

Summary
Identifying drugs targeting p53 remains a major focus of precision oncology, with over twenty compounds that can rescue p53 mutants reported. Here, we suggest three easily accessible assays to determine the thermostability, protein folding, and transcriptional activity of p53 mutants—the go-to criteria for evaluating a rescue compound that acts by increasing p53 thermostability. Because of the diversity of p53 mutants, a compound that meets the criteria of one assay does not necessarily meet the criteria of the other assays.
For complete details on the use and execution of this protocol, please refer to Chen et al. (2021).
Subject areas: Cell-based Assays, Cancer, Antibody, Protein Biochemistry
Graphical abstract
Highlights
-
•
Optimized and sensitive protocols to evaluate structural mutant p53-rescuing compounds
-
•
Choose appropriate p53 mutants in a specific assay to avoid false negatives
-
•
Mutant p53 thermostabilizations in DSF assay do not necessarily lead to cellular rescue
Identifying drugs targeting p53 remains a major focus of precision oncology, with over twenty compounds that can rescue p53 mutants reported. Here, we suggest three easily accessible assays to determine the thermostability, protein folding, and transcriptional activity of p53 mutants—the go-to criteria for evaluating a rescue compound that acts by increasing p53 thermostability. Because of the diversity of p53 mutants, a compound that meets the criteria of one assay does not necessarily meet the criteria of the other assays.
Before you begin
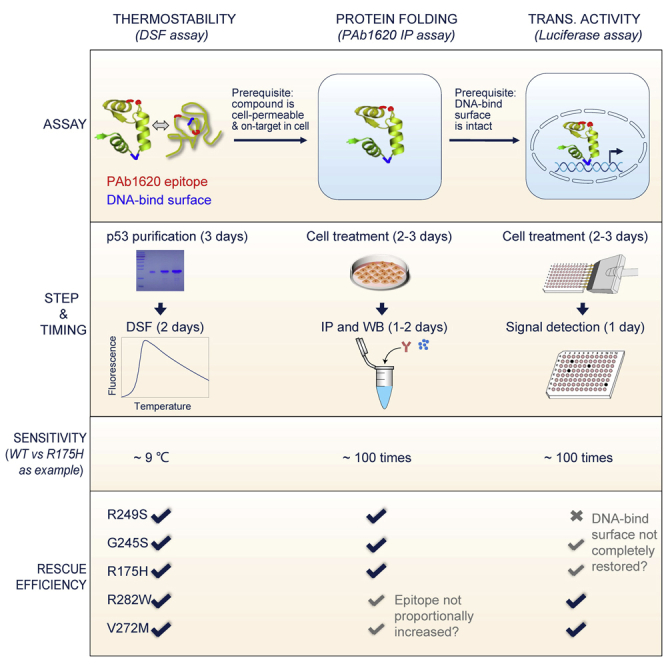
Over ten assays for the determination of p53 activity at different levels have been used to evaluate compounds that can rescue p53 mutants. These include the differential scanning fluorimetry (DSF) assay (Zhang et al., 2018), PAb1620 immunoprecipitation (IP) assay (Xirodimas and Lane, 1999), luciferase reporter assay (Doffe et al., 2020), PAb1620 ELISA assay, PAb1620 immunostaining assay, electrophoretic mobility-shift assay, biotin-DNA pull-down assay for assessing DNA-binding ability, various p53 target expression assays, etc. Some assays such as PAb1620 immunostaining assay are not highly sensitive in our hands, presumably due to the challenge in maintaining structural PAb1620 epitope during paraformaldehyde fixation. In contrast, the first three assays that are used to determine thermostability, folding status, and transcriptional activity of p53, respectively, are easily accessible and sensitive in our hands. Below we suggest these three assays.
Note: we think the three suggested assays are sensitive because, in these assays, the difference between two samples can be much larger than the one among replicates. Consequently, low p values can be frequently observed when comparing two different samples.
Note: Over twenty compounds that can rescue p53 mutants have been reported and most of them are reported to act by increasing the protein’s thermostability. The DSF assay and PAb1620 IP assay described can only be used to evaluate rescue compounds that act by increasing the thermostability of p53.
Note: Compounds that are active in the cell-free DSF assay do not necessarily to be active in the cell-based PAb1620 IP assay or luciferase reporter assay since they may fail to efficiently permeate the cell membrane or fail to bind mutant p53 with sufficient affinity due to off-target effects inside the cell (Figure 1).
Note: Compounds that are active in the PAb1620 IP assay do not necessarily to be active in the luciferase reporter assay since the refolded mutant may fail to regain intact DNA-binding surface or the compound-binding site that overlaps with DNA-binding surface (Figure 1).
Note: Because of the great diversity of inactivation mechanisms that affect different p53 mutants, the same rescue compound can show different effects in the same assay when different mutants are tested. Details are shown in the graphic abstract and the limitations section.
Figure 1.

