

Abstract
Multiplexed proteomics is a powerful tool to assay cell states in health and disease, but accurate quantification of relative protein changes is impaired by interference from co-isolated peptides. Interference can be reduced by using MS3-based quantification, but this reduces sensitivity and requires specialized instrumentation. An alternative approach is quantification by complementary ions, the balancer group-peptide conjugates, which allows accurate and precise multiplexed quantification at the MS2 level and is compatible with most proteomics instruments. However, complementary ions of the popular TMT tag form inefficiently and multiplexing is limited to five channels. Here, we evaluate and optimize complementary ion quantification for the recently released TMTpro tag (TMTproC), which increases complementary ion plexing capacity to eight channels. Furthermore, the beneficial fragmentation properties of TMTpro increase sensitivity for TMTproC resulting in ~65% more proteins quantified compared to TMTpro-MS3, and ~18% more when compared to real-time-search TMTPro-MS3 (RTS-SPS-MS3). TMTproC quantification is more accurate than TMTpro-MS2 and even superior to MS3-SPS-RTS. We provide the software for quantifying TMTproC data as an executable that is compatible with the MaxQuant analysis pipeline. Thus, TMTproC advances multiplexed proteomics data quality and widens access to accurate multiplexed proteomics beyond laboratories with MS3-capable instrumentation.
Keywords: multiplexing, complementary ions, shotgun proteomics, TMTpro, CID, isobaric labelling, accurate quantification, interference-free, FAIMS, prefractionation
Graphical Abstract
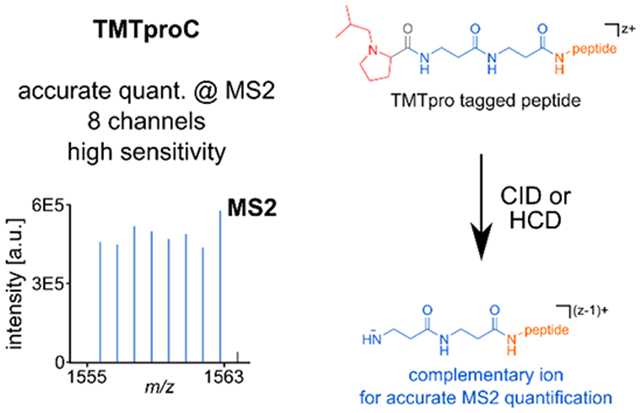
Introduction:
Quantitative multiplexed proteomics has become a powerful tool to analyze the proteome across various biological conditions. Proteins from multiple samples are enzymatically digested and the resulting peptides are labelled with one of several isobaric tags. The samples are then combined before analysis on a mass spectrometer in a single run. The different quantification channels are encoded by the distribution of heavy isotopes between the reporter and balancer region of the reagents. Because the overall number of heavy isotopes is constant between the different tags, they add the same total mass to the peptides, and hence are isobaric. However, during gas-phase fragmentation, the reporter and balancer group are separated, revealing differences in the masses of each region. This information is used for determining which condition the ions stem from and permits relative quantification of peptides in the MS2 or MS3 spectrum (Fig. 1A).1-3 Multiplexing is especially attractive because of the increased sample throughput - with current commercial isobaric tags up to 16 conditions can be compared in a single experiment, thus helping to save expensive mass spectrometer instrument time.2,4
Figure 1: Complementary Ion Quantification with TMT and TMTpro.

A) TMT- and TMTpro-tags are comprised of a reporter region (red), a balancer region (blue) and an amine-reactive NHS-ester moiety (green rectangle). The carboxyl group lost as CO during fragmentation is part of the balancer and highlighted in an ellipse (grey-blue). While the TMT-tag can incorporate a maximum of five heavy isotope labels (asterisks) in its balancer group, this number increases to nine in the TMTpro balancer region. To make use of the larger plexing potential, the reporter group in TMTpro utilizes an iso-butylpyrrolidine-moiety, which can incorporate nine heavy isotope labels. B) When analyzing complex samples via shotgun proteomics, in addition to the peptide of interest (orange), other peptides with similar m/z-ratio (interferents, green) will be co-isolated (grey box). If MS2 reporter ions are used for quantification, the interfering peptides lead to a distortion of the measured ratios, as the source of reporter ions (red squares) cannot be distinguished. However, because the masses of complementary ions are peptide-dependent and include the heavy isotope labels of the balancer-region (blue rectangles), they can be used for interference-free, accurate MS2 quantification. C) During fragmentation of a TMTpro-modified peptide, the positively charged reporter ion is separated from the ion and a neutral CO-molecule is lost. This leads to an ion where the balancer part is still attached to the peptide. Because the balancer region encodes the complementary heavy isotope labels of the reporter ion, the balancer-peptide conjugate is called the TMTproC or complementary ion. In this process, the charge state of the precursor ion is reduced by one. D) Example MS2 spectra of the peptide ASNTAEVFFDGVR2+, labeled with TMT (left) or with TMTpro (right) and fragmented with HCD. As the inserts show, TMTpro-labeled peptides generate eight instead of five complementary ions, extending the plexing capability from TMT.
An inherent advantage of multiplexed proteomics is that the samples are co-analyzed, avoiding problems with missing values that are common in label-free experiments. The co-analysis of all samples in the same MS experiment enables exquisite measurement reproducibility and precision. Because isobaric tags are attached after sample lysis, data collection is compatible with essentially any protein sample, avoiding the need for isotopic labeling in living systems, which is common in methods that depend on heavy isotopes (e.g. SILAC).5 These advantages have resulted in the ever increasing popularity of multiplexed proteomics, leading to a wide variety of findings in breast cancer treatment,6 lung cancer metastasis,7 as well as fundamental research into translation regulation,8 among many others.
A major challenge inherent to multiplexed proteomics quantification is measurement accuracy. In its simple implementation, co-eluting peptides with similar mass to charge ratios are co-isolated and co-fragmented with the peptide of interest. The resulting quantification is typically significantly distorted (Fig. 1B).1,3,9,10 The most widely used method to overcome ratio distortion uses an additional MS3 scan.1,11 In these methods, commercialized on Thermo Fisher Scientific tribrids,12 several b- and y-ions from the MS2 scan are simultaneously co-isolated and fragmented in an ion-trap (multi-notch or SPS-MS3). The extra gas-phase purification step leads to a significant decrease of ratio distortion. However, this advantage comes at the cost of decreased sensitivity and the need for highly specialized instrumentation with SPS-MS3 capabilities, which have been used in less than a quarter of proteomics studies deposited in PRIDE in 202013.
