

Abstract
The neuronal dynamin1 functions in the release of synaptic vesicles by orchestrating the process of GTPase-dependent membrane fission. Dynamin1 associates with the plasma membrane–localized phosphatidylinositol-4,5-bisphosphate (PIP2) through the centrally located pleckstrin homology domain (PHD). The PHD is dispensable as fission (in model membranes) can be managed, even when the PHD-PIP2 interaction is replaced by a generic polyhistidine- or polylysine-lipid interaction. However, the absence of the PHD renders a dramatic dampening of the rate of fission. These observations suggest that the PHD-PIP2–containing membrane interaction could have evolved to expedite fission to fulfill the requirement of rapid kinetics of synaptic vesicle recycling. Here, we use a suite of multiscale modeling approaches to explore PHD–membrane interactions. Our results reveal that 1) the binding of PHD to PIP2-containing membranes modulates the lipids toward fission-favoring conformations and softens the membrane, and 2) PHD associates with membrane in multiple orientations using variable loops as pivots. We identify a new loop (VL4), which acts as an auxiliary pivot and modulates the orientation flexibility of PHD on the membrane—a mechanism that we believe may be important for high-fidelity dynamin collar assembly. Together, these insights provide a molecular-level understanding of the catalytic role of PHD in dynamin-mediated membrane fission.
INTRODUCTION
Dynamin is a multidomain GTPase that self-assembles into a helical collar and catalyzes membrane fission, leading to the release of nascent clathrin-coated vesicles during endocytosis (Koenig et al., 1983; Van der Bliek et al., 1993; Faelber et al., 2011; Morlot and Roux, 2013; Dar and Pucadyil, 2017). The GTPase domain (G-domain), bundle-signaling element (BSE), and stalk domain are the three conserved domains in dynamin that are required to evoke stimulated GTPase activity upon self-assembly on the membrane (Chappie and Dyda, 2013). In addition to these domains, classical dynamins contain a proline-rich domain (PRD) that interacts with SH3 domain–containing partner proteins and a pleckstrin-homology domain (PHD) that binds to plasma membrane–localized lipid phosphatidylinositol-4,5-bisphosphate (PIP2). GTP hydrolysis is proposed to orchestrate a series of conformational changes in the self-assembly to surmount the activation barrier to membrane fission (Chernomordik and Kozlov, 2003; Schneck et al., 2012).
Structurally, the ubiquitous dynamin1 PHD has a C-terminal α-helix and a core β-sandwich with variable loops between the β-strands that form the binding pocket for PIP2. The crystal structure of dynamin1 PHD (PDB: 1DYN) suggests a β-sandwich structure with two β-sheets, one with four and the other with three β-strands, oriented in an antiparallel arrangement. The structure defines four loops referred to as variable loops (VLs) as shown in Figure 1. These loops significantly differ from each other in hydrophobicity and electrostatics. VL1 (531IGIMKGG537) contains a hydrophobic stretch that inserts in the membrane and assists dynamin’s subcellular localization (Ramachandran and Schmid, 2008). VL3 (590NTEQRNVYKDY600) is highly polar and has also been shown to be important for membrane association (Liu et al., 2011; Francy et al., 2015). While the PHD helps classical dynamins to engage with the membrane (Okamoto et al., 1997; Ferguson and De Camilli, 2012), numerous lines of evidence suggest that it functions more than a generic membrane anchor (Klein et al., 1998; Achiriloaie et al., 1999; Vallis et al., 1999; Ramachandran et al., 2009; Mehrotra et al., 2014; Reubold et al., 2015; Terzi and Deserno, 2016; Dar and Pucadyil, 2017). Genetic neurological disorders such as centronuclear myopathies and Charcot–Marie–Tooth disease are linked to mutations in the PHD that map to regions distinct from those involved in membrane binding (Durieux et al., 2010; Kenniston and Lemmon, 2010; Haberlová et al., 2011). Cellular assays combined with biochemical and microscopic analysis of membrane fission with point mutants in the PHD of dynamin1 suggest that this domain might induce local membrane curvature by shallow insertion of one of its