

Abstract
Purpose:
The incidence of retinopathy of prematurity (ROP) has increased continuously in recent years. However, the therapeutic effects of current treatments still remain undesired. This study aims to investigate the role of C-CBL in retinal angiogenesis in ROP and its potential as a therapeutic target.
Methods:
Mouse retina microvascular endothelial cells (mRMECs) and induced experimental ROP/ oxygen-induced retinopathy (OIR) mice were employed to investigate the role of C-CBL in angiogenesis with combined molecular and cellular approaches, and histopathology methods. OIR mouse pups at postnatal day 12 (P12) were either injected intravitreally with adenovirus overexpressing c-Cbl or c-Cbl siRNA. Retinal neovascularization and avascular status were evaluated by retinal immunofluorescence (IF) staining, whole-mounts and hematoxylin and eosin (H&E) staining.
Results:
C-CBL inhibits neovascularization by negatively regulating JAK2/STAT3/VEGF signaling axis in a ubiquitination-dependent manner. Knockdown of c-Cbl by siRNA reduced ubiquitin-mediated JAK2 degradation and increased levels of p-JAK2, p-STAT3, VEGF, and neovascularization in mRMECs, which can be reversed by JAK2 inhibitor treatment. While knockdown of c-Cbl significantly increased neovascular (NV) zone in the retinas, c-Cbl overexpression inhibited neovascularization in the retinal tissues in OIR mice.
Conclusion:
We found that C-CBL is required for anti-neovascularization process in ROP development by inhibiting JAK2/STAT3-dependent angiogenesis. Thus, our finding strongly suggest that C-CBL may be a potential novel therapeutic target for treating ROP.
Keywords: Retinopathy of prematurity, c-Cbl, JAK2/STAT3/VEGF signaling pathway, Angiogenesis
1. Introduction
Retinopathy of prematurity (ROP) is a proliferative retinal vascular disease, which is a leading cause of potential childhood blindness. An estimated 15 million babies are born prematurely worldwide [1]. With improved survival for premature infants, however, ROP incidence has increased continuously in recent years [2]. Once premature babies are exposed to high oxygen levels, pro-angiogenic factor production decreases, leading to the cessation of normal blood vessel growth. However, after the infants return to room air, relative retinal hypoxia occurs, which causes multiple ROP pathological conditions, including retinal neovascularization, intravitreal hemorrhages, retinal detachment, and potential blindness [3,4]. Current treatments for ROP include freezing, laser photocoagulation, anti-VEGF drugs, and vitreous surgery as well. These treatments, however, are all invasive and tissue destructive [5]. Although anti-VEGF injection is the only treatment available thus far for treating neovascularization in infants with ROP, whether systemically reduced VEGF level could cause lifelong side effects on neurodevelopment remains in question [6,7]. These therapies also induce undesired long-term outcome on visual acuity and fields. In particular, the inherently destructive side effect to the retina caused by those treatments shouldn’t be neglected [8]. There have been very limited studies that focused on the targets related to neurovascular protection and neuroprotection in ROP prevention, such as VEGF, EPO, HIF-1α, and NOS [9,10]. Therefore, additional research is needed to better understand the exact cause and etiopathogenesis of the disease, which may lead to the identification of new therapeutic targets and open an avenue for better treating children with ROP.
Angiogenesis plays a pivotal role in a broad array of physiologic and pathologic conditions. Aberrant activation of Janus kinase 2/signal transducer and activator of transcription 3 (JAK2/STAT3) signaling pathway is involved in abnormal angiogenesis through upregulating VEGF expression in many ocular diseases, including ROP [11–13], suggesting that inhibition of JAK2/STAT3 activation may lead to a promising therapeutic strategy for ROP treatment. Although previous reports showed that both PEDF and UCP-2 suppress VEGF expression through negative regulation of the JAK2/STAT3 signaling pathway [14–16], the molecular mechanism underlying JAK2/STAT3 regulation still remains to be further unveiled.
C-CBL protein is encoded by the c-Cbl gene (named after Casitas B-lineage Lymphoma), which is an E3 ubiquitin-protein ligase and ubiquitously expressed in mammalian cells with the highest level of expression in hematopoietic tissues. C-CBL is a multifunctional protein involved in cell survival, migration and proliferation [17–20]. For instance, C-CBL is capable of degrading various receptor-tyrosine kinases (RTKs) and diverse sets of proteins through its E3 ubiquitin-protein ligase activity [19,20]. We previously reported that C-CBL is associated with the development of age-related macular degeneration (AMD), an eye disease caused by aberrant angiogenesis [21]. Husain et al. found that loss of C-CBL results in enhanced choroidal neovascularization (CNV) [22], which might be associated with elevated PLCγ1 activation. However, the possible link between C-CBL and JAK2/STAT3 activity in ocular pathologic angiogenesis remains unknown.