A brief summary of the consistence between outcomes from the DSF, PAb1620 IP, and luciferase reporter assays
.
Prepare purified recombinant p53 DNA-binding domain (DBD)
Timing: 3 days
-
1.Prepare purified recombinant p53 DBD from bacteria (Chen et al., 2021).
-
a.Clone Human p53 DBD (residues 94–293) with R175H, G245S, R249S, R282W, or V272M mutation into the pET28a vector (Novagen) between the NcoI and XhoI restriction sites.
-
b.Transform the constructs into Escherichia coli BL21-Gold (DE3) cells (Stratagene).
-
c.Inoculate a single colony into LB medium containing 50 μg/mL kanamycin and 0.1 mM zinc chloride, grown at 37°C, 180 rpm until the A600 reaches 0.3–0.4.
-
d.Switch the culture to 16°C, 180 rpm until the A600 reaches 0.6–0.8, followed by addition of isopropyl 1-thio-β-D-galactopyranoside (IPTG) with a final concentration of 0.5 mM. Protein was then expressed at 16°C, 180 rpm for 16–20 h.
-
e.Collect the bacteria pellet by centrifugation, resuspended in lysis buffer A (50 mM Tris, 50 mM NaCl, and 10 mM DTT, pH 7.0), followed by sonication on ice.
-
f.Supernatant was separated from cell debris by centrifugation at 10,000 × g for 30 min at 4°C. Load the supernatant onto an SP-FF column (GE Healthcare) that was equilibrated with lysis buffer A. Wash the column successively with 10 column volumes of lysis buffer A with additional 0, 50, 150, 250, or 450 mM NaCl by gradient dilution of lysis buffer A and B using ÄKTApurifier™ 10.
-
g.Conduct SDS–PAGE analysis to confirm the p53 DBD is eluted.
-
h.Concentrate and further polish the purified protein using a Superdex 75 column (GE Healthcare).
-
i.Concentrate the purified protein in storage buffer (20 mM Tris, 150 mM NaCl, and 10 mM DTT, pH 7.6), flash freeze protein in liquid nitrogen, and stored at −80°C.
-
a.
Note: it is challenging to purify full-length structural p53 mutants due to their thermodynamic instability, aggregation propensity, and high content of disordered N-terminal and C-terminal regions (Cho et al., 1994; Joerger and Fersht, 2007). Since the stability of full-length p53 is predominantly dictated by its DBD (Joerger and Fersht, 2007), the purified DBD was used instead of full-length p53 in the following DSF assay.
Maintenance of cell cultures
-
2.
Culture human lung cancer cell line H1299 (p53 null) (passage number < 15) in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1 × glutamine, 100 U/mL penicillin, and 100 mg/mL streptomycin. Culture the cells at 37°C in a humidified atmosphere comprising 5% CO2.
Note: Confirm cell line to be free of mycoplasma using MycoAlert™ PLUS Mycoplasma Detection Kit as this can affect the transfection efficiency. Other p53-null cell lines such as Saos-2 can also be used.
Key resources table
| REAGENT | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| p53, clone DO1, mouse monoclonal Ab (1:1000 dilution) | Abcam | Cat# ab1101, RRID: AB_297667 |
| p53 (wild type), clone PAb1620, mouse monoclonal Ab (1–3 μg per IP assay) | Merck Millipore | Cat# MABE339 |
| Anti-mouse kappa-light chain (HRP), clone H139-52.1, Rat monoclonal Ab (1:3000 dilution) | Abcam | Cat# ab99632, RRID: AB_10697414 |
| Bacterial and virus strains | ||
| Escherichia coli BL21-Gold (DE3) cells | Stratagene | Cat# 230132 |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM, high glucose, pyruvate | Thermo Fisher | Cat# 11995073 |
| Fetal Bovine Serum | Moregate | Cat# FBSF-500 |
| Opti-MEM® I Reduced Serum Medium, GlutaMAX™ | Thermo Fisher | Cat# 51985-034 |
| Lipofectamine 2000 | Thermo Fisher | Cat# 11668019 |
| Penicillin-Streptomycin | Thermo Fisher | Cat# 15140-122 |
| 100 × L-Glutamine | Sangon | Cat# E607004 |
| SYPRO Orange | Invitrogen | Cat# S6650 |
| NP40 | Sangon | Cat# A100109 |
| NaCl | Sangon | Cat# A610476-0001 |
