North Am 32: 701–712. 10.1016/j.hoc.2018.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, He X, Sui Y, Yu R, Xu G. 2015. Transcription factor IKZF1 is degraded during the apoptosis of multiple myeloma cells induced by kinase inhibition. FEBS Lett 589: 2233–2240. 10.1016/j.febslet.2015.06.044 [DOI] [PubMed] [Google Scholar]
- Mirabello L, Macari ER, Jessop L, Ellis SR, Myers T, Giri N, Taylor AM, McGrath KE, Humphries JM, Ballew BJ, et al. 2014. Whole-exome sequencing and functional studies identify RPS29 as a novel gene mutated in multicase Diamond–Blackfan anemia families. Blood 124: 24–32. 10.1182/blood-2013-11-540278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirabello L, Khincha PP, Ellis SR, Giri N, Brodie S, Chandrasekharappa SC, Donovan FX, Zhou W, Hicks BD, Boland JF, et al. 2017. Novel and known ribosomal causes of Diamond–Blackfan anaemia identified through comprehensive genomic characterisation. J Med Genet 54: 417–425. 10.1136/jmedgenet-2016-104346 [DOI] [PubMed] [Google Scholar]
- Morgan B, Sun L, Avitahl N, Andrikopoulos K, Ikeda T, Gonzales E, Wu P, Neben S, Georgopoulos K. 1997. Aiolos, a lymphoid restricted transcription factor that interacts with Ikaros to regulate lymphocyte differentiation. EMBO J 16: 2004–2013. 10.1093/emboj/16.8.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narla A, Ebert B. 2010. Ribosomopathies: human disorders of ribosome dysfunction. Blood 115: 3196–3205. 10.1182/blood-2009-10-178129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne KJ, Nicolas JH, Zhu JY, Barsky LW, Crooks GM. 2001. Cutting edge: predominant expression of a novel Ikaros isoform in normal human hemopoiesis. J Immunol 167: 1867–1870. 10.4049/jimmunol.167.4.1867 [DOI] [PubMed] [Google Scholar]
- Popescu M, Gurel Z, Ronni T, Song C, Hung KY, Payne KJ, Dovat S. 2009. Ikaros stability and pericentromeric localization are regulated by protein phosphatase 1. J Biol Chem 284: 13869–13880. 10.1074/jbc.M900209200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran VG, Ghazvinian R, Do R, Thiru P, Vergilio JA, Beggs AH, Sieff CA, Orkin SH, Nathan DG, Lander ES, et al. 2012. Exome sequencing identifies GATA1 mutations resulting in Diamond–Blackfan anemia. J Clin Invest 122: 2439–2443. 10.1172/JCI63597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Yang XR, Ballew B, Rotunno M, Calista D, Fargnoli MC, Ghiorzo P, Bressac-de Paillerets B, Nagore E, Avril MF, et al. 2014. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet 46: 482–486. 10.1038/ng.2941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura A, Alter BP. 2010. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev 24: 101–122. 10.1016/j.blre.2010.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speedy HE, Di Bernardo MC, Sava GP, Dyer MJ, Holroyd A, Wang Y, Sunter NJ, Mansouri L, Juliusson G, Smedby KE, et al. 2014. A genome-wide association study identifies multiple susceptibility loci for chronic lymphocytic leukemia. Nat Genet 46: 56–60. 10.1038/ng.2843 [DOI] [PubMed] [Google Scholar]
- Stanulla M, Cave H, Moorman AV. 2020. IKZF1 deletions in pediatric acute lymphoblastic leukemia: still a poor prognostic marker? Blood 135: 252–260. 10.1182/blood.2019000813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajuddin SM, Schick UM, Eicher JD, Chami N, Giri A, Brody JA, Hill WD, Kacprowski T, Li J, Lyytikainen LP, et al. 2016. Large-scale exome-wide association analysis identifies loci for white blood cell traits and pleiotropy with immune-mediated diseases. Am J Hum Genet 99: 22–39. 10.1016/j.ajhg.2016.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nieuwenhove E, Garcia-Perez JE, Helsen C, Rodriguez PD, van Schouwenburg PA, Dooley J, Schlenner S, van der Burg M, Verhoeyen E, Gijsbers R, et al. 2018. A kindred with mutant IKAROS and autoimmunity. J Allergy Clin Immunol 142: 699–702 e612. 10.1016/j.jaci.2018.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachos A, Rosenberg PS, Atsidaftos E, Kang J, Onel K, Sharaf RN, Alter BP, Lipton JM. 2018. Increased risk of colon cancer and osteogenic sarcoma in Diamond–Blackfan anemia. Blood 132: 2205–2208. 10.1182/blood-2018-05-848937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Ahlford A, Järvinen TM, Nordmark G, Eloranta ML, Gunnarsson I, Svenungsson E, Padyukov L, Sturfelt G, Jönsen A, et al. 2013. Genes identified in Asian SLE GWASs are also associated with SLE in Caucasian populations. Eur J Hum Genet 21: 994–999. 10.1038/ejhg.2012.277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Yoshida K, Toki T, Sawada T, Uechi T, Okuno Y, Sato-Otsubo A, Kudo K, Kamimaki I, Kanezaki R, et al. 2015. Loss of function mutations in RPL27 and RPS27 identified by whole-exome sequencing in Diamond–Blackfan anaemia. Br J Haematol 168: 854–864. 10.1111/bjh.13229 [DOI] [PubMed] [Google Scholar]
- Wojcik H, Griffiths E, Staggs S, Hagman J, Winandy S. 2007. Expression of a non-DNA-binding Ikaros isoform exclusively in B cells leads to autoimmunity but not leukemogenesis. Eur J Immunol 37: 1022–1032. 10.1002/eji.200637026 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genomic data are available through controlled access in dbGaP per the National Institutes of Health (NIH) genomic data sharing policy (Study Accession: phs001481.v1.p1). The IKZF1 variant was submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and can be found under accession number VCV000988782.1.