In this study, we demonstrated that C-CBL is involved in JAK2/STAT3-dependent ROP pathogenesis. By using cultured mouse retina microvascular endothelial cells (mRMECs) and induced ROP/oxygen-induced retinopathy (OIR) mouse model [23,24], we demonstrated that C-CBL inhibits angiogenesis by negatively regulating JAK2/STAT3/VEGF signaling axis in a ubiquitination-dependent manner. While knockdown of c-Cbl significantly increased neovascular (NV) zone in the retina tissues, c-Cbl overexpression inhibited retinal neovascularization in OIR mice. Thus, our study provides strong evidence for the first time that C-CBL could be a potential therapeutic target for treating ROP.
2. Materials and methods
2.1. SiRNA and adenoviruses
SiRNA SMARTpool targeting mouse c-Cbl (5′-GAGAAUCAACUCAGAACGA-3′, 5′-CGUUUGGGUCAGUGGGCUA-3′, 5′–CCGGAUUACUAA AGCUGAU-3′, and 5′-CGUCAGUCAUAUAUAGAUU-3′) and non-targeting pool control (5′-UGGUUUACAUGUCGACUAA-3′, 5′-UGGUUUACAUGUUGUGUGA-3′, 5′-UGGUUUACAUGUUUUCUGA-3′, and 5′-UGGUUUACAUGUUUUCCUA-3′) were purchased from Dharmacon. Mouse adenoviruses (Ad) Ad-Cbl overexpressing mouse c-Cbl (GenBank NM_007619) and vehicle control Ad-MCMV-MCS-3FLAG were purchased from OBiO Technology (Shanghai, China).
2.2. MRMEC culture and genetic manipulation
MRMEC line was purchased from Cell Biologies. MRMECs were cultured in ECM supplemented with 5% fetal bovine serum, 100U/mL penicillin, and 100U/mL streptomycin at 37°C in a humidified incubator containing 5% CO2 and 95 % air. For siRNA knockdown, mRMECs were transfected with either c-Cbl siRNA or non-targeting control siRNA using lipofectamine RNAiMAX (Invitrogen) as instructed in the manual. For Ad infection, mRMECs were infected with either Ad overexpressing mouse c-Cbl or Ad vehicle control at a multiplicity of infection (MOI) of 50. 24 h post-transfection or infection, cells were exposed to hypoxic gas environment with 1% O2, 94 % N2 and 5% CO2 for 24 h as a hypoxic model. For inhibition, 10μM MG-132 (MedChemExpress) and 2.5μM JAK2-specific inhibitor TG101209 (MedChemExpress) were used individually or in combination with siRNA.
2.3. ROP/OIR mouse model and intravitreal injection
C57BL/6J mice were purchased from the Animal Laboratory of Shanghai Institute of Materia Medica. All procedures with animals in this study were performed in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the Institutional Animal Care and Use Committee. Neonatal mice and nursing mother were exposed to high oxygen environment (75 % O2) from P7 to P12, and then returned to room air to induce ROP model. On P12, animals were divided into four groups: 1) OIR mice treated with Ad-Cbl; 2) OIR mice treated with vehicle control Ad-MCMV-MCS-3FLAG; 3) OIR mice treated with SMARTpool siRNAs targeting c-Cbl; and 4) OIR mice treated with non-targeting pool control. The pupils were dilated with a topical application of 2.5 % phenylephrine and 1% tropicamide (weight/volume). A glass injector (~33 gauge) connected to a syringe filled with 1 μl of adenovirus (1 × 1011 pfu/mL) or 0.5μl of siRNA (1μg/μl) was injected into the vitreous cavity. All mice were sacrificed on P17.
2.4. Tube formation assay
24 h post-siRNA transfection, mRMECs were harvested and seeded (2 × 104 cells/well) in 96-well plates pre-coated with 50μl/well Matrigel (Corning). Cells were then incubated with 10 μM MG-132 and 2.5 μM TG101209 individually or in combination with siRNA in serum-free ECM medium for 4 h, followed by tube formation assay under microscope. Control groups were incubated with inhibitor-free medium. Images were acquired for each well using inverted microscope with 10X Objective Len (Olympus), and were analysed using ImageJ software (National Institutes of Health).
2.5. Western blot analysis
Cultured mRMECs or retinal samples isolated from mice were harvested and lysed in RIPA lysis buffer (Cell signaling) containing 1X protease/phosphatase inhibitor cocktail (Roche) and 1 mM PMSF. Protein concentrations of lysates were measured using BCA protein assay reagent (Thermo Scientific), and were used for Western blot analyses (30–50 μg/sample). C-CBL, phospho-JAK2 (p-JAK2), JAK2, phospho-STAT3 (p-STAT3), STAT3, HIF-1α antibodies were purchased from Cell Signaling, β-actin was purchased from Sigma.