| LB Broth (premixed powder) | Beyotime | Cat# ST156 |
| Arsenic trioxide (ATO) | Sigma | Cat# 202673 |
| Nutlin | Cayman Chemical | Cat# 10004372 |
| Kanamycin | Sangon | Cat# A100408-0025 |
| Zinc chloride | Sangon | Cat# A501003-0250 |
| Isopropyl 1-thio-β-D-galactopyranoside (IPTG) | Sangon | Cat# A600168-0005 |
| DTT | Sangon | Cat# A620058-0025 |
| HEPES | Sangon | Cat# A100511-0250 |
| Tris | Sangon | Cat# A600194-0005 |
| EDTA | Sangon | Cat# A100322-0500 |
| Critical commercial assays | ||
| FuGENE®6 Transfection Reagent Kit | Promega | Cat# E2691 |
| Dual Luciferase Reporter Assay Kit | Vazyme | Cat# DL101-01 |
| MycoAlert™ PLUS Mycoplasma Detection Kit | Lonza | Cat# LT07-705 |
| SP-FF column | GE Healthcare | Cat# 17505401 |
| Superdex 75 column | GE Healthcare | Cat# 17517401 |
| Deposited data | ||
| Raw image and data | This study | Mendeley Data https://doi.org/10.17632/y742f9j8v5.1 |
| Experimental models: cell lines | ||
| H1299 | ATCC | CRL-5803 |
| A549 | ATCC | CCL-185 |
| SK-MEL37 | MSKCC | CVCL_3878 |
| A431 | ATCC | CRL-1555 |
| Recombinant DNA | ||
| pGL3-PUMA-luc | (Lu et al., 2013) | N/A |
| Renilla | (Lu et al., 2013) | N/A |
| pcDNA3.1-human p53, various mutants | (Lu et al., 2013) | N/A |
| pET28a | Novagen | N/A |
| Software and algorithms | ||
| ImageJ | (Schneider et al., 2012) | https://imagej.nih.gov/ij/index.html |
| LightCycler® 480 Software | Roche | N/A |
| Other | ||
| ÄKTApurifier™ 10 | GE Healthcare | N/A |
| Protein G agarose | Thermo Fisher | Cat# 15920010 |
| ExpressPlus PAGE Gel, 10×8, 4–20% | GenScript | Cat# M42015C |
| White 96-well plates | SPL | Cat# 31396 |
| SpectraMax® L Microplate Reader | Molecular Devices | SpectraMax L |
| LightCycler® 480 MultiWell Plate (White) | Roche | Cat# 04729749001 |
| LightCycler® 480 Sealing Foil | Roche | Cat# 04729757001 |
| LightCycler® 480 RT-PCR machine | Roche | N/A |
| Ultrasonic Homogenizer | Scientz | JY92-IIN |
| Refrigerated microcentrifuge | Eppendorf | Cat# 5415 R |
| Small benchtop centrifuge | Eppendorf | Cat# 5810 R |
Materials and equipment
Alternatives: The DSF protocol describes the procedure using a LightCycler® 480 RT-PCR Detection system and corresponding reagents, in 10 μL reaction volumes. Alternative qPCR systems can be used as well. We recommend using a 384-well format, since this allows examination of multiple samples across timepoints in the same plate, thus minimizing potential batch effects.
HEPES buffer for DSF assay
| Reagent | Final concentration | Volume (mL) |
|---|---|---|
| 4 M NaCl | 150 mM | 18.75 |
| 2 M HEPES pH 7.5 | 20 mM | 5 |
| ddH2O | N/A | 476.25 |
| Total | N/A | 500 |
Keep the HEPES buffer at 2°C–8°C for up to 6 months.
NP40 buffer for PAb1620 IP
| Reagent | Final concentration | Volume (mL) |
|---|---|---|
| 1 M Tris-HCl pH 8.0 | 50 mM | 25 |
| 4 M NaCl | 150 mM | 18.75 |
| 0.5 M EDTA | 1 mM | 1 |
| NP40 | 1% (v/v) | 5 |
| ddH2O | N/A | 450.25 |
| Total | N/A | 500 |
Keep the NP40 buffer at 2°C–8°C for up to 6 months.
LB medium
| Reagent | Final concentration | Volume (mL) |
|---|---|---|
| Tryptone | 10 g/L | N/A |
| Yeast extract | 5 g/L | N/A |
| NaCl | 10 g/L | N/A |
| ddH2O | N/A | 1000 |
| Total | N/A | 1000 |
Sterilize in autoclave at 121°C for 15 min. Store at 2°C–8°C for 1–3 months.
Lysis buffer A
| Reagent | Final concentration | Volume (mL) |
|---|---|---|
| 1 M Tris-HCl pH 7.0 | 50 mM | 25 |
| 4 M NaCl | 50 mM | 6.25 |
| 1 M DTT | 10 mM | 5 |
| ddH2O | N/A | 463.75 |
| Total | N/A | 500 |
Keep the lysis buffer A at 2°C–8°C for up to 6 months.
Lysis buffer B
| Reagent | Final concentration | Volume (mL) |
|---|---|---|
| 1 M Tris-HCl pH 7.0 | 50 mM | 25 |
| 4 M NaCl | 500 mM | 62.5 |
| 1 M DTT | 10 mM | 5 |
| ddH2O | N/A | 407.5 |
| Total | N/A | 500 |
Keep the lysis buffer B at 2°C–8°C for up to 6 months.