An alternative method for accurate multiplexed proteomics is to make use of the complementary ions.14,15 This method was designed as a quantification strategy that does not require higher order scans and can be employed on a wide variety of instruments. When peptides are fragmented in the MS2 spectra, the loss of the isobaric reporter ion leaves the balancer region with a complementary isotope distribution behind. These balancer-peptide-conjugates, the so-called complementary ions, have peptide-dependent m/z ratios that are typically slightly different than co-isolated peptides (Fig. 1A, C). Therefore, using the complementary ions for quantification reduces ratio distortion effects compared to both MS2 reporter ion quantification as well as multi-notch MS3 approaches. Further improvements to the method, including a narrower isolation window and modelling of the isolation window shape in the deconvolution algorithm, further improved measurement precision.15 This approach, called TMTc+ when used with the TMT isobaric tag, has been successfully applied to multiple biological research studies.16-19
Despite its attractiveness, remaining challenges hinder widespread application of TMTc+. First, the plexing capacity of TMTc+ is limited to five channels because the small mass differences between 13C and 15N cannot be resolved in the high m/z-regime of the complementary ions. In addition, the loss of CO during TMT-fragmentation reduces the number of heavy isotopes available for encoding quantification channels by one. Furthermore, commercial isobaric tags were not designed for this approach and complementary ion formation is poor compared to reporter ion formation. High energies are necessary to separate the reporter from the balancer region, but the peptide backbone is also amenable to breaking at these levels, leading to reduced complementary ion intensity.
Easy to cleave sulfoxide tags, like the SO-tag and EASI-tag, were designed to improve complementary ion formation efficiency.20,21 However, fragmentation with these tags seems to happen too readily, typically leading to additional fragmentations and MS2 spectra of very high complexity, which hinders identification and leads to low identification success-rates and distributing much of the signal away from the intact peptide complementary ions. Though we believe these processes generate complement fragment b- and y-ions that might be very attractive for multiplexed Data-Independent Acquisition or targeted Data-Dependent Acquisition approaches,3 they provide a severe hindrance for shotgun-proteomics.
Recently Thermo Fisher Scientific released a new isobaric tag, named TMTpro.2 This tag was primarily designed for Thermo Fisher Scientific’s MS3 methods and is commercially available as encoding up to 16 different conditions in a single experiment. We noticed that this proline-based tag breaks easier than the previous TMT-tag while not having a detrimental effect on identification rates. We reasoned that this tag could be well-suited for the complement reporter ion approach. Here, we optimize data acquisition strategies for quantification of complement reporter ions with TMTpro in a method termed TMTproC. We find that TMTpro significantly improves the efficiency of complementary ion formation compared to TMT and increases plexing from five to eight with one Dalton separation. TMTproC also reduces ratio compression more than MS3 methods while maintaining sensitivity levels equivalent to TMTpro-MS2.
To facilitate use of the method, the deconvolution software used to analyze TMTproC data has been packaged into a stand-alone version that is compatible with the MaxQuant analysis pipeline22 and made available on Github (https://github.com/wuhrlab/TMTproC). We have also made the source code available in the same Github directory.
Methods:
Proteomics sample preparation
Samples were mostly prepared as previously described.15,23,24 Human peptides from HeLa cell lysate and yeast peptides from Saccharomyces cerevisiae lysate were both used for method optimization. For description of lysis conditions and further information, please reference the supporting information.
Briefly, lysates were reduced by 5 mm DTT (20 min, 60°C), alkylated with 20 mm NEM (20 min, RT), and quenched with 10 mm DTT (10 min, RT). Proteins were purified by methanol-chloroform precipitation25 and afterwards resuspended in 10 mm EPPS pH 8.0 with 6 M guanidine hydrochloride (GuHCl). They were then diluted to 2 M GuHCl with 10 mM EPPS pH 8.0 and digested with 10 ng/μL LysC (Wako) at room temperature overnight. Samples were further diluted to 0.5 M GuHCl with 10 mM EPPS pH 8.0 and digested with an additional 20 ng/μL LysC and 10 ng/μL sequencing grade Trypsin (Promega) at 37°C for 16 hours.
Samples were then vacuum dried and re-suspended in 200 mM EPPS at pH 8.0. TMTpro tags were mixed at the appropriate ratios prior to labelling peptides. The TMTpro mixture was added at a mass ratio of 5:1 tag:peptide and allowed to react for 2 hours at room temperature. The reaction was quenched with 1% hydroxylamine (30 min, RT). Peptides were then acidified to pH~2 with phosphoric acid.
Unfractionated samples were centrifuged at 24k rcf for 10 minutes at 4 °C before desalting via SepPak-cartridges (Waters). Samples were vacuum-dried and re-suspended in 1% formic acid before mass spectrometer analysis. For pre-fractionation, samples were spun at 50krcf for 1 hour at 4°C and separated by reverse-phase HPLC at pH 8. For samples used to evaluate ratio accuracy and the extent of peptide interference, human and yeast peptides were mixed prior to mass spectrometry analysis.
LC-MS experiments
Samples were analyzed on an EASY-nLC 1200 (Thermo Fisher Scientific) HPLC coupled to an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific) with Tune version 3.3. Peptides were separated on an Aurora Series emitter column (25 cm x 75 μm ID, 1.6 μm C18) (ionopticks, Australia), held at 60°C during separation by an in-house built column oven, over 120 min for unfractionated and 90 min for fractionated samples, applying nonlinear acetonitrile-gradients at a constant flow rate of 350 nL/min. Samples were analyzed with either a MS2-CID-method or MS2-HCD-method for TMTproC, or with conventional MS2-HCD- and SPS-MS3-methods for reporter ion-based quantification adjusted from J. Li et al.26
To compare the results of RTS-SPS-MS3 and TMTproC methods, several samples were analyzed on an nLC-1200 HPLC (Thermo Fisher Scientific) coupled to an Orbitrap Eclipse (Thermo Fisher Scientific) running Tune version 3.3. Peptides were separated on an EASY-Spray column (Thermo Fisher Scientific, 2 μm, ID 75 μm x 25 cm) held at 60 °C during separation by an EASY-Spray Source (Thermo Fisher Scientific). 120 min nonlinear acetonitrile-gradients at a constant flow rate of 300 nL/min were utilized.