2.6. Coimmunoprecipitation (Co-IP)
Co-IP was performed using Pierce Co-IP kit (Invitrogen) as instructed in the manual. MRMECs were infected with Ad-Cbl and Ad-Ubiquitin-FLAG for 24 h, then treated with 10 μM MG-132 for 6 h and lysed in RIPA lysis buffer containing 1X protease/phosphatase inhibitor cocktail and 1 mM PMSF. Anti-FLAG antibody (Invitrogen) was immobilized with AminoLink Plus coupling resin for 2 h at room temperature. Cell lysates were incubated with anti-FLAG-conjugated agarose resin at 4°C overnight. After washing, the eluted Co-IP samples were analyzed by Western blotting.
2.7. ELISA assay
Mouse retinal tissues or mRMECs were homogenized or lysed in an ice-cold lysis buffer containing a 1X protease inhibitor cocktail and 1 mM PMSF by sonication on ice. Protein concentrations were determined using BCA protein assay reagent. The VEGF levels were measured using mouse VEGF Quantikine ELISA kit (R&D Systems) as instructed in the manual. The amount of VEGF was normalized with total protein concentrations.
2.8. Retinal isolation and retinal whole-mounts
Eyes were fixed in 4% paraformaldehyde (Sigma) for 2 h at 4°C. Retinas were isolated under an operating microscope, and were then permeabilized by incubating each retina with 500μl of 1X PBS containing 0.5 % Triton X-100 (Sigma) at 4°C overnight. After three washes with 1X PBS, the retinas were stained with 10μg/mL isolectin GS-IB4 (Invitrogen) in a light-proof container at 4°C overnight. The retinas were then wholly mounted, and images were taken under Leica TCS SP8 confocal microscope. Images were analyzed using Adobe Photoshop CS5 as previously described [25]. Amount of avascular or neovascular area = number of pixels in the avascular or neovascular area / number of pixels in the total retina.
2.9. Haemotocylin and Eosin (H&E) staining
Eyes were enucleated from mice on P17 and fixed in 10 % neutral buffered formalin overnight. Fixed eyes were paraffin-embedded, and axial sectioned at 6μm thickness, and then subjected to H&E staining. Retinal neovascularization was quantified by counting the vascular nuclei extending beyond the inner limiting membrane. For accuracy, hyaloid vessels near the optic disc and lens were excluded. Results were presented as the mean of neovascular nuclei per ocular cross section.
2.10. Immunofluorescence (IF) staining
Eyes were enucleated from mice on P17 and fixed in 4% paraformaldehyde for 24 h at 4 °C. Fixed retinal tissues were paraffin-embedded, and axial sectioned at 6μm thickness through the optic disk. The paraffin sections were dewaxed with xylene and rehydrated by sequential dipping into different concentration of ethanol and water. Antigens were retrieved in boiled 1X target retrieval solution (Dako) for 40 min and blocked in Tris-NaCl-Tween (TNT) buffer containing 10 % goat serum and 1% BSA for 2 h at room temperature. The slides were then incubated with primary C-CBL antibody (1:50 dilution in TNT buffer) at 4 °C overnight, and then subjected to secondary antibody Alexa Fluor® 488 conjugate (1:200 dilution, Cell Signaling) incubation for 1 h at room temperature. After each incubation, three washes were performed. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Images of the retinas were taken with confocal microscope.
2.11. Statistical analysis
All data are presented as mean ± SD of at least three independent experiments. Differences among experimental groups were analyzed by two-tailed Student’s t-test using GraphPad Prism 5.0 software or SPSS22.0 (IBM). P values of less than 0.05 were considered statistically significant.
3. Results
3.1. C-CBL controls JAK2/STAT3-mediated angiogenesis in mRMECs under normoxia through its ubiquitination activity
To examine whether C-CBL regulates JAK2/STAT3 activity under normoxia, c-Cbl siRNA was employed to knockdown c-Cbl expression in mRMECs. We found JAK2 level was increased compared to vehicle control. Correspondingly, both p-JAK2 and p-STAT3 protein levels were markedly elevated (Fig. 1A). Next, we sought to explore whether C-CBL is involved in JAK2 stability through ubiquitin-mediated protein degradation. After co-transducing mRMECs with Ad-Ubiquitin-FLAG and Ad-Cbl, the cell lysates were co-precipitated with anti-FLAG antibody. Co-IP experiment showed that the ubiquitination level of JAK2 was clearly increased comparing to vehicle control, and p-STAT3 level was well maintained when ubiquitination was inhibited by MG-132, a proteasome inhibitor (Fig. 1B). These data suggest that knockdown of c-Cbl expression by siRNA reduced ubiquitin-mediated JAK2 degradation, which is responsible for increased p-JAK2 and p-STAT3 levels. ELISA and tube formation assays further validated our finding that C-CBL affects angiogenesis through modulating JAK2/STAT3 activity, as knockdown of c-Cbl by c-Cbl siRNA significantly increased VEGF level and promoted tube formation in mRMECs (Fig. 1C&D). Taken together, these results demonstrate that C-CBL plays a pivotal role in angiogenesis through regulating JAK2/STAT3 activity under normoxia in a ubiquitination-dependent manner.