Storage buffer
| Reagent | Final concentration | Volume (mL) |
|---|---|---|
| 1 M Tris-HCl pH 7.6 | 20 mM | 10 |
| 4 M NaCl | 150 mM | 18.75 |
| 1 M DTT | 10 mM | 5 |
| ddH2O | N/A | 466.25 |
| Total | N/A | 500 |
Keep the storage buffer at 2°C–8°C for up to 6 months.
Step-by-step method details
This protocol is divided into three sections, each of which will offer a unique readout for evaluating the thermostability, protein folding, and transcriptional activity of p53.
Differential scanning fluorimetry
DSF can be used to measure the thermostability of a protein based on the melting temperature (Tm) as readout (Chen et al., 2021; Niesen et al., 2007). This assay provided evidence that arsenic trioxide (ATO) stabilized five mutant p53 DBDs (Figure 2). In this assay we compared the stabilizing effect of ATO on five frequent DBD mutants, including four structural hotspot mutants and the V272M mutant.
-
1.
Thaw the purified p53 DBD protein solution and place it on ice.
-
2.
Dilute p53 DBD using 4°C storage buffer (20 mM Tris, 150 mM NaCl, and 10 mM DTT, pH 7.6) to a final concentration of 10 μM (about 0.2 mg/mL). Exchange the buffer with HEPES buffer (20 mM HEPES, 150 mM NaCl, pH 7.5) by dialysis for 16–20 h.
Note: Because DTT competes with cysteines of p53 for ATO binding, it should be removed from the DSF system when evaluating ATO. To avoid precipitation, the DBD was maintained at low concentration during dialysis.
-
3.
Dilute ATO with HEPES buffer to a final concentration of 1.25 mM.
Note: Because of the thermodynamic instability of the p53 DBD, the HEPES buffer used throughout the protocol must be pre-cooled to 4°C.
Note: A stock solution of 1 mg/mL ATO was prepared by diluting the ATO powder in 1 mol/L NaOH and then adjusting pH to 7.2 with HCl.
-
4.
Add the purified p53 DBD, ATO, and HEPES buffer into a pre-cooled PCR tube as listed in the table below. The final molar ratio of p53 DBD to ATO is 1:20.
| Reagent | Final concentration | Volume |
|---|---|---|
| p53 DBD | 6.25 μM | Dependent on stock concentration of DBD |
| 1.25 mM ATO | 125 μM | 3.2 μL |
| HEPES buffer | N/A | To 32 μL |
-
5.
Mix gently and spin down the solution using refrigerated microcentrifuge (100 × g, 4°C, 5 s).
-
6.
Incubate the solution at 4°C for 16–20 h.
CRITICAL: For some compounds such as PK11007, incubation for shorter periods (e.g., 15 min) is sufficient to achieve the maximal change of Tm (indicated by the ΔTm value). However, for some covalently reacting compounds such as ATO, incubation for > 2–8 hrs is required to result in significant changes of ΔTm. Thus, this step is suggested to be performed for 16–20 h for ATO, see problem 2.
-
7.
Prepare a 25× working solution of SYPRO Orange by diluting the 5000 × stock solution 1:200 with HEPES buffer.
-
8.
Add 8 μL of 25× SYPRO Orange to the 32 μL solution and mix gently.
-
9.
Pipette 30 μL of the resulting solution into three wells of a pre-cooled PCR plate, i.e., 10 μL for each replicate, and seal the plate with optical sealing foil.
-
10.
Spin the solution to the bottom of the plate using small benchtop centrifuge (64 × g, 4°C, 5 s).
-
11.
Set the detection format to the SYPRO® Orange format (FAM (492 nm) and ROX (610 nm), for excitation and emission, respectively).
-
12.
Place the plate into an RT-PCR instrument and run a temperature gradient from 20°C to 85°C at 2°C per min, reading the SYPRO® Orange fluorescence intensity every 0.2°C.
-
13.Data analysis.
-
a.Click the ‘Export’ button and export the .ixo file from LightCycler® 480 software.
-
b.In Roche Protein Melting software, choose the ‘folder’ and open the exported .ixo file.
-
c.Choose ‘Analyse’ tab and click ‘Add Analysis’ button. The calculated Tm values that analyzed by Boltzmann method can be obtained in the .csv file. A representative melting temperature curve of p53 DBD (R175H and G245S) with or without ATO treatment was shown in Figure 3.
-
d.Export the calculated Tm in .csv file.
-
a.
Figure 2.

Calculated Tm values of the indicated p53 DBDs
Protein was incubated with or without ATO treatment for 16–20 h at the indicated ratios in 20 mM HEPES buffer (pH 7.5) containing 150 mM NaCl. Bars represent mean ± SD (n = 3) and p-values are shown.
Figure 3.

The representative melting curves for p53 DBD without or with ATO
Recombinant p53 DBD was mixed without or with ATO at a molar ratio of 1:20 in 20 mM HEPES (pH 7.5) containing 150 mM NaCl for 16–20 h. Melting curves were then measured in the DSF assay and the calculated Tm is shown.