To investigate the effect on ratio distortion when utilizing field asymmetric ion mobility spectrometry, a FAIMS Pro device (Thermo Fisher Scientific) was used in some experiments.27
For detailed information about the gradients and methods utilized, as well as data analysis procedures, please see the supporting information.
Proteomics data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE13 partner repository with the dataset identifier PXD021661 and 10.6019/PXD021661.
Software access
The deconvolution software used to analyze the TMTproC data sets has been packaged into a stand-alone version that is compatible with the MaxQuant analysis pipeline22 and made available on Github (https://github.com/wuhrlab/TMTproC). The only inputs required of users are a Thermo Scientific .raw file and the output of the MaxQuant search. Complementary peak assignment and extraction of signal to Fourier transform noise (S:N) are done through a Python script. The complementary ion deconvolution algorithm and other data processing are performed by an executable file that requires the use of a free version of Matlab Runtime. We have also made the source code for these Matlab scripts available in the same Github directory.
Results and Discussion:
TMTpro increases plexing for complementary reporter ion quantification
A major shortcoming of the previous complementary reporter ion approach (TMTc+) was the limit of five encoded channels.14,15 Thermo Fisher Scientific recently released an isobaric tag (TMTpro) with a new proline-based reporter group and a longer balancer region that can accommodate up to nine heavy isotopes, compared to five heavy isotopes accessible in TMT (Fig. 1A).2 The current set of commercially available TMTpro tags increase plexing for the complement reporter ion approach from five (TMT) to eight (Fig. 1D, Sup. Fig. S1A). A structure without heavy isotopes in the balancer region could increase plexing capacity to nine while maintaining the same total number of heavy isotopes (Sup. Fig. S1B).
The slight mass difference between isotopomeric structures incorporating 13C vs 15N allows up to 16 distinguishable channels with reporter ion quantification in high-resolution mass analyzers (50k resolution at m/z 200).2 However, at the higher mass range of complementary reporter ions, these small mass differences cannot be resolved while maintaining high acquisition speed. Super-resolution acquisition is a very active area of research and once a resolution of >500k at the high m/z-ranges of complementary ions becomes available with acceptable transient times,28,29 twelve channels could be distinguished with the current set of TMTpro tags (see Sup. Fig. S1A). Theoretically, isotopomeric structures with a total of eight heavy isotopes could encode up to 21 different conditions using TMTproC (see Sup. Fig. S1C). The higher plexing capacity of TMTpro for complementary ions using fewer heavy isotopes, compared to low m/z reporter ion quantification, is due to the two nitrogen atoms in the balancer region, while there is only a single nitrogen atom in the reporter region. A structure with the same number of total heavy isotopes in the complementary ion could therefore be split into three distinguishable peaks.
TMTproC generates highly accurate quantitative proteomics data
To evaluate the accuracy of measured ratios and distortion due to interfering peptides, we prepared a sample of mixed HeLa lysate and Saccharomyces cerevisiae (yeast) lysate (Fig. 2A).1 HeLa peptides were labelled with TMTpro in ratios of 1:1 in all eight channels, while yeast peptides were labelled in ratios of 0:1:5:10:10:5:1:0. The two lysates were combined after labelling with ten parts HeLa for every one part yeast peptides. This sample simulates the quantification of lower abundant peptides that change concentration between conditions (yeast) in a background of highly abundant peptides that do not change concentration (HeLa). When isolating yeast peptides, co-isolation of HeLa peptides will tend to bias the measured channel ratios towards 1, making quantification less accurate. The mixed HeLa-yeast sample was analyzed with a 120-minute run using three different quantification methods: TMTpro-MS2, TMTpro-MS3, and TMTproC. In this section all experiments are performed on an Orbitrap Lumos. Later we will compare TMTproC with TMTpro-MS3 RT search on an Orbitrap Eclipse. Isotopic impurities in the reporter and complementary regions of each tag were measured using heavy-labelled arginine reacted with TMTpro (see Supporting Information. Fig. S2). In addition, we assessed the effectiveness of a High-Field Asymmetric Waveform Ion Mobility Spectrometry (FAIMS) device to reduce the effect of ratio distortion using each method.27
Figure 2. Evaluating ratio distortion for TMTproC and alternative multiplexed quantification strategies.

A) Yeast lysate labelled with TMTpro in ratios of 0:1:5:10:10:5:1:0 was mixed with HeLa lysate labelled with TMTpro in ratios of 1:1:1:1:1:1:1:1 at a mixing ratio of 1 (yeast):10 (HeLa). B-D) Histograms showing the measured ratios of the 10:5 (C), 5:1 (D), and 10:1 (E) channels for yeast peptides with various quantification strategies. Peptides with a sum S:N of less than 200 were removed from this analysis. Measured ratios outside the histogram were set to the closest ratio shown. MS2 quantification of the 10:1 and 5:1 channels is plagued by interference, which MS3 and TMTproC reduce. TMTproC outperforms TMTpro-MS3, since fewer peptides are distorted (shoulders on the left side of the 5:1 and 10:1 histogram). E) The cumulative distribution function of measured ratios for each peptide of the two 10:0 channel pairs was calculated using three different quantification methods, each with and without ion-mobility prefractionation (FAIMS). The measured ratio of the 10:0 channels is plotted against the summed fraction of peptides showing a ratio less than or equal to that ratio. In absence of interference, the ratio would be infinite, but with interference, the ratio is reported as smaller. Therefore, the lower the cumulative fraction of peptides at the high 100 ratio cut-off, the better the method deals with interfering peptides. Peptides for which the sum of the quantifiable ions signal to noise ratio (S:N) was less than 500 were removed for all methods. Other signal to noise cutoffs can be found in Sup Fig. S5. Measured ratios greater than 100 were set to 100. TMTpro-MS3 reduces interference compared to TMTpro-MS2. TMTproC reduces interference even further compared to TMTpro-MS3.