Fig. 1. C-CBL regulates JAK2/STAT3-dependent angiogenesis under normoxia through its ubiquitination activity.
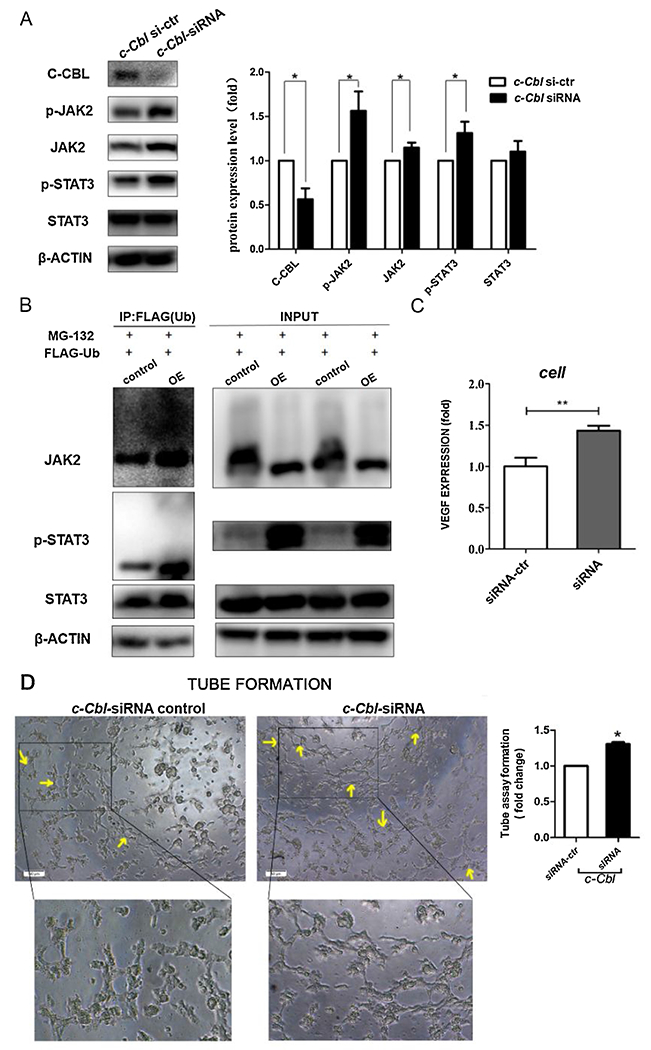
A) Western blot analysis of p-JAK2, JAK2, p-STAT3, and STAT3 protein levels in mRMECs with siRNA transfection as indicated, β-actin was used as a loading control. Right panel shows densitometric analysis of Western blots. Fold change in protein expression is expressed relative to the respective control. Data are representative of three independent experiments. *P < 0.05; t test.
B) Cell lysates were co-precipitated with anti-FLAG antibody (left). Right: input by Western blot.
C) VEGF levels in mRMECs by ELISA. Data represent mean ± SD (three repeats). **P < 0.01; t test.
D) Tube formation assay. The tube formation in mRMECs was indicated by arrows. Average length of tube formation for each field was statistically analyzed. Data represent mean ± SD (three repeats). *P < 0.05; t test. Scale bar: 50 μm.
3.2. Hypoxia-mediated C-CBL reduction promotes tube formation in mRMECs through elevating JAK2/STAT3 activity
Next, we asked if JAK2/STAT3/VEGF signaling cascade is amplified under hypoxia condition. Indeed, VEGF level was robustly increased when mRMECs were exposed to hypoxia preconditioning (1% O2) for 24 h in a tri-gas incubator (Fig. 2A). Given the role of C-CBL in angiogenesis and its potential involvement in ROP pathogenesis, we naturally went on to examine whether C-CBL responds to hypoxia treatment. As expected, hypoxia treatment did cause reduced C-CBL level in mRMECs (Fig. 2B). Correspondingly, we observed significantly increased levels of p-JAK2, p-STAT3, and HIF-1α in mRMECs in response to hypoxia treatment (Fig. 2B). This observation was further verified by knockdown of c-Cbl in mRMECs under hypoxia, which also resulted in markedly increased levels of JAK2/STAT3 phosphorylation, HIF-1α expression, and tube formation (Fig. 2C&D). However, JAK2 inhibitor TG101209 treatment reversed the phenotype, as both JAK2/STAT3 phosphorylation and tube formation were blocked (Fig. 2C&D). Notably, c-Cbl knockdown-mediated more robust tube formation under hypoxia compared to that under normoxia, which clearly demonstrates a bona fide role for C-CBL in suppression of JAK2/STAT3 activity and subsequent neovascularization inhibition under hypoxia (Figs. 1D, 2 D).