PAb1620 immunoprecipitation
Folded p53 can be recognized by the PAb1620 antibody (Wang et al., 2001). This IP protocol assesses the immunoreactivity of p53 mutants with the PAb1620 antibody, indicating level of folded p53 (Figure 4).
-
14.
One day before starting the protocol, seed H1299 cells into 6-well plates with 1.0 x 106 cells and 3 mL of complete DMEM medium per well, incubate the cells for 16–20 h . The cells should reach around 50% confluence the next day.
-
15.
On day 1, transfect the cells with the p53 expression plasmid (2.5 μg plasmid per well) according to the manufacturer’s protocol of Lipofectamine 2000 reagent.
Alternatives: Other transfection reagents can also be used. No transfection is required if working with cell lines that carry endogenous p53 mutations.
-
16.
On day 2, dilute ATO stock resolution with growth medium to a concentration of 100 μg/mL, and add diluted ATO to each well to a final concentration of 1 μg/mL. After gentle shaking, incubate the cells for 16–20 h.
Note: incubation for 2 h is also acceptable for ATO.
-
17.
On day 3, wash the cells with cold PBS, lyse them with 1 mL of 4°C NP40 buffer, and collect the lysate into a 1.5 mL centrifuge tube.
Pause point: at this point the sample can be directly used or stored at −80°C (stable for 1–3 weeks).
-
18.
Sonicate the cell lysate on ice for 3 cycles with 2 s ON and 2 s OFF at 200 watts.
-
19.
Shake the cell lysate on ice for 10 min.
-
20.
Spin the lysate in a pre-cooled refrigerated microcentrifuge (16,100 × g, 20 min, 4°C) and collect the supernatant in a 1.5 mL centrifuge tube (20 μL supernatant should be taken out as an input sample).
-
21.
To immunoprecipitate p53, add 20 μL protein G beads and 1–3 μg of PAb1620 antibody to 450 μL of supernatant, and rotate the mixture for 2 h at 4°C.
-
22.
Centrifuge the mixture at 16,100 × g for 5 s using refrigerated microcentrifuge, and carefully remove the supernatant with a pipette.
-
23.
Wash the beads with 1 mL of 20°C–25°C NP40 buffer, centrifuge at 16,100 × g for 5 s using refrigerated microcentrifuge, and carefully remove the supernatant with a pipette. Repeat this step once.
-
24.
Add 20 μL of 2× SDS loading buffer to the pellet.
-
25.
Heat the sample at 95°C–100°C for 5 min, followed by immunoblotting.
Note: To probe the immunoprecipitated p53 on the PVDF membrane, incubate the membrane with anti-mouse kappa light chain (HRP) secondary antibody which does not recognize the heavy chain of PAb1620 on the membrane.
Figure 4.

PAb1620 IP assay of p53 mutants
p53 mutants were expressed in transfected H1299 cells. The input and immunoprecipitated p53 was probed. Relative band intensity was measured by ImageJ and normalized for each immunoblotting. Bars represent mean ± SD derived from three PAb1620 immunoprecipitation experiments. Representative images from one immunoprecipitation experiment are shown.
Luciferase reporter assay
This luciferase reporter assay assesses the transcriptional activity of p53 on a defined response element in H1299 cells. The readout is relative luciferase units (RLU), indicating the transcriptional activity of p53.
-
26.
One day before starting the protocol, seed H1299 cells into 96-well plates with 3.0 x 104 cells and 100 μL of medium per well, followed by incubation for 16–20 h. The cells should reach around 30% confluence the next day.
Note: Before starting, make sure the H1299 cells used for seeding are in the rapid multiplication stage, typically corresponding to 80–90% confluence.
-
27.On day 1, co-transfect the cells with the p53-expression plasmid, luciferase reporter plasmid and Renilla plasmid using FuGENE®6 transfection reagent (Promega, E1960). 3 replicates should be set for each sample. The instructions below list the reagents for 10 wells in a 96-well plate.
-
a.Prepare the followed mixture of plasmids (total volume can be about 1–20 μL dependent on the plasmid concentration) in a 1.5 mL tube:
Reagent Amount for 10 wells in 96-well plate p53 expression plasmid 750 ng Luciferase reporter plasmid 250 ng Renilla plasmid 12 ng -
b.Mix 3 μL FuGENE®6 transfection reagent and 50 μL Opti-MEM in a 300 μL tube by gently pipetting and incubate for 5 min.
-
c.Add the 53 μL solution to the plasmid mixture, and mix by gently pipetting. Incubate the resulting transfection mixture for 15 min at 20°C–25°C.
-
d.Add 1 mL of 20°C–25°C DMEM medium to the transfection mixture and pipette the solution thoroughly. The resulting solution is then used to replace all the culture medium in the wells of the 96-well plate with pre-seeded cells (100 μL per well).CRITICAL: Different from some protocols in which the small-volume transfection mixture is added into 96-wells plate directly, in this protocol, large-volume DMEM is firstly added to the transfection mixture, followed by pipetting to achieve thorough mixing. The thoroughly mixed solution is then used to replace the DMEM in the 96-well plate with the pre-cultured cells, see problem 5.