In the analyzed sample, ratio distortion from interfering HeLa peptides can be assessed by calculating the ratio of the signal to Fourier transform noise (S:N) of the 10 and 0 channels on each side of the reporter or complementary ion envelope for each yeast peptide. The lower the measured ratios, the more co-eluted HeLa peptides are interfering, while a ratio approaching infinity would be the expected outcome without interference (Sup. Fig. S9). Peptides with less than 500 total S:N in the complementary or reporter envelope were removed, regardless of the quantification method, so that a S:N ratio of 100 could theoretically be detected from the 10:0 channel. The cutoff ensures that peaks at the noise level are not misinterpreted as a measured infinite ratio due to low signal.
The cumulative distribution function of the relative S:N ratio in the 10:0 channels is presented in Fig. 2B. TMTpro-MS3 (orange) outperforms TMTpro-MS2 (blue), with 39% of peptides having a measured ratio greater than 100, as compared to 4.2% for TMTpro-MS2. Decreasing spectra complexity by utilizing a FAIMS device moderately reduced interference for TMTpro-MS2, but not to the level of TMTpro-MS3. The highest reduction of interference is observed with TMTproC, yielding even less interference than TMTpro-MS3, both for runs with and without a FAIMS device. Using TMTproC (green), more than 66% of peptides had a measured ratio greater than 100, which agrees with the results using TMTc+.15 FAIMS consistently reduced ratio distortion for all three quantification methods, although the method used had a stronger effect in all cases.
We also evaluated the effect of ratio distortion on the accuracy and precision of the remaining channel ratios by determining the measured ratios for the 10:5 channels, the 5:1 channels, and the 10:1 channels (Fig. 2C-E). Ratios were normalized by the measured ratios of a yeast-only measurement using the same quantification method (Sup. Fig. S10). TMTpro-MS3 and TMTproC were able to reproduce the expected 10:5 ratio of the innermost channels with only small variations between the methods (Fig. 2C). Even TMTpro-MS2 performed reasonably well for this ratio, but ratio distortion is clearly observable.
Ratio distortion greatly reduced the measured ratio of the 5:1 and 10:1 ratios using TMTpro-MS2. The median ratios for this method were 2.6 and 4.6, respectively (Fig. 2D, E). The use of FAIMS slightly improved the median measured ratios of the channels to 3.1 and 5.9, respectively, for MS2 reporter ion quantification (Sup. Fig. 4).
TMTpro-MS3 and TMTproC reduce interference for both the measured 5:1 and 10:1 ratios. We observe that the mode for TMTpro-MS3 and TMTproC is slightly lower than the expected ratio (Fig. 2D, E). This could be due to interference or slightly inaccurate data normalization. The two methods differ moderately in the tails of their distributions. The TMTproC distribution is approximately symmetric relative to mode, including a tail extending further to the right. The TMTpro-MS3 method is slightly asymmetric, with 18% of peptides having a ratio less than 2.5:1 for the 5:1 channels (Fig. 2D) and 15% of peptides having a ratio less than 5:1 in the 10:1 channels (Fig. 2E) due to interference from co-eluting peptides. With TMTproC, the fraction of these inaccurate peptides was nearly cut in half, with 7% of 5:1 channels having a ratio less than 2.5:1 (Fig. 2D) and 10% having a ratio less than 5:1 for the 10:1 channels (Fig 2E). For those cases, TMTproC found no signal in the lower abundant channel for ~2% peptides, likely due to interference from peptides with slightly different complementary masses than the peptide of interest, thus shifting the true complementary peak. This effect was negligible for the 10:5 ratio. Overall, these results show that TMTproC reduces interference from contaminating peptides as well as or better than MS3 quantification, resulting in highly accurate quantification even in highly complex samples.
TMTpro efficiently forms complementary ions
So far, we have shown that TMTpro increases plexing and maintains superior measurement accuracy for the complementary reporter ion strategy. Next, we evaluated how efficiently TMTpro forms complementary ions, which is a major shortcoming of TMT for this strategy.14 For TMT, this is particularly challenging for higher charge states and for ions that contain highly mobile protons, i.e. more charges than are localized on arginine, lysine, and histidine. In previous studies we only isolated precursors with a 2+ charge state for complementary reporter ion quantification with TMT,14-17,23 due to inefficient complementary ion formation for more highly charged ions. We wondered if TMTpro would break more easily and increase the flux into complementary ions which would further increase the method's sensitivity.
First, we optimized various fragmentation methods and energies for TMTpro complementary reporter ion formation. To do so, we labeled HeLa peptides with TMTpro without heavy isotopes (TMTpro0). For each quantified peptide we calculated the ratio of the ion flux in the precursor peak of the MS1 spectrum and the ion flux of complimentary ions in the MS2 spectrum (see Supporting Information).
Using beam-type HCD fragmentation30, we find that the optimal normalized collision energy for complementary ion generation is around 29% for both 2+ and 3+ ions, although 3+ complementary ion formation efficiency shows less variation at 27% HCD energy while retaining similar signal. Optimal energies were lower than those used for TMT owing to the relatively facile fragmentation of the TMTpro reagent. Peptides with a 2+ charge transmitted on average more than 13% of precursor ion flux in the MS1 into the complementary ion peak of the MS2. Efficiencies were significantly higher than those of TMT-tagged peptides with a 2+ charge state at only 4% median efficiency (Student’s t-test, p-value=0.002).
Peptides with a 3+ charge formed complementary ions at around a 7.5% median efficiency. With the increase in complementary ion formation efficiency, many more peptides with a 3+ charge state have enough signal for quantification compared to TMT. However, we would like to note that 3+ ions are more prone to systematic biases if the actual quadrupole isolation window differs slightly from the modeled isolation window in the deconvolution algorithm. The narrower m/z selection afforded by the QR5 Segmented Quadrupole, e.g. available on the Orbitrap Eclipse might be particularly attractive for quantifying 3+ peptides with the TMTproC method.12
We also explored resonance collision induced dissociation (CID)31 of TMTpro. Complementary ion formation was 15% higher for 2+ ions and 45% higher for 3+ ions compared to HCD fragmentation (Student’s t-test p-value=0.31 and 0.008, respectively). The combination of TMTpro and CID fragmentation increased total complementary ion formation efficiency by more than 3-fold over TMT fragmented with the optimized TMTc+ method (32% HCD, Student’s t-test, p-value=0.002). This major improvement brings TMTproC close to the efficiencies of TMTpro-MS3 low m/z reporter ion-based quantification, greatly increasing the sensitivity of the complementary reporter ion quantification approach.