loops (Ramachandran et al., 2009) and that such point mutations alter the dynamics and orientation of the PHD on the membrane (Mehrotra et al., 2014). Experimentally guided modeling has shown that tilting of the PHD could be responsible for creating a low-energy pathway toward reaching the hemi-fission state (Shnyrova et al., 2013). GTP hydrolysis is required to initiate fission through constriction (Antonny et al., 2016), but whether this is adequate to achieve complete and leakage-free vesicle scission is still under debate (Shnyrova et al., 2013; Mattila et al., 2015). Recent models indicate that the GTPase-driven scaffold constriction brings the membrane in close proximity but not close enough to cross the energy barrier for fission and that stochastic fluctuations are responsible for the cross-over from the constricted stage to the hemi-fission state (Terzi and Deserno, 2016; McDargh and Deserno, 2018). Indeed, analysis of membrane fission at the single-event resolution indicates that replacement of the PHD-PIP2 interaction with a generic lipid–protein interaction forms long-lived, highly constricted prefission intermediates on the membrane that results in dramatic dampening of fission kinetics (Dar and Pucadyil, 2017). Recent cryo electron microscopy (cryo-EM) data of the dynamin polymer assembled on tubular membranes reveal complex and variable geometries of the PHD (Shnyrova et al., 2013; Sundborger et al., 2014; Mattila et al., 2015; Kong et al., 2018). The variable PHD configurations may reflect an evolutionary requirement for generating membrane torque necessary for traversing a pathway whereby membrane constriction guarantees leakage-free fission as shown in some of the recent models (Pannuzzo et al., 2018).
FIGURE 1:
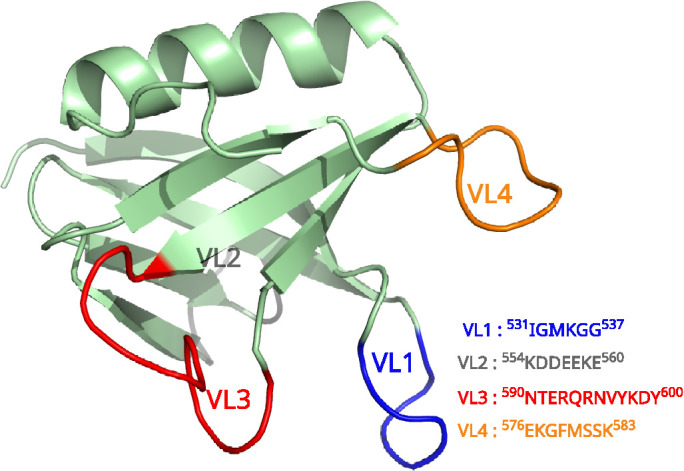
PHD and its variable loops.
Guided by these observations, and to circumvent the difficulty of observing short-lived transition states of PHD-PIP2 interaction in an experimental scenario, we apply multiscale molecular simulations to study the membrane association behavior of dyn-PHD in great molecular detail. Undulation spectra extracted from Martini coarse-grained (CG) trajectories of large membrane systems (with and without PHD) were used to observe the effect on membrane physical properties. These methods also test the hypothesis of local curvature generation by the PHD. We then explore the conformational changes induced by PHD in lipids of the membrane using all-atom molecular dynamics (AAMD) simulations. Mixed-resolution highly mobile membrane-mimetic model (HMMM)-based MD simulations initiated from different orientations were used to explore possible membrane association pathways. Membrane association geometries were deciphered using data from both AAMD simulations and by free energy difference calculations from enhanced sampling methods such as metadynamics and umbrella sampling simulations. PHD is a ubiquitous membrane adaptor, and PHD–membrane interactions have been studied thoroughly for several proteins in great detail both in experiments and using molecular simulations (Ferguson et al., 1995; Klein et al., 1998; Lemmon and Ferguson, 2000; Lai et al., 2013; Srivastava and Voth, 2014; Jian et al., 2015; Vonkova et al., 2015; Yamamoto et al., 2016, 2020; Naughton et al., 2016, 2018; Chan et al., 2017; Pant and Tajkhorshid, 2020; Soubias et al., 2020). In our work, we also look at the membrane association mechanism and intricate binding geometries and interactions of dyn-PHD in light of other available studies and highlight the similarities and differences while connecting the PHD–membrane interactions to dynamin-assisted fission process (Achiriloaie et al., 1999; Lee et al., 1999; Ramachandran et al., 2009; Shnyrova et al., 2013; Mehrotra et al., 2014; Dar et al., 2015; Dar and Pucadyil, 2017). Together, our results indicate that the presence of the PHD causes significant changes in lipid conformations and the membrane-bending rigidity, thus providing mechanistic insights into its catalytic effect on membrane fission due to dynamin. These effects are managed by flexible pivoting of the previously described variable loop1 (VL1) and the yet-unexplored variable loop (VL4) in the PHD with the membrane.