-
a.
-
28.
On day 2, dilute ATO stock resolution with growth medium to a concentration of 3 μg/mL, and add 50 μL diluted ATO to each well to a final concentration of 1 μg/mL.
-
29.On day 3, detect luciferase signal using a Dual Luciferase Reporter Assay Kit (Vazyme, DL101-01). In current protocol, 1 μg/mL ATO treatment killed about 10% H1299 cells (Chen et al., 2021).
-
a.Remove growth medium from cultured cells.
-
b.Rinse cells using 1 x PBS twice.
-
c.Add 20 μL 1 x lysis buffer to each well of the 96-well plate.
-
d.Shake the plate for 15 min at 20°C–25°C.Pause point: at this point the sample can be directly used or stored at −80°C (stable for 1 week).
-
e.Transfer 4 μL lysates to a white 96-well plate, and place the plate into a luminometer (SpectraMax® L Microplate Reader) with injectors.
-
f.Add 20 μL Reaction Buffer II into each well and measure the firefly luciferase signal.
-
g.Add 20 μL Stop & Reaction Buffer to each well and measure the Renilla luciferase signal.Alternatives: Other dual luciferase reporter assay kits, such as the Dual-Luciferase® Reporter Assay System (Promega, E1910), can also be used.
-
a.
Expected outcomes
In the DSF assay, the relatively thermostable G245S and R249S DBDs are expected to have higher Tm values than the other three DBDs before ATO treatment (Figure 2) (Chen et al., 2021; Joerger and Fersht, 2007). ATO treatment of the highly thermolabile R175H DBD produces a ΔTm value > 5.0°C, while the values are smaller for the relatively thermostable DBDs (such as G245S and R249S DBDs) (Figure 2).
In the PAb1620 IP assay, wild-type p53 and the DNA-contact p53-R273H/C can be efficiently immunoprecipitated by PAb1620, while the highly thermolabile p53-R175H and p53-R282W can only be weakly immunoprecipitated (Figure 4). It is notable the DNA-contact p53-R248Q/W is also somewhat thermolabile and thus cannot be immunoprecipitated by PAb1620 as efficiently as wild-type p53 (Figure 4) (Chen et al., 2021; Joerger and Fersht, 2007). In this assay, the PAb1620 IP efficiency of p53-R175H can be increased by ATO over 50 times (Figures 5 and 6).
Figure 5.

PAb1620 IP assay of p53 mutants upon ATO treatment
p53 mutants were expressed in transfected H1299 cells upon treatment with 1 μg/mL ATO. The input and immunoprecipitated p53 was probed. Relative band intensity was measured by ImageJ and normalized for each immunoblotting. Bars represent mean ± SD derived from three PAb1620 immunoprecipitation experiments. Representative images from one immunoprecipitation experiment are shown. Figure reprinted with permission from (Chen et al., 2021)
Figure 6.

PAb1620 IP assay of p53-R175H upon ATO treatment by using NP40 buffer at different temperature
p53-R175H were expressed in transfected H1299 cells upon treatment with 1 μg/mL ATO. The input and immunoprecipitated p53 was probed. The beads were washed with either 20°C–25°C or 4°C NP40 buffer (step 21). Relative band intensity was measured by ImageJ and normalized for each immunoblotting. Bars represent mean ± SD derived from three PAb1620 immunoprecipitation experiments. Representative images from one immunoprecipitation experiment are shown. Figure reprinted with permission from (Chen et al., 2021)
In the luciferase reporter assay, the transcriptional activities of p53-R282W and p53-V272M on the PUMA promoter are expected to be increased over 10 and 30 times, respectively (Figure 7). However, the transcriptional activities of p53-R249S, p53-G245S and p53-R175H are less efficiently promoted by ATO in this assay. The rescue efficiencies differ when different plasmid ratios are used (Figure 8).
Figure 7.

Luciferase reporter assay of the indicated p53 mutants on the target BBC3 (PUMA)
p53 mutants were expressed in H1299 cells upon treatment with 1 μg/mL ATO for 24 h. The fold-change of RLU upon ATO treatment is indicated. An optimized plasmid ratio was used. Bars represent mean ± SD (n = 3) and p-values are shown.
Figure 8.

Luciferase reporter assay of p53-R282W on the target BBC3 (PUMA)
p53-R282W was expressed in H1299 cells upon treatment with 1 μg/mL ATO for 24 h. The amounts of plasmids used for one well in the 96-well plate are shown. Bars represent mean ± SD (n = 3). p-values and fold changes of RLU are shown.