Efficient TMTproC ion formation results in high sensitivity for complex quantitative proteomics studies
A significant advantage of TMTproC over TMTpro-MS3 is the increased depth of proteome coverage. Using a sample of TMTpro0-tagged HeLa peptides, we quantified, on average, 3,842 proteins in a single 2-hour run at 1% protein false discovery rate (FDR) using CID fragmentation. The equivalent run with TMTpro-MS2 quantified 3,234 proteins on average, while TMTpro-MS3 only quantified 2,321 proteins on average (Fig. 4A). These differences were statistically significant using a Student’s two-sample t-test (p-value<10−3 for both). TMTproC with HCD fragmentation performed similarly to CID fragmentation and was still superior to TMTpro-MS2 (3,756 quantified proteins on average, Student’s t-test p-value=0.026). Similarly, TMTproC quantifies more peptides (18,390) than TMTpro-MS3 (11,051) and TMTpro-MS2 (14,924) (Fig. 4B, Student’s t-test p-value<10−3 for both comparisons). It should be noted that these sensitivity measurements do not account for the lower number of accessible channels in TMTproC compared to reporter ion-based quantification. The reduction in sensitivity for TMTpro-MS3 is almost entirely due to the higher overhead times of performing an additional MS scan. We expected that TMTpro-MS2 would be more sensitive than TMTproC because reporter ions are formed at higher efficiency, thereby lowering injection times and increasing duty cycles. However, this effect is offset by the need to filter peptides from the TMTpro-MS2 run where the isolation specificity is less than 75% to diminish interference effects, as peptides with low isolation specificity are subject to more interference than those with higher isolation specificity (Sup. Fig. S6).11 Filtering TMTproC data for this criterion is unnecessary because interfering peptides are inherently excluded from quantification.
Figure 4. Evaluating TMTproC sensitivity for the analysis of a sample proteome.

A, B) Number of quantified HeLa proteins (A) and peptides (B) in replicates of a 120 min unfractionated analysis of a TMTpro0-tagged HeLa lysate sample. Both TMTproC measurements were analyzed five times, while TMTpro-MS2 and MS3 quantification were each done four times. Error bars show a single standard deviation from the mean. TMTproC using CID fragmentation is more sensitive than either TMTpro-MS3 or TMTpro-MS2 in the number of quantified peptides and proteins (Student’s t-test p-value<10−3 for all four comparisons). The sensitivity of TMTproC using HCD fragmentation also outperforms TMTpro-MS3 and TMTpro-MS2, and was not significantly different from CID fragmentation (Student’s t-test p-value=0.60 at the peptide level and p-value=0.61 at the protein level). C) Average peptide spectral matches (PSM), quantifications and effects of filter criteria for the analyses in A) and B). Percentages in parentheses in each cell represent the proportion of PSM that passed that filter. With TMTproC, isolation specificity filters do not have to be applied due to higher interference resistance, but stringent sum S:N thresholds for the ions used for quantification lead to some peptide removal. Still, after filtering, TMTproC outperforms the reporter ion methods in both number of quantified peptides and overall quantification rate. D) Number of human and yeast proteins quantified from a mixed sample shot with TMTproC (S:N>40, 1% Protein FDR, max ppm deviation<10) as a function of the number of fractions used. Only a handful of fractions were necessary to quantify more than 10,000 proteins, and the full 24 fractions led to 13,960 quantified proteins.
We chose to consider a peptide as quantified if its complementary ion envelope signal to FT noise (S:N) summed to at least 40. Recent advancements in integrating ion statistics with peptide concordance allow for peptides with nearly any S:N to improve ratio estimates.23 We therefore used 40 S:N as a conservative cutoff for this study. To use this Bayesian inference method (BACIQ), we need to be able to convert signal to noise into pseudo-counts. We have done so for peptides with 2+ and 3+ charge states at various Orbitrap resolutions as shown in Supplemental Figure S3.
To test the limits of TMTproC sensitivity, we prefractionated a 1:1 mixture of human and yeast peptides by mid-pH reverse-phase HPLC into 24 fractions. Each fraction was analyzed with a 90-minute gradient. Across all fractions, we quantify 13,290 proteins at a 1% protein FDR and signal:noise cutoff of 40 (Fig. 4D). Peptides were also removed if the measured m/z value of any complementary peak disagreed with their expected value by more than 10 ppm. 4,610 of these quantified proteins were yeast and 8,680 were from HeLa. A similar sample of HeLa peptides, also separated into 24 fractions, which was analyzed using TMTc+, resulted in 8,943 protein quantifications.15 A mixed human and yeast sample using the EASI tag quantified 9,760 proteins using four-fold more instrument time and less stringent quantification filtering criteria.21 These results demonstrate the improved sensitivity of TMTproC over other complementary ion-based quantification strategies.
Benchmarking TMTproC against Real-Time Search TMTpro-MS3
Recently, it has been shown that combining a real-time search (RTS) algorithm with MS3-based methods can improve measurement accuracy and sensitivity.32 This approach obviates the need for MS3 scans when MS2 spectra could not be matched to peptides and improve ratio accuracy by avoiding isolation and fragmentation of interfering peptide fragments for MS3 scans. Thermo Fisher Scientific has made this method available exclusively on the Orbitrap Eclipse, which our lab does not own. We sent a TMTpro0-tagged HeLa sample and the mixture of HeLa and yeast peptides to the Kirschner Lab at Harvard Medical School that kindly allowed us to use their Orbitrap Eclipse to benchmark TMTproC relative to MS3-SPS-RTS.