Fig. 2. Hypoxia-mediated C-CBL reduction promotes tube formation in mRMECs through elevating JAK2/STAT3 activity.

A) Comparison of VEGF expression in mRMECs between normoxia and hypoxia conditions by ELISA. Data represent mean ± SD (three repeats). ***P < 0.001; t test.
B) Comparison of endogenous C-CBL, p-JAK2, JAK2, p-STAT3 and STAT3 protein levels in mRMECs between normoxic and hypoxia conditions by Western blot. Right panel shows densitometric analysis of Western blots. Fold change in protein expression is expressed relative to the respective control. Data are representative of three independent experiments. *P < 0.05; **P < 0.01; t test.
C) Western blot analysis of effects of c-Cbl knockdown or in combination with JAK2 inhibitor TG101209 on JAK2/STAT3 pathway under hypoxia. Right panels show densitometric analysis of Western blots. Fold change in protein expression is expressed relative to the respective control. Data are representative of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001; t test.
D) Tube formation assay. The tube formation in mRMECs was indicated by arrows. Average length of tube formation for each field was statistically analyzed. Data represent mean ± SD (three repeats). ***P < 0.001; t test. Scale bar: 50 μm.
3.3. Ubiquitination activity of C-CBL is required for preventing hypoxia-mediated angiogenesis
To examine if ubiquitination activity of C-CBL is also required for preventing hypoxia-mediated angiogenesis, we treated mRMECs under hypoxia preconditioning with MG-132. MG-132 treatment significantly elevated the levels of p-JAK2, p-STAT3, and HIF-1α (Fig. 3A), along with increased tube formation (Fig. 3B). Meanwhile, a synergetic effect on JAK2/STAT3 activity and tube formation was observed when MG-132 treatment and c-Cbl knockdown were combined, as the levels of p-JAK2, p-STAT3 and HIF-1α, and the amount of tube formation were all significantly higher than c-Cbl-siRNA treatment only (Fig. 3A&C). These data demonstrated that ubiquitination activity of C-CBL is indispensable for preventing hypoxia-mediated angiogenesis.
Fig. 3. Ubiquitination activity of C-CBL is required for preventing hypoxia-mediated angiogenesis.

A) Comparison of p-JAK2, JAK2, p-STAT3, and STAT3 protein levels in mRMECs with different treatment by Western blot, β-actin was used as a loading control. Right panels show densitometric analysis of Western blots. Fold change in protein expression is expressed relative to the respective control. Data are representative of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001; t test.
B&C) Tube formation assay. The tube formation in mRMECs was indicated by arrows. Average length of tube formation for each field was statistically analyzed. Data represent mean ± SD (three repeats). *P < 0.05; **P < 0.01; t test. Scale bar: 50 μm.
3.4. OIR mice exhibit reduced C-CBL level and increased JAK2/STAT3 activity in the retinal tissues
To further investigate the physiological relevance between C-CBL and JAK2/STAT3 in OIR mice, we examined the levels of these proteins, including JAK2/STAT3 phosphorylation status. IF staining revealed that C-CBL was widely present in the normal retinal, including ganglion cell layer (GCL), inner plexiform layer (IPL), inner nuclear layer (INL), outer plexiform layer (OPL), and outer nuclear layer (ONL). The most prevalent cell type in these retinal layers is vascular endothelial cell. In line with our finding in hypoxia-treated mRMECs, IF staining showed significantly reduced C-CBL expression in pre-retinal neovascular vessels (abnormal vessels) in retinal cross-sections of OIR mice (Fig. 4A). In contrast, C-CBL level was markedly increased in those different retinal layers of normal mice, particularly in GCL (Fig. 4A). Meanwhile, the levels of p-JAK2, JAK2, p-STAT3, and STAT3 are increased significantly in the retinal tissues of OIR mice (Fig. 4B). Consistently, the levels of p-JAK2, p-STAT3 and VEGF were also robustly increased in the retinal tissues of OIR mice injected with c-Cbl siRNA (Fig. 4C&D). These data demonstrated that hypoxia-mediated C-CBL reduction contributes to aberrantly activated JAK2/STAT3/VEGF signaling cascade in OIR mice.
Fig. 4. C-CBL reduction by hypoxia increases JAK2/STAT3 activity in the retinas of OIR mice.

A) IF staining of C-CBL in retina sections of C57BL/6 J mice in room air and induced C57BL/6 J OIR mice. Arrowheads show the infiltrated cells positive for C-CBL. Each group contains eight mice. Scale bar: 50 μm.
B) Western blot assay revealed that JAK2/STAT3 pathway is activated in the retinas of OIR mice. Right panel shows densitometric analysis of Western blots. Fold change in protein expression is expressed relative to the respective control. Data are representative of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001; t test.