Quantification and statistical analysis
The luminescence signal of firefly luciferase is normalized to that of Renilla luciferase for each sample. Statistics analysis (unpaired two-tailed Student’s t test, with a 95% confidence interval under the untested assumption of normality) can be performed in Prism 6 (GraphPad). Data should be presented as means ± SD. Group size is indicated in the main text. Experiments are not performed in a blinded manner. The quantification of band intensity in immunoblotting was performed using ImageJ according to the guideline (https://imagej.nih.gov/ij/index.html) (Schneider et al., 2012). Briefly, membranes were inverted and the band intensity was measured by selecting the band with rectangle tool. Blank area of membranes was selected as base signal to eliminate the background of the selected rectangle area. The calculated signal intensity was then normalized according to the first band of each membrane.
Limitations
Because of the diverse inactivation mechanisms of p53 mutants, different outcomes may be generated by the same rescue compound in the same assay when different mutants are being tested (see graphical abstract). Listed below are some examples and our explanations.
When evaluating the effects of ATO in the DSF assay, we found that there is a trend in which the relatively thermostable mutants (such as G245S) benefit less from ATO-induced thermal stabilization. Thus, we suggest using the relatively thermolabile mutants, such as p53-R175H and p53-V272M, to increase the sensitivity of the DSF assay (Figure 2).
For the PAb1620 IP assay, we found that the PAb1620 epitope of some mutations, such as those occurring at the LSH and β-sandwich of p53, cannot efficiently restored (Figure 5, R282W and V272M mutants as examples) despite being highly rescued in terms of thermostability and transcriptional activity (Figures 2 and 7). We speculate that this is because of the DBD topology (i.e., strand 10 connecting a loop in the vicinity of the PAb1620 epitope to the C-terminal helix). In addition, mutations occurring near the PAb1620 epitope are not suggested for the PAb1620 IP assay, since these mutations may directly destroy the PAb1620 epitope regardless of the global folding status. Thus, we suggest not to use such mutants to test the rescue potency of a compound in the PAb1620 IP assay.
In the luciferase reporter assay, we found that the transcriptional activity of some mutants, such as R249S, G245S, and R175H, cannot be completely restored (Figure 7) despite successfully increased thermostability and a restored PAb1620 epitope. Based on the crystal structures solved to date, we speculate that these mutants do not regain a completely intact DNA-binding surface upon global refolding. Thus, we suggest using more appropriate mutants, such as p53-V272M, which is thought to retain an intact DNA-binding surface upon global refolding, to increase the sensitivity of the luciferase reporter assay.
Troubleshooting
Problem 1
High starting point of melting curve in DSF assay (steps 1–10)
Potential solution
As p53 is a protein with poor thermodynamic stability, melting curves are easily affected by improper temperature control. Thus, all the steps, before loading the PCR plate into the RT-PCR instrument, should be performed on ice or at 4°C. It is also recommended to avoid directly touching the edges of the PCR plate with fingers, since the fingers may warm the samples at the edge of the plate.
Problem 2
Compound has a weaker effect than expected in the DSF assay (step 6)
Potential solution
Increase the incubation time, since some compounds, particularly those that lead to covalent modifications, react with p53 slowly.
Problem 3
Low efficiency of p53 IP by PAb1620 (steps 14, 15, and 21)
Potential solution
It is expected that structural p53 mutants cannot be efficiently immunoprecipitated by PAb1620 without rescue compound treatment. We thus suggest using wild-type p53 or non-structural mutant p53 as positive control. When wild-type p53 is used, it is suggested to boost its expression level using MDM2 inhibitor since endogenous wild-type p53 is normally low. When DNA-contacting mutant p53 is used, it is suggested to use R273H/C mutant rather than R248Q/W mutant since the latter is structurally instable and thus loss some PAb1620 reactivities. Nevertheless, PAb1620 IP assay is more sensitive than PAb1620 immunostaining assay in our hands. In our PAb1620 immunostaining assay, PAb1620 failed to distinguish between wild-type p53 (or p53-R273H) and p53-R175H (Figure 9).
Figure 9.

Immunostaining results of three cancer cell lines using p53 antibodies DO1 or PAb1620
The indicated cancer lines A549 (A), SK-MEL37 (B), and A431 (C) were pretreated with DMSO or 10 μM Nutlin for 24 h, followed by immunofluorescence staining using p53 antibodies DO1 or PAb1620, respectively. Scale bar = 50 μm .The right graph (mean ± SD) shows percentage of staining-positive cells. Three scopes (> 50 cells in each scope) for each sample were randomly picked for cell counting. The detail procedure of the experiment can be found in (Chen et al., 2021).
Problem 4
Compound leads to weaker reconstitution of the PAb1620 epitope in the IP assay than expected (step 23)
Potential solution
It is critical to use 20°C–25°C NP40 buffer to wash the beads. For example, p53-R175H has a Tm value much higher than 4°C. Thus, before ATO treatment p53-R175H is detectably folded and thus retains PAb1620 reactivity at 4°C, which will greatly decrease the ΔTm for p53-R175H before and after ATO treatment (Figure 6).
