
Figure 5. Working model of hepatic lipid accumulation-induced changes in hepatic metabolism and resulting changes in hepatic vagal nerve signaling to affect insulin secretion and sensitivity.
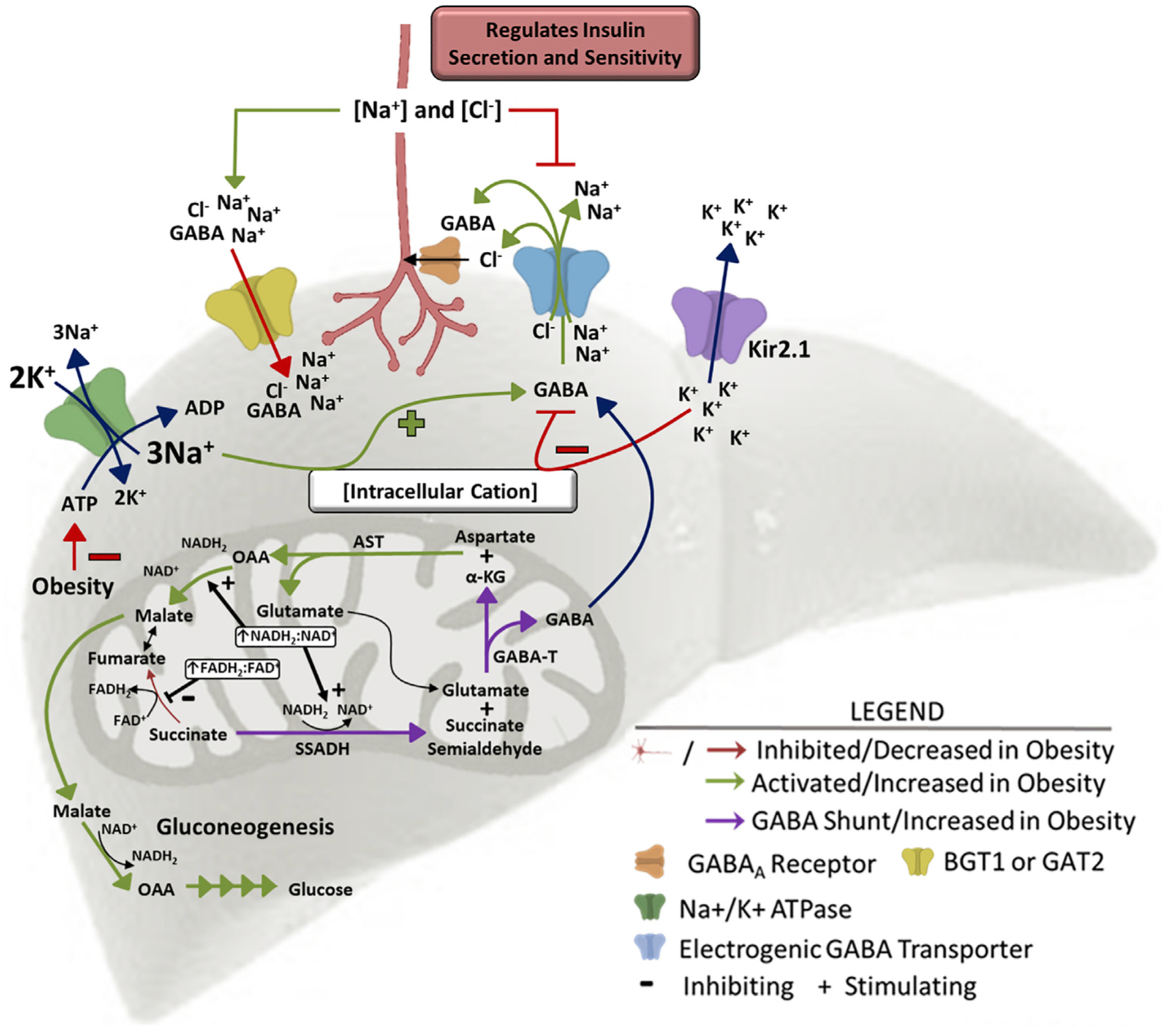
High levels of β-oxidation in the obese liver increase the mitochondrial NADH2:NAD+ and FADH2:FAD+ ratios driving succinate to succinate semialdehyde, generating substrate for GABA-transaminase. GABA-transaminase produces GABA and α-ketoglutarate, a substrate for aspartate aminotransferase. Increased gluconeogenic flux in obesity drives the mitochondrial export of OAA as malate. The increased GABA release is encouraged by the depolarized membrane in obesity. GABA is co-transported with 3 Na+ and 1 Cl− ion, so an increase in intracellular cation concentration (hepatocyte depolarization) encourages GABA export, while a decrease in intracellular cation concentration (hepatocyte hyperpolarization) limits GABA export. Kir2.1 expression induces hepatic K+ efflux and hyperpolarization, inhibiting GABA export. Obesity decreases hepatic ATP concentrations, impairing activity of the Na+/K+-ATPase pump and increasing intracellular Na+ concentrations, driving GABA export. This mechanism explains how hepatic lipid accumulation increases hepatic GABA release. AST, aspartate aminotransferase; GABA-T, GABA-transaminase; α-KG, α-ketoglutarate; OAA, oxaloacetate; SSADH, succinate semialdehyde dehydrogenase.