RESULTS AND DISCUSSION
Significance
Dynamin, a large multidomain GTPase, remodels the membrane by self-assembling onto the neck of a budding vesicle and induces fission by its energy-driven conformational changes. In this work, we use multiscale molecular simulations to probe the role of dynamin’s PHD, which facilitates membrane interactions. Notably, PHD is dispensable for fission, as is the case with extant bacterial and mitochondrial dynamins. However, reconstitution experiments suggest that the functional role of PHD in neuronal membrane goes beyond that of an adaptor domain as it possibly “expedites” the fission reaction during synaptic vesicle recycling. We provide a MD picture of how PHDs make membranes more pliable for fission and suggest new insights into the molecular-level processes driving the expedited fission behavior.
Binding of the PHD renders the membrane pliable for fission
To determine the effect of the PHD on the mechanical properties of the membrane, we performed 4-μs-long CG simulations of a large lipid bilayer patch (∼625 nm2) using the Martini force field (Marrink et al., 2004). This bilayer is composed of 2048 lipids with DOPC:DOPS:PIP2 in a 80:19:1 molar ratio. Fourteen CG PHD units were placed randomly onto one of the leaflets of the bilayer. The bending moduli calculated by fitting undulation spectra to regular Helfrich theory (see Figure 2) show that the presence of the PHDs reduces the bending modulus (BM) of the lipid bilayer. We notice that the BM (Kc) of the membrane with PHDs is ∼4.5 kBT lower than that without PHDs. To gain insights into the molecular structures and changes related to the two-dimensional density structure factor of the membrane, we plotted the full height undulation spectra (see Supplemental Figure S1) with larger q-regimes (Brandt et al., 2011). We observed some differences between the two spectra in the intermediate q regime but observed no noticeable difference in in-plane molecular structure fluctuations in the larger q regime while interpreting and comparing the full spectra. Analyses of trajectories did not reveal any noticeable protrusion modes, which we had anticipated from the differences that we saw in the intermediate q-regimes.
FIGURE 2:

Height-height undulation spectra for the two Martini CG systems. Spectra (A) with 2048 lipids and (B) with membrane and 14 CG PHDs distributed randomly on the bilayer surface. The schematics of the system, rendered from a snapshot of the trajectory, are shown in the inset. The undulation spectra follow the standard Helfrich-type q4 scaling, and the effect of curvature on the BM is not significant. Full fluctuation spectra for CG systems comprising 2048 lipids with pure membrane (no PHD) and membrane with 14 PHD are shown in Supplemental Figure S1. Time-averaged mean curvature observed in (C) pure membrane and (D) membrane with PHDs. Time-averaged Gaussian curvature observed in (E) pure membrane and (F) membrane with PHDs.