C) Knockdown c-Cbl by siRNA intravitreal injection evaluates p-JAK2/p-STAT3/t-STAT3 levels in retinal tissues by western blot. Right panel shows densitometric analysis of Western blots. Fold change in protein expression is expressed relative to the respective control. Data are representative of three independent experiments. *P < 0.05; t test.
D) Comparison of VEGF levels in retinal tissues by ELISA. Data represent mean ± SD (ten repeats). *P < 0.05; t test.
3.5. C-Cbl overexpression inhibits neovascularization in OIR mice
To better understand the role of C-CBL in controlling neovascularization, isolectin B4-stained retina flat mounts and serial paraffin cross-sections from OIR mice at P17 were used to evaluate the pathological changes in retinal tissues. Not surprisingly, mice in room air showed a normal retinal vasculature. Although the avascular regions were similar in the retinas of OIR mice with different treatments, we observed robust retinal neovascularization changes (Fig. 5A). While c-Cbl siRNA injection resulted in a larger area of neovascularization, it was much reduced in OIR mice injected with Ad-c-Cbl-OE that overexpresses c-Cbl (Fig. 5A). Meanwhile, we also examined the status of pre-retinal neovascular vessels, which was defined by vascular tufts extending from the retinal surface into the vitreous cavity using H&E stained retinal sections. Compared with the room air group, the number of tufts was significantly increased in both OIR mice and OIR mice injected with c-Cbl siRNA, whereas c-Cbl overexpression markedly reduced number of neovascular nuclei (Fig. 5B). These data suggest that C-CBL could be a promising therapeutic target for ROP treatment.
Fig. 5. C-Cbl overexpression inhibits neovascularization in OIR mice.

A) Retinal vascular development in normal and OIR mice (P17). Representative retinal whole mounts showing avascular area and NV after intravitreal injection of c-Cbl-OE, c-Cbl siRNA and respective vehicle controls as indicated. Data represent mean ± SD. n = 8; ns, not significant; *P < 0.05; **P < 0.01; t test. Scale bar: 1000 mm.
B) H&E staining of retinas. Neovascular nuclei are indicated by arrows. The number of nuclei per retinal area was quantitated. Data represent mean ± SD. n = 8; **P < 0.01; ***P < 0.001; t test. Scale bar: 50 mm.
4. Discussion
ROP is a vasoproliferative retinopathy in preterm infants, which is now a major cause of preventable childhood blindness worldwide. Oxygen inhalation treatment for preterm infants often causes hyperoxia-induced vessel regression with subsequent hypoxia-induced retinal neovascularization, which is the cause of ROP development [26,27]. We are particularly interested in understanding the mechanisms regulating retinal neovascularization, giving its crucial role in the progression of ROP. Thus far, C-CBL is known as an important molecule for the inhibition of angiogenesis and tumorigenesis by regulating many fundamental cellular processes [17–20]. Our study demonstrated for the first time that loss of C-CBL promotes angiogenesis in cells under hypoxia and in OIR mice. We found that knockdown of c-Cbl significantly increases capillary-like tube numbers in mouse retina microvascular endothelial cells and promotes neovascularization in OIR mouse retinas. Importantly, we found that endogenous C-CBL expression level is reduced dramatically in both hypoxia-treated mRMECs and the retinas of OIR mice, underscoring the role of C-CBL in ROP development.
CBL-mediated modulation of JAK2/STAT3 signaling pathway could result from ubiquitination-mediated protein degradation. Lv et al. reported that the E3 ligase activity of CBL protein is important for regulating cytokine-mediated cell growth through modulating JAK2 protein levels, which is promptly ubiquitinated and degraded by CBL and CBL-B [28]. In agreement with this observation, we also found that C-CBL regulates JAK2/STAT3 activity through its E3 ubiquitin-protein ligase activity under normoxia condition for multiple reasons: First, C-CBL determines the level of JAK2. Second, the effect of knockdown of c-Cbl on increased JAK2/STAT3 activity can be reversed by JAK2 inhibitor treatment. And third, JAK2 interacts with ubiquitin, which is further enhanced by MG-132 treatment. Last but not least, MG-132 treatment increases the amount of tube formation.
Our study further demonstrated that C-CBL/JAK2/STAT3 signaling pathway has a profound effect on aberrant angiogenesis under hypoxia. The activation of JAK2/STAT3 signaling is commonly involved in angiogenic function in various ocular diseases [14,29]. Knockdown of Stat3 in retinal endothelial cells in rat pup eyes efficiently inhibits intravitreal neovascularization (IVNV) [11], and inhibition of JAK2/STAT3-mediated VEGF upregulation by PEDF halts the development of complications in diabetic retinopathy [15]. Consistently, we confirmed that the levels of p-JAK2/p-STAT3/VEGF are significantly increased in both hypoxia-treated mRMECs and the retinas of OIR mice. Similarly, these altered protein/protein phosphorylation levels in hypoxia-treated mRMECs is reversible by JAK2 inhibitor treatment. Javadi et al. found that JAK2 inhibitor may be applied to the treatment of myeloid malignancies that has association with CBL linker region and RING finger mutations [30], which further supports our findings.