The RTS step removed some of the ratio distortion effect from co-eluting peptides, justifying the removal of the isolation specificity filter of 0.75 that was used for MS2 and traditional MS3 reporter ion quantification (Sup. Fig. S8). For the 10:1 channels, MS3-SPS-RTS measured a ratio below 5 for 18% of peptides across both runs (Fig. 5A). The median reported ratio was shifted away from the true value of 10 to 7.5, indicating that some interference was still present in many spectra (Sup. Fig. S11). These metrics nearly match those achieved by traditional MS3 quantification, but without the need to filter peptides from the analysis due to limited isolation specificity. For TMTproC, the median ratio of the 10:1 channels was shifted to 8.3, and 8% of peptides had a measured ratio below 5. These results validate the data presented in Fig. 2E for TMTproC performance in measuring a 10:1 ratio in a complex background of interfering peptides and demonstrate that TMTproC reduces the distorting effect of co-eluting peptides even better than the MS3-SPS-RTS method.
Figure 5. Comparison of the sensitivity and accuracy of TMTproC against TMTpro-MS3 Real-Time Search.

A) Identical experimental design as in Fig. 2. Yeast lysate labelled with TMTpro in ratios of 0:1:5:10:10:5:1:0 was mixed with HeLa lysate labelled with TMTpro in ratios of 1:1:1:1:1:1:1:1 at a mixing ratio of 1 (yeast):10 (HeLa) and this very challenging interference sample analyzed on an Orbitrap Eclipse. Although the distribution of the measured ratios of yeast peptides’ 10:1 channels is similar for both methods, RTS-SPS-MS3 (orange) shows a larger shift of the median away from the real ratio of 10 after normalization (median: 7.5) compared to TMTproC (green, median: 9.1). For both methods, a minimum summed S:N value of 200 was required for the quantifying ions. B) Cumulative fraction plot of the measured yeast peptides’ ratios of the 10:0 channels. Without interference, a value of infinity is expected, while lower values indicate increasing levels of interference. While both methods behave very similarly on the left side of the distribution, a larger portion of peptides quantified with RTS-SPS-MS3 show less severe, but present levels of interference than TMTproC. Measured ratios greater than 100 were set to 100, and a minimum S:N threshold of 500 was required for quantifying ions of both methods. C-D) TMTpro0-tagged HeLa analyzed on an Orbitrap Eclipse Tribrid Mass Spectrometer. Despite the increased efficiency of the real-time search algorithm, and dropping the isolation specificity filter of 0.75 which is no longer necessary, TMTproC is still able to quantify more proteins (C) and peptides (D) in back-to-back analyses of the same sample on the Eclipse.
The cumulative distribution plot (Fig. 5B) displays the even more challenging case of determining the ratio of the innermost channels labelled in 10 parts to the unlabeled channels at the end of the envelope for yeast peptides, as was also shown in Fig. 2B. To avoid misinterpreting low signals as an absence of interference, peptides were only considered when they showed a very conservative summed S:N value of at least 500 in their ions used for quantification. Both methods show a similar behavior on the left side of the distribution, with ~19% of peptides reporting severe interference with measured ratios between 1 and 10 of the 10:0 channels. However, a higher proportion of peptides quantified by RTS-SPS-MS3 had a measured ratio between 10 and 100 than peptides quantified by TMTproC. This is in accordance with Fig. 5A, since a small level of interference for most peptides will lead to a downward shift of the median ratio. Overall, only 20% of yeast peptides quantified with RTS-SPS-MS3 report a ratio of greater than 100, with the majority of peptides being distorted by varying levels of interference. In contrast, TMTproC produces more accurate quantification results, measuring a ratio of greater than 100 for 40% of yeast peptides.
Sensitivity comparisons of TMTpro-RTS-SPS-MS3 and TMTproC were conducted with TMTpro0-tagged HeLa with back-to-back duplicates analyzed in 2-hour gradients. The number of quantified proteins and peptides are presented in Fig. 5C,D. Compared to Fig. 4A,B and the non-RTS MS3 method, the difference in quantified species between RTS-MS3 and TMTproC is reduced, partly because the RTS-method avoids the time-wasting acquisition of MS3-scans which cannot be matched to a peptide, and partly because the more accurate quantification removes the need to filter for isolation specificity. Despite these improvements, TMTproC is still able to quantify 18% more proteins and 25% more peptides than the current state-of-the-art method RTS-SPS-MS3, which is currently available on one mass spectrometer model.
Conclusion:
In this paper we have demonstrated that the combination of TMTpro with complementary ion quantification for multiplexed proteomics, termed TMTproC, has superb accuracy, sensitivity, higher plexing capacity, and is subject to relatively little interference from co-eluting peptides. Its inherent flexibility makes it a competitive method in a wide variety of research areas, including investigation of cellular organization, bioengineering, and disease states. We show that TMTproC maintains the ratio precision of TMTc+, and owing to more facile fragmentation, forms complementary ions for quantification at more than 3-fold the efficiency of TMT. Resonance CID and beam-type HCD fragmentation are both considered, with CID fragmentation narrowly outperforming HCD fragmentation for complementary ion formation, although the differences were not statistically significant. HCD has the further advantage of being compatible with nearly all fragmentation-capable mass spectrometers. In addition, using HCD allows the simultaneous acquisition of both low m/z reporter ions and high m/z complementary ions, which could be beneficial for peptides that inefficiently form complementary ions. Use of a FAIMS device slightly reduced interference from co-eluting peptides in all cases, but the effect of the quantification method was more pronounced. We utilized the optimized method to quantify 13,290 proteins in 24 fractions each analyzed with a 90-minute gradient.
Furthermore, TMTproC can be performed on relatively simple instruments compared to (RTS)-multi-notch MS3 methods. We have made the TMTproC deconvolution software accessible as source code and as executable files that can perform quantification based on the popular MaxQuant analysis pipeline22 (https://github.com/wuhrlab/TMTproC). TMTproC is compatible with multiple mass spectrometers, e.g. with the QExactive/Exploris platform or QTOF-platforms, encompassing the majority of commercial proteomics instruments. Therefore, TMTproC opens up the possibility of high-quality proteomics measurements to many research laboratories that were previously reliant on inaccurate, interference prone MS2 reporter ion quantification.