On the basis of our finding that the presence of the PHD lowers the BM of the membrane, we wanted to explore the molecular origin of decrease in BM. For this, we generated a microsecond-long atomistic trajectory of a single PHD on a lipid bilayer composed of DOPC:DOPS:PIP2 (80:19:1) and observed the membrane thickness and lipid conformations (tilt and splay) of lipids proximal and distal to the PHD on the membrane. Figure 3A shows a snapshot of the bilayer-PHD system with the proximal and distal lipids marked in different colors. We define lipid splay as the distance between the terminal carbon atoms on each tail of the lipid. Lipid tail tilt is defined by the angle between the membrane normal and the vector formed by the glycerol carbon and the last tail atom on the chains. We report the average of the two values per lipid for tail tilt. Lipid head tilt is defined by the angle between the P-N vector (vector formed by phosphorus and nitrogen atom in the lipid) and the membrane normal. Figure 3B shows the variation of membrane thickness across the cross-section of the bilayer observed at the end of 500 ns (converged state). Remarkably, the PHD influences the underlying membrane by thinning it by as much as 0.4 nm (Figure 3B), which would have a bearing on the BM (Deserno, 2007). Similar analyses for different 250-ns-long time-averaged plots show that membrane thinning is tightly coupled to the location of the PHD (Supplemental Figure S2, top panel). The change in thickness comes due to changed molecular configurations of lipids proximal to the PHD. Indeed, the presence of the PHD leads to a positive lipid splay in proximal lipids (Agrawal et al., 2016) and is seen by measurements of the lipid tail (Figure 3C) and head (Figure 3D) tilt angles for a 25 ns time period. As seen in Figure 3, C and D, the presence of the PHD induces significant fluctuations in the tail and head tilt angles in lipids proximal to the PHD than in the distal lipids or in lipids in a membrane without the PHD. Snapshots of tilt and splay for a proximal and distal lipid are shown in Supplemental Figure S2 and highlight the extent to which the PHD influences local lipid dynamics. The ability of PHD to induce such noticeable changes in tilt fluctuations of lipids strongly suggests the possibility that its engagement with the membrane could prime proximal lipids to attain nonbilayer intermediates and thereby lower the energy barrier for fission (Shnyrova et al., 2013; Mattila et al., 2015; McDargh and Deserno, 2018; Agrawal and Ramachandran, 2019). Importantly, the changes seen in the tilt values and the higher populations of tilt angles deviating from the average emphasize the need to go beyond the simple Helfrich-like elastic sheet models of membranes, including augmented models such as the ones with undulation-curvature coupling (Bradley and Radhakrishnan, 2016), to understand the energetics of membrane remodeling in terms of finer molecular degrees of freedom such as lipid tilt and splay (Nagle, 2017; McDargh and Deserno, 2018).
FIGURE 3:

(A) Snapshot from an all-atom DOPC-DOPS-PIP2 bilayer system with a single PHD (shown in light green) on the leaflet at the top. The lipids proximal to the protein are marked in blue. The distal lipids are marked in red. The lipids were tracked for 500 ns for the analysis. (B) Thickness profile of the bilayer with the PHD, showing thinner regions in red and thicker regions in blue. (C) Average tail angle distribution of lipids proximal to the protein (blue), distal to the protein (red), and in pure bilayer with no proteins (black). (D) Average head angle distribution of lipids. In Supplemental Figure S2, we provide some more data with thickness profiles of replicates and visuals of lipid conformations near and far away from the PHD.
Together these results confirm that PHDs soften the bilayer and possibly create favorable lipid configurations that induce or stabilize the nonbilayer intermediate topologies in the course of membrane fission. The implications of these molecular degrees of freedom in terms of the two paradigmatic models of membrane fission, namely the “constriction/ratchet model” (Leibler, 1986; Shlomovitz et al., 2011; McDargh and Deserno, 2018) and the “catalytic model” (Shnyrova et al., 2013; McDargh and Deserno, 2018) are discussed in detail later in the text.
In addition to bending modulus calculation, we performed local curvature calculations on CG lipid bilayer patch systems to observe spatial correlation between position of induction of curvatures and position of PH domains. The local curvatures observed in these membrane systems are quite dynamic in nature (see supporting data : mean-curvature.mp4 and gaussian-curvature.mp4). To observe curvatures that are persistent over time (and not simply thermal fluctuations), we performed time-averaging of mean (Figure 2, C and D) and Gaussian curvatures (Figure 2, E and F) observed at each point of the membrane surface. As seen in Figure 2D the lipid bilayer patch with PHDs does exhibit small scattered patches of both positive (green) and negative (purple) Gaussian curvatures (though small in magnitude) while pure membrane exhibits predominantly zero curvature (white color). In particular, there is one particular region on the membrane where a noticeable purple patch is seen and this corresponds to the region where a few PH domains are closely positioned. To some extent, this observation suggests that PH domains may be inducing a negative Gaussian curvature (saddle regions) on the membrane. To further test for curvature induction, we also created a hypothetical system where the PH domains were arranged in double collar – this was done to crudely model the scaffold and with an intention to accentuate the effect of PH Domain on curvature induction (see Figures R1-R4 in the supporting information).