Hypoxia damage causes the production of oxidative stress and inadequate blood supply, which triggers constant inflammatory responses and activates various genes involved in ROP pathogenesis. In particular, ROS can activate JAK2/STAT3 signaling directly by tissue damage or inflammation [12,31]. In this study, we provided strong new evidence that C-CBL acts as a molecular switch for controlling JAK2/STAT3/VEGF signaling axis during ROP development. It is likely that hypoxia-mediated C-CBL reduction may promote the proliferation of abnormal blood vessels, leading to rapid progression of the disease. Thus, C-CBL/JAK2/STAT3/VEGF signaling axis would be of interest for developing new anti-angiogenesis therapies against ROP. Although the underlying mechanism in terms of hypoxia-mediated C-CBL reduction is not clear, it should be addressed in a separate study in the future. We hypothesize that VEGF compensatory elevation may occur through increased JAK2/STAT3 activity, which can be negatively regulated by inducing C-CBL-mediated JAK2 degradation in a ubiquitination-dependent manner.
Current conventional treatments for ROP can only partially cure or alleviate ROP symptom because aberrant neovascularization has formed already by the time of treatment, which certainly make the disease more complex to deal with, even not mention the side effects caused by treatments. However, high ROP risk premature infants could benefit from early intervention via targeting neovascularization. As such, it is of importance to develop a novel gene therapy through combination of multiple gene targets involved in anti-angiogenesis process, such as combination of targeting c-Cbl with other genes involved in regulation of JAK2/STAT3 pathway. On the other hand, since ROP is also considered as one of the neurodegeneration diseases, whether C-CBL has the protective effect on retinal neurons should be explored in the future.
Acknowledgements
We thank all the members in Shanghai Key Laboratory of Ocular Fundus Diseases and Chinese Academy of Sciences Shanghai Institute of Materia Medica for their expertise and technical assistance. The work was supported by Natural Science Foundation of China (Grant No. 81700842, 81970812), Excellent Medical Young Talent Projects of Shanghai General Hospital (Grant No. 06N1702019), National Key R&D Program of China (2016YFC0904800, 2019YFC0840607) and National Science and Technology Major Project of China (2017ZX09304010).
Funding information
Natural Science Foundation of China (Grant No. 81700842, 81970812), Excellent Medical Young Talent Projects of Shanghai General Hospital (Grant No. 06N1702019), National Key R&D Program of China (2016YFC0904800, 2019YFC0840607) and National Science and Technology Major Project of China (2017ZX09304010).
Footnotes
Declaration of Competing Interest
The authors report no declarations of interest.
References
- [1].Blencowe H, Cousens S, Oestergaard MZ, et al. , National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: a systematic analysis and implications, Lancet (379) (2012) 2162–2172. [DOI] [PubMed] [Google Scholar]
- [2].Aclimandos W, Seventy years of retinopathy of prematurity, Br. J. Ophthalmol 95 (2011) 899–900. [DOI] [PubMed] [Google Scholar]
- [3].Sapieha P, Joyal JS, Rivera JC, et al. , Retinopathy of prematurity: understanding ischemic retinal vaseulopathies at an extreme of life, J. Clin. Invest 120 (2010) 3022–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hellström A, Smith LEH, Dammann O, Retinopathy of prematurity, Lancet 382 (2013) 1445–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lau CML, Yu Y, Jahanmir G, Chau Y, Controlled release technology for anti-angiogenesis treatment of posterior eye diseases: current status and challenges, Adv. Drug Deliv. Rev 126 (2018) 145–161. [DOI] [PubMed] [Google Scholar]
- [6].Cavallaro G, Filippi L, Bagnoli P, et al. , The pathophysiology of retinopathy of prematurity: an update of previous and recent knowledge, Acta Ophthalmol. 92 (2014) 2–20. [DOI] [PubMed] [Google Scholar]
- [7].Mintz-Hittner HA, Kennedy KA, Chuang AZ, Group B-RC, Efficacy of intravitreal bevacizumab for stage 3+ retinopathy of prematurity, N. Engl. J. Med 364 (2011) 603–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rivera JC, Holm M, Austeng D, et al. , Retinopathy of prematurity: inflammation, choroidal degeneration, and novel promising therapeutic strategies, J. Neuroinflammation 14 (165) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tsang JKW, Liu J, Lo ACY, Vascular and neuronal protection in the developing retina: potential therapeutic targets for retinopathy of prematurity, Int. J. Mol. Sci (2019) 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Caprara C, Grimm C, From oxygen to erythropoietin: relevance of hypoxia for retinal development, health and disease, Prog. Retin. Eye Res 31 (2012) 89–119. [DOI] [PubMed] [Google Scholar]