Supplementary Material
Figure S1. Tag structures and plexing using TMTproC
Figure S2. Isotopic impurities for the commercially available TMTpro-tags
Figure S3. Converting FT signal:noise ratios to pseudo-counts for Bayesian inference
Figure S4. Evaluating ratio distortion for TMTproC and alternative multiplexed quantification strategies with a FAIMS
Figure S5. Evaluating ratio distortion for TMTproC and alternative multiplexed quantification strategies with various S:N cutoffs
Figure S6. Evaluating ratio distortion for TMTproC and alternative multiplexed quantification strategies for peptides with an Isolation Specificity of less than 0.75
Figure S7. Evaluating ratio distortion of TMTproC and MS3-SPS-RTS
Figure S8. Evaluating ratio distortion of TMTproC and MS3-SPS-RTS for peptides with an isolation specificity of less than 0.75
Figure S9. Measured ratio of 10:0 channels of the yeast peptides analyzed without interfering HeLa peptides
Figure S10. Evaluating ratio distortion for TMTproC and alternative multiplexed quantification strategies with a direct comparison of a yeast sample measured alone to the yeast peptide quantified in a background of HeLa peptides
Figure S11. Evaluating ratio distortion for TMTproC and SPS-MS3-RTS quantification strategies on an Orbitrap Eclipse with a direct comparison of the yeast sample measured alone to the yeast peptide quantified in a background of HeLa peptides
Figure 3. Optimization of TMTproC ion formation and comparison with ion formation efficiencies in alternative multiplexed proteomics methods.

A) Evaluation of various fragmentation methods for TMTproC ion formation. HeLa peptides were labelled with TMTpro0, subjected to various fragmentation schemes and analyzed five times across several weeks of normal instrument use. For each run, the median proportion of MS1 ion flux from the precursor peak that was converted into complementary ions was calculated. Shown are the mean of five runs along with error bars of one standard deviation. On average, CID fragmentation produced 15% more signal in the complementary ion cluster than HCD fragmentation, but this difference was not significant (Student’s t-test, p-value=0.31). B) Comparison of TMTproC ion efficiencies with alternative methods. HeLa peptides labeled with TMTpro0 or TMT0 were subjected to various quantification methods. For each, we calculate the median fraction of precursor ion flux that is converted into the relevant ion used for quantification and then calculated the mean across multiple runs (five for TMTproC methods, four for all others) along with error bars of one standard deviation. TMTproC improves complementary ion formation efficiency to nearly the same level as MS3 reporter ion-based quantification methods (Student’s t-test, p-value=0.27).
Acknowledgments
We would like to thank Lance Martin (supported by the Princeton Catalysis Initiative) for technical assistance. We thank Matthew Sonnett, Elizabeth Van Itallie, and Marc Kirschner for help and access to their Orbitrap Eclipse. We also thank Meera Gupta, Thao Nguyen and Felix Keber for helpful comments on the manuscript, and Graeme McAlister for helpful suggestions and discussions. This work was supported by the Eric and Wendy Schmidt Transformative Technology Fund, NIH grant R35GM128813, and the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research under award number DE-SC0018420.
References
- 1.Ting L, Rad R, Gygi SP, Haas W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Methods. 2011;8(11):937–940. doi: 10.1038/nmeth.1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thompson A, Wo N, Koncarevic S, et al. TMTpro: Design, Synthesis, and Initial Evaluation of a Proline-Based Isobaric 16-Plex Tandem Mass Tag Reagent Set. Anal Chem. 2019;91:15941–15950. doi: 10.1021/acs.analchem.9b04474 [DOI] [PubMed] [Google Scholar]
- 3.Pappireddi N, Martin L, Wühr M. A Review on Quantitative Multiplexed Proteomics. ChemBioChem. 2019;20:1210–1224. doi: 10.1002/cbic.201800650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ross PL, Huang YN, Marchese JN, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3(12):1154–1169. doi: 10.1074/mcp.M400129-MCP200 [DOI] [PubMed] [Google Scholar]
- 5.Ong S, Blagoev B, Kratchmarova I, et al. Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.M200025-MCP200 [DOI] [PubMed] [Google Scholar]
- 6.Jordan NV, Bardia A, Wittner BS, et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature. 2016;537. doi: 10.1038/nature19328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lignitto L, Leboeuf SE, Homer H, et al. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1 Article Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell. 2019;178(2):316–329.e18. doi: 10.1016/j.cell.2019.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simsek D, Tiu GC, Flynn RA, et al. The Mammalian Ribo-interactome Reveals Ribosome Functional Diversity and Heterogeneity Article The Mammalian Ribo-interactome Reveals Ribosome Functional Diversity and Heterogeneity. Cell. 2017;169(6):1051–1057.e18. doi: 10.1016/j.cell.2017.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ow SY, Salim M, Noirel J, Evans C, Rehman I, Wright PC. iTRAQ Underestimation in Simple and Complex Mixtures : “The Good , the Bad and the Ugly.” J Proteome Res. 2009;8:5347–5355. doi: 10.1021/pr900634c [DOI] [PubMed] [Google Scholar]
