
Figure 1. Machine learning in antibiotic discovery.
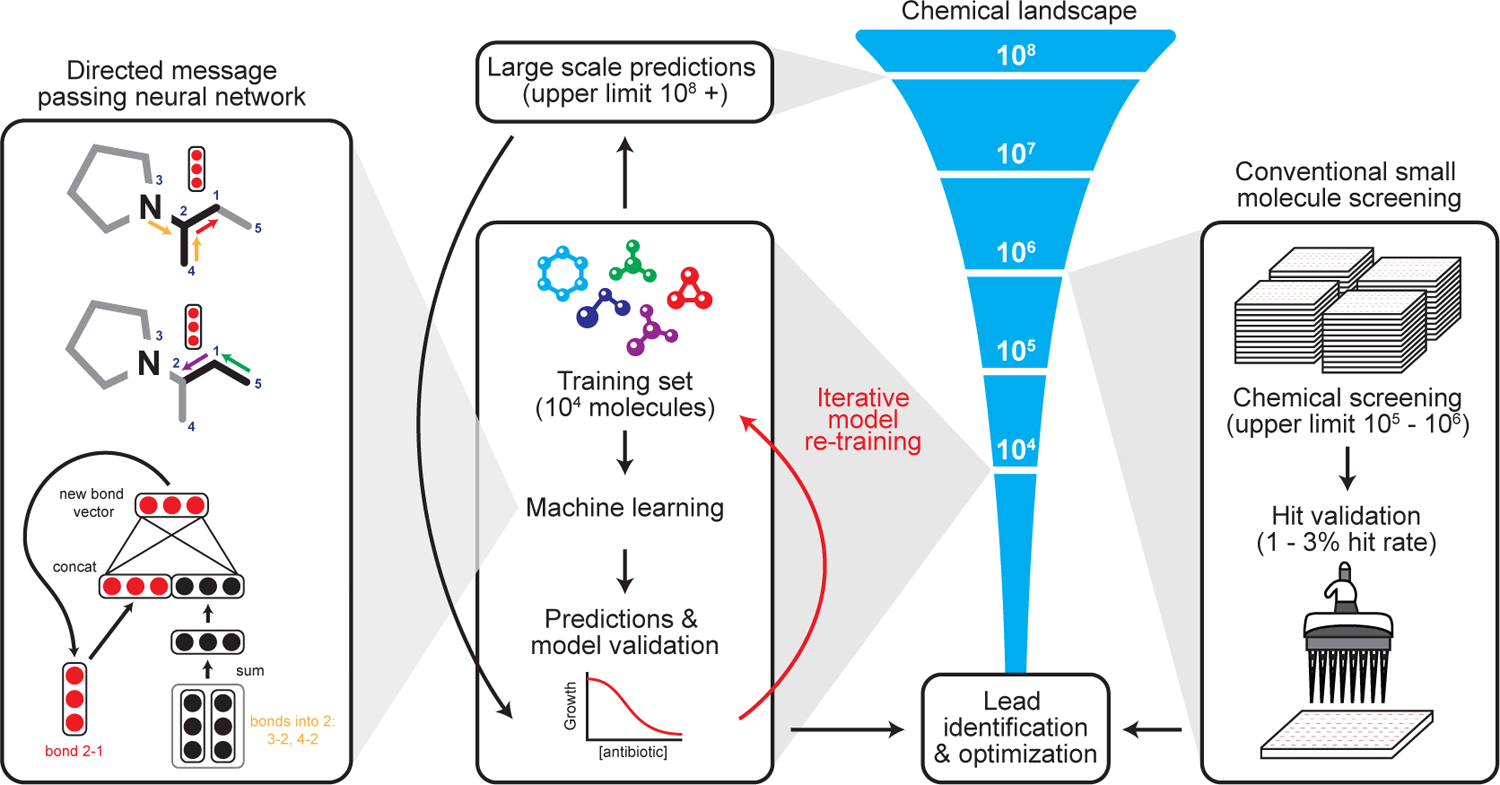
Modern approaches to antibiotic discovery often include screening large chemical libraries for those that elicit a phenotype of interest. These screens, which are upper bound by hundreds of thousands to a few million molecules, are expensive, time consuming, and can fail to capture an expansive breadth of chemical space. In contrast, machine learning approaches afford the opportunity to rapidly and inexpensively explore vast chemical spaces in silico. Our deep neural network model works by building a molecular graph based on a specific property, in our case the inhibition of the growth of E. coli, using a directed message passing approach. We first trained our neural network model using a collection of 2,335 diverse molecules for those that inhibited the growth of E. coli, augmenting the model with a set of molecular features, hyperparameter optimization, and ensembling. Next, we applied the model to multiple chemical libraries, comprising >107 million molecules, to identify potential lead compounds with activity against E. coli. After ranking the candidates according to the model’s predicted score, we selected a list of promising candidates.