Dynamin PHD engages the VL4 loop as an auxiliary pivot that regulates its orientation flexibility
Recent cryo-EM reconstructions of the dynamin polymer assembled on a membrane report a super-constricted state at 10.1 Å resolution, which highlights localized conformational changes at the BSE and GTPase domains that drive membrane constriction on GTP hydrolysis (Sundborger et al., 2014; Kong et al., 2018; Dandey et al., 2020). We reconstructed these data and focused on the PHD orientation by taking slices across the collar, which provided us some interesting observations (Figure 4A). PHD orientation about a ring of the dynamin polymer was measured by plotting the distribution of the angle between the major inertial axis of each PHD and the vector that connects the center of the ring with the centroid of the PHD (Figure 4B; see inset in Figure 4A for a schematic of the angle measurement for a single slice). This analysis revealed that the membrane-bound PHDs adopt a wide range of orientations in the polymer. This is also apparent in other slices analyzed in the polymer (Supplemental Figure S3). The wide range of PHD orientations, even in the highly packed constricted state of the dynamin polymer, suggests that the PHD has an inherent ability to associate with the membrane in multiple orientations.
FIGURE 4:

(A) End view of the 3D density map of dynamin polymer assembled on the membrane. The radial densities colored in purple show the PHDs on the collar. The inset shows the angle that the end-to-end vector of a given PHD makes with the radial line passing through the centers of the tube and the given PHD. The density map is redrawn from the available cryo-EM density data by Jennifer Hinshaw (National Institute of Diabetes and Digestive and Kidney Diseases, NIH) and coworkers. (B) Probability distribution of angle described above in the cryo-EM map. The angle θ has a wide range for the constricted collar. In Supplemental Figure S3, we show reconstruction for six other slices.
To test this and to obtain molecular insights into the association between the PHD and the lipid bilayer, we carried out advanced mixed-resolution molecular simulations using the HMMM (Ohkubo et al., 2012; Vermaas and Tajkhorshid, 2014a; Baylon et al., 2016). This model utilizes a biphasic setup comprising lipids with short-chain fatty acyl chains (typically 3–5 carbon atoms long) organized around an organic solvent, 1,1-dichloro-ethane (DCLE), which mimics the hydrophobic core of the membrane. Many studies have shown remarkable success in probing membrane-protein interactions using HMMM model in MD simulations without compromising on the atomic details of the protein–lipid headgroup interface, because lipid diffusion in the bilayer is accelerated by an order of magnitude due to the absence of tail friction (Baylon et al., 2013; Blanchard et al., 2014; Vermaas and Tajkhorshid, 2014b; Pant and Tajkhorshid, 2020).We ran 12 different HMMM simulations with different initial orientations of the PHD on a bilayer composed of DOPC:DOPS:PIP2 (80:19:1 mol%) and a DCLE hydrophobic core. Each system was run for at least 500 ns, with a total simulation time of ∼9.7 μs.
An important observation stands out from these simulations. In all our simulations, we consistently see that PHD associates with the membrane using both VL1 (531IGIMKGG537) and VL4 (576EKGFMSSK583). VL3 (590NTEQRNVYKDY600) does not show direct membrane anchoring but is always proximal to the PIP2 lipid headgroup that tends to “stick out” of the membrane plane. We explore this feature of VL3 more and put it in the context of existing literature (Liu et al., 2011; Mehrotra et al., 2014) when we discuss the binding geometries using the all-atom trajectories. In Figure 5 we have picked the extreme example from our HMMM runs where the initial PHD configuration is set up such that all the loops face away from the membrane (Figure 5A). The final configuration is shown in Figure 5B, where the VL1 and VL4 clearly partition into the membrane. Figure 5D shows the time evolution of residues on the various loops, and Figure 5C shows the initial and final z-distances for each residue with respect to the membrane phosphate plane. We have made a movie file for the full trajectory of this system, which is shared in Supplemental Movie-HMMM. The VL1, VL3, and VL4 loops are colored blue, red, and orange, respectively. The VL4 loop shows highly stable membrane association, and this observation is consistent across different starting configurations. In Figure 5E, we show the convergence data for each of the 12 systems by plotting the distances of the two loops from the membrane surface. Supplemental Figure S4 reports the initial and final membrane association profiles, in terms of z-distance of each residue away from the membrane plane for all 12 systems. The initial and the final orientations with respect to the membrane are also shown in insets of Supplemental Figure S4 for each of the 12 systems.