- 10.Wenger CD, Lee MV, Hebert AS, et al. Gas-phase purification enables accurate , multiplexed proteome quantification with isobaric tagging. Nat Methods. 2011;8(11):1–5. doi: 10.1038/nmeth.1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McAlister GC, Nusinow DP, Jedrychowski MP, et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem. 2014;86(14):7150–7158. doi: 10.1021/ac502040v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu Q, Paulo JA, Naverrete-perea J, et al. Benchmarking the Orbitrap Tribrid Eclipse for Next Generation Multiplexed Proteomics. Anal Chem. 2020;92:6478–6485. doi: 10.1021/acs.analchem.9b05685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perez-Riverol Y, Csordas A, Bai J, et al. The PRIDE database and related tools and resources in 2019 : improving support for quantification data. Nucleic Acids Res. 2019;47:442–450. doi: 10.1093/nar/gky1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wühr M, Haas W, McAlister GC, et al. Accurate multiplexed proteomics at the MS2 level using the complement reporter ion cluster. Anal Chem. 2012;84(21):9214–9221. doi: 10.1021/ac301962s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sonnett M, Yeung E, Wühr M. Accurate , Sensitive , and Precise Multiplexed Proteomics using the Complement Reporter Ion Cluster. Anal Chem. 2018;90:5032–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hart EM, Gupta M, Wühr M, Silhavy TJ. The gain-of-function allele bamA E470K bypasses the essential requirement for BamD in β -barrel outer membrane protein assembly. Proc Natl Acad Sci. 2020;117(31). doi: 10.1073/pnas.2007696117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cao WX, Kabelitz S, Gupta M, et al. Precise Temporal Regulation of Post-transcriptional Repressors Is Required for an Orderly Drosophila Maternal-to-Zygotic Transition. Cell Rep. 2020;31(107783). doi: 10.1016/j.celrep.2020.107783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li A, Mao D, Yoshimura A, et al. Multi-Omic Analyses Provide Links between Low-Dose Antibiotic Treatment and Induction of Secondary Metabolism in Burkholderia thailandensis. Mol Biol Physiol. 2020;11(1):e03210–19. doi: 10.1128/mBio.03210-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hart EM, Gupta M, Wühr M, Silhavy TJ. The synthetic phenotype of Δbamb Δbame double mutants results from a lethal jamming of the bam complex by the lipoprotein RcsF. Mol Biol Physiol. 2019;10(3):1–12. doi: 10.1128/mBio.00662-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stadlmeier M, Bogena J, Wallner M, Martin W, Carell T. A Sulfoxide-Based Isobaric Labelling Reagent for Accurate Quantitative Mass Spectrometry Angewandte. Angew Chemie. 2018;08544:2958–2962. doi: 10.1002/anie.201708867 [DOI] [PubMed] [Google Scholar]
- 21.Winter SV, Meier F, Wichmann C, Cox J, Mann M, Meissner F. EASI-tag enables accurate multiplexed and quantification. Nat Methods. 2018;15(July):527–530. doi: 10.1038/s41592-018-0037-8 [DOI] [PubMed] [Google Scholar]
- 22.Tyanova S, Temu T, Cox J. The MaxQuant computational platform for mass spectrometry – based shotgun proteomics. Nat Protoc. 2016;11(12):2301–2319. doi: 10.1038/nprot.2016.136 [DOI] [PubMed] [Google Scholar]
- 23.Peshkin L, Gupta M, Ryazanova L, Wühr M. Bayesian Confidence Intervals for Multiplexed Proteomics Integrate Ion-statistics with Peptide Quantification Concordance. Mol Cell Proteomics. 2019;18(10):2108–2120. doi: 10.1074/mcp.TIR119.001317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gupta M, Sonnett M, Ryazanova L, Presler M, Wühr M. Quantitative Proteomics of Xenopus Embryos I, Sample Preparation. In: Xenopus. ; 2018:175–194. doi: 10.1007/978-1-4939-8784-9_13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wessel D, Flugge UI. A Method for the Quantitative Recovery of Protein in Dilute Solution in the Presence of Detergents and Lipids. Anal Biochem. 1984;143:141–143. [DOI] [PubMed] [Google Scholar]
- 26.Li J, Vranken Van JG, Vaites LP, et al. TMTpro reagents: a set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nat Methods. 2020;17:399–404. doi: 10.1038/s41592-020-0781-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hebert AS, Prasad S, Belford MW, et al. Comprehensive Single-Shot Proteomics with FAIMS on a Hybrid Orbitrap Mass Spectrometer. Anal Chem. 2018;90:9529–9537. doi: 10.1021/acs.analchem.8b02233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kelstrup CD, Aizikov K, Batth TS, et al. Limits for Resolving Isobaric Tandem Mass Tag Reporter Ions Using Phase-Constrained Spectrum Deconvolution. J Proteome Res. 2018;17:4008–4016. doi: 10.1021/acs.jproteome.8b00381 [DOI] [PubMed] [Google Scholar]
- 29.Kozhinov AN, Tsybin YO. Filter diagonalization method-based mass spectrometry for molecular and macromolecular structure analysis. Anal Chem. 2012;84(6):2850–2856. doi: 10.1021/ac203391z [DOI] [PubMed] [Google Scholar]
- 30.Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M. Higher-energy C-trap dissociation for peptide modification analysis. Nat Methods. 2007;4(9):709–712. doi: 10.1038/NMETH1060 [DOI] [PubMed] [Google Scholar]
- 31.Schwartz JC, Senko MW. A Two-Dimensional Quadrupole Ion Trap Mass Spectrometer. Am Soc Mass Spectrom. 2002;13:659–669. [DOI] [PubMed] [Google Scholar]
- 32.Schweppe DK, Eng JK, Yu Q, et al. Full-Featured, Real-Time Database Searching Platform Enables Fast and Accurate Multiplexed Quantitative Proteomics. J Proteome Res. 2020;19:2026–2034. doi: 10.1021/acs.jproteome.9b00860 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Tag structures and plexing using TMTproC
Figure S2. Isotopic impurities for the commercially available TMTpro-tags
Figure S3. Converting FT signal:noise ratios to pseudo-counts for Bayesian inference
Figure S4. Evaluating ratio distortion for TMTproC and alternative multiplexed quantification strategies with a FAIMS
Figure S5. Evaluating ratio distortion for TMTproC and alternative multiplexed quantification strategies with various S:N cutoffs
Figure S6. Evaluating ratio distortion for TMTproC and alternative multiplexed quantification strategies for peptides with an Isolation Specificity of less than 0.75
Figure S7. Evaluating ratio distortion of TMTproC and MS3-SPS-RTS
Figure S8. Evaluating ratio distortion of TMTproC and MS3-SPS-RTS for peptides with an isolation specificity of less than 0.75
Figure S9. Measured ratio of 10:0 channels of the yeast peptides analyzed without interfering HeLa peptides
Figure S10. Evaluating ratio distortion for TMTproC and alternative multiplexed quantification strategies with a direct comparison of a yeast sample measured alone to the yeast peptide quantified in a background of HeLa peptides
Figure S11. Evaluating ratio distortion for TMTproC and SPS-MS3-RTS quantification strategies on an Orbitrap Eclipse with a direct comparison of the yeast sample measured alone to the yeast peptide quantified in a background of HeLa peptides