Problem 5
Compound induces a weaker increase of transcriptional activity in the luciferase reporter assay than expected (step 27)
Potential solution
Optimize the ratio of the p53-expression plasmid, luciferase reporter (PUMA) plasmid, and Renilla plasmid. In our laboratory, a ratio of 75:25:1.2 yields the highest fold-change of transcriptional activity upon ATO treatment (Figure 8). However, the ratio yielding the highest fold-change may vary in different laboratories dependent on the reagents used.
Resource availability
Lead contact
Further information and requests for resources, plasmids and reagents should be directed to and will be fulfilled by the lead contact, Min Lu (min.lu@shsmu.edu.cn).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate any unique datasets or code. The authors declare that the data supporting
the findings of this study are available within the paper. The raw image and data of this study have been deposited in the Mendeley Data and available at https://doi.org/10.17632/y742f9j8v5.1.
Acknowledgments
M.L. was funded by the National Key R&D Program of China (2017YFA0506200), the NSFC (82073292, 81622002, 81861130368), the SJTU Trans-med Awards Research, the Gaofeng Clinical Medicine Grant (828318), the Shanghai Excellent Youth Academic Leader Program (20XD1422700), the Shanghai Medical and Health Excellent Discipline Leader Development Plan (2018BR36), the Shanghai Collaborative Innovation Center for Translational Medicine (TM201902), the Foundation of the National Facility for Translational Medicine (Shanghai) (TMSK-2020-003), the Newton Advanced Fellowship, and the Samuel Waxman Cancer Research Foundation. We thank the staff at the BL17U/BL19U1 beamlines of the National Center for Protein Science Shanghai (NCPSS) at Shanghai Synchrotron Radiation Facility (SSRF) and high-throughput screening core of the National Research Center for Translational Medicine (Shanghai). J.W. was sponsored by Shanghai Sailing Program (21YF1427200).
Author contributions
M.L. developed and optimized the protocol and wrote the manuscript. J.W., H.S., and Z.W. performed PAb1620 IP assay, luciferase reporter assay, and DSF assay, respectively.
Declaration of interests
M.L., J.W., and H.S. are co-authors of the pending patents “PANDA as a novel therapeutic” (PCT/CN2018/085190) and “mp53 rescue compounds and methods of treating a p53 disorder” (PCT/CN2019/070117). The authors declare no other competing interests.
References
- Chen S., Wu J.L., Liang Y., Tang Y.G., Song H.X., Wu L.L., Xing Y.F., Yan N., Li Y.T., Wang Z.Y. Arsenic trioxide rescues structural p53 mutations through a cryptic allosteric site. Cancer Cell. 2021;39:225–239.e8. doi: 10.1016/j.ccell.2020.11.013. [DOI] [PubMed] [Google Scholar]
- Cho Y., Gorina S., Jeffrey P.D., Pavletich N.P. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- Doffe F., Carbonnier V., Tissier M., Leroy B., Martins I., Mattsson J.S.M., Micke P., Pavlova S., Pospisilova S., Smardova J. Identification and functional characterization of new missense SNPs in the coding region of the TP53 gene. Cell Death Differ. 2020;28:1477–1492. doi: 10.1038/s41418-020-00672-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joerger A.C., Fersht A.R. Structure-function-rescue: the diverse nature of common p53 cancer mutants. Oncogene. 2007;26:2226–2242. doi: 10.1038/sj.onc.1210291. [DOI] [PubMed] [Google Scholar]
- Lu M., Breyssens H., Salter V., Zhong S., Hu Y., Baer C., Ratnayaka I., Sullivan A., Brown N.R., Endicott J. Restoring p53 function in human melanoma cells by inhibiting MDM2 and cyclin B1/CDK1-phosphorylated nuclear iASPP. Cancer cell. 2013;23:618–633. doi: 10.1016/j.ccr.2013.03.013. [DOI] [PubMed] [Google Scholar]
- Niesen F.H., Berglund H., Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protocol. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.L., Sait F., Winter G. The 'wildtype' conformation of p53: epitope mapping using hybrid proteins. Oncogene. 2001;20:2318–2324. doi: 10.1038/sj.onc.1204316. [DOI] [PubMed] [Google Scholar]
- Xirodimas D.P., Lane D.P. Molecular evolution of the thermosensitive PAb1620 epitope of human p53 by DNA shuffling. J. Biol. Chem. 1999;274:28042–28049. doi: 10.1074/jbc.274.39.28042. [DOI] [PubMed] [Google Scholar]
- Zhang Q., Bykov V.J.N., Wiman K.G., Zawacka-Pankau J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018;9:439. doi: 10.1038/s41419-018-0463-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate any unique datasets or code. The authors declare that the data supporting
the findings of this study are available within the paper. The raw image and data of this study have been deposited in the Mendeley Data and available at https://doi.org/10.17632/y742f9j8v5.1.