- [11].Simmons AB, Bretz CA, Wang H, Kunz E, Hajj K, Kennedy C, et al. , Gene therapy knockdown of VEGFR2 in retinal endothelial cells to treat retinopathy, Angiogenesis 21 (2018) 751–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hartnett ME, Pathophysiology and mechanisms of severe retinopathy of prematurity, Ophthalmology 122 (2015) 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bressler NM, Antiangiogenic approaches to age-related macular degeneration today, Ophthalmology 116 (2009) 15–23. [DOI] [PubMed] [Google Scholar]
- [14].Zheng Z, Chen H, Ke G, et al. , Protective effect of perindopril on diabetic retinopathy is associated with decreased vascular endothelial growth factor-to-pigment epithelium-derived factor ratio: involvement of a mitochondria-reactive oxygen species pathway, Diabetes 58 (2009) 954–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zheng Z, Chen H, Zhao H, et al. , Inhibition of JAK2/STAT3-mediated VEGF upregulation under high glucose conditions by PEDF through a mitochondrial ROS pathway in vitro, Invest. Ophthalmol. Vis. Sci 51 (2010) 64–71. [DOI] [PubMed] [Google Scholar]
- [16].Solomon SD, Lindsley K, Vedula SS, et al. , Anti-vascular endothelial growth factor for neovascular age-related macular degeneration, Cochrane Database Syst. Rev 3 (2019), Cd005139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schmidt MHH, Dikic I, The Cbl interactome and its functions, Nat. Rev. Mol. Cell Biol 6 (2005) 907–918. [DOI] [PubMed] [Google Scholar]
- [18].Blake TJ, Heath KG, Langdon WY, The truncation that generated the v-cbl oncogene reveals an ability for nuclear transport, DNA binding and acute transformation, EMBO J. 12 (1993) 2017–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lyle CL, Belghasem M, Chitalia VC, c-Cbl: an important regulator and a target in angiogenesis and tumorigenesis, Cells (2019) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Singh AJ, Meyer RD, Navruzbekov G, et al. , A critical role for the E3-ligase activity of c-Cbl in VEGFR-2-mediated PLCgammal activation and angiogenesis, Proc. Nad. Acad. Sci. U. S. A 104 (2007) 5413–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang J, Jiang M, Yuan F, et al. , Identification of age-related macular degeneration related genes by applying shortest path algorithm in protein-protein interaction network, Biomed Res. Int 2013 (2013), 523415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Husain D, Meyer RD, Mehta M, et al. , Role of c-Cbl-dependent regulation of phospholipase Cgamma1 activation in experimental choroidal neovascularization, Invest. Ophthalmol. Vis. Sci 51 (2010) 6803–6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Campochiaro PA, Molecular pathogenesis of retinal and choroidal vascular diseases, Prog. Retin. Eye Res 49 (2015) 67–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Smith LE, Wesolowski E, McLellan A, Oxygen-induced retinopathy in the mouse, Invest. Ophthalmol. Vis. Sci 35 (1994) 101–111. [PubMed] [Google Scholar]
- [25].Connor KM, Krah NM, Dennison RJ, et al. , Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis, Nat. Protoc 4 (2009) 1565–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hartnett ME, Penn JS, Mechanisms and management of retinopathy of prematurity, N. Engl. J. Med 367 (2012) 2515–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chan-Ling T, Gole GA, Quinn GE, et al. , Pathophysiology, screening and treatment of ROP: a multi-disciplinary perspective, Prog. Retin. Eye Res 62 (2018) 77–119. [DOI] [PubMed] [Google Scholar]
- [28].Lv K, Jiang J, Donaghy R, et al. , CBL family E3 ubiquitin ligases control JAK2 ubiquitination and stability in hematopoietic stem cells and myeloid malignancies, Genes Dev. 31 (2017) 1007–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Al-Shabrawey M, Bartoli M, El-Remessy AB, et al. , Role of NADPH oxidase and Stat3 in statin-mediated protection against diabetic retinopathy, Invest. Ophthalmol. Vis. Sci 49 (2008) 3231–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Javadi M, Richmond TD, Huang K, et al. , CBL linker region and RING finger mutations lead to enhanced granulocyte-macrophage colony-stimulating factor (GM-CSF) signaling via elevated levels of JAK2 and LYN, J. Biol. Chem 288 (2013) 19459–19470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wilkinson-Berka JL, Rana I, Armani R, et al. , Reactive oxygen species, Nox and angiotensin II in angiogenesis: implications for retinopathy, Clin. Sci. (Lond.) 124 (2013) 597–615. [DOI] [PubMed] [Google Scholar]