FIGURE 5:

Top panel shows the data for one of the 12 HMMM runs. In this case, the PHD is initially positioned such that all loops face away from the membrane. The PC, PS, and PIP2 mimetic lipids are shown in gray, green, and red, respectively, and phosphorus is marked in light blue. DCLE is shown in white in the core of the bilayer. The VL1 of the PHD is shown in blue, VL3 in red, and VL4in orange color. The top panel shows the initial (A) and final (B) PHD configurations. (C) Initial and final z-distance of each of the residues averaged over the last 100 ns. (D) Time evolution of signature residues on different loops. The entire 800 ns trajectory data for this system are also reported in Supplemental Movie-HMMM). In the left panel, (E) shows the convergence data for all 12 HMMM systems, where the distance from the membrane surface for the VL1 loop (I533) on the x-axis and VL4 (F579) on the y-axis is tracked with time. Almost all systems converge to a configuration with VL1 and VL4 physically engaging with the membrane. Plot akin to one on the right panel is shown for all 12 systems in Supplemental Figure S4 (i–xii)
Movie S1.
All atom simulation data
Movie S2.
Mixed resolution movie file
Movie S3.
Gaussian Curvature movie file
Movie S4.
Mean Curvature movie file
Results on VL1 are consistent with previous studies indicating that it acts as a membrane anchor, and the I533A mutation on VL1 is known to destabilize this interaction and reduce the stability of the scaffold on the membrane in the wake of GTP hydrolysis (Klein et al., 1998; Achiriloaie et al., 1999; Lemmon and Ferguson, 2000; Bethoney et al., 2009; Ramachandran et al., 2009; Dar et al., 2015; Dar and Pucadyil, 2017). To gain further insights into how the I533A mutant affects the membrane association, we carried out AAMD simulations for wild-type (WT)-PHD and I533A-PHD on a bilayer composed of DOPC:DOPS:PIP2 (80:19:1 mol%). Each system was run for at least 500 ns. Figure 6A shows the residue-wise z-distance for initial and final configurations for the WT system. Figure 6B shows the same data for the I533A system. To our surprise, the membrane association profile for the I533A did not show noticeable changes and the mutant does not seem to be membrane defective. We carried out similar AAMD runs on mutations on VL4 residues such as F579A and K583A (Figure 6, C and D), which also showed association behavior similar to that of WT. We anticipated these to be facile associations and carried out umbrella sampling calculations on the WT and various mutant systems to extract the binding free energy (ΔG) information. Figure 6E shows the membrane dissociation “potential of mean force” (PMF) profile for the WT and various mutant systems. As anticipated and reassuringly, there is a significant difference in membrane-binding free energies for the WT and I533A mutant system. This suggests that while the mutant I533A is nondefective with respect to membrane association, it binds superficially to the membrane and this explains the compromised assembly and reduced stability of the dynamin on the membrane.
FIGURE 6:

(A–D) Data for four of the AAMD simulation runs with respect to z-distance of each residue on the protein away from the membrane phosphate plane. The simulations were run for 500 ns each. The four systems are (A) WT, (B) I533A, (C) F579A, and (D) K583A. The gray curve on each figure indicates the initial configuration, and the blue curve is the final configuration averaged over the last 100 ns. Similar plots for K339A and Y600L mutant systems are shown in Supplemental Figure S7. (E) PMF profiles of the five systems obtained through umbrella sampling simulations. Right-most end of the graph denoted PHD in solution. The zero of the free energy difference plot is set at the phosphate plane of the membrane.
The analyses on F579A and K583A mutations shed some light on the role of VL4 as an auxiliary/secondary pivot. We chose to test the F579A mutation first because our HMMM simulations as well as our AAMD simulations consistently show that F579 has the deepest penetration for VL4. Interestingly, F579A hardly shows any change in binding free energies from the calculations done using umbrella sampling. K583A shows a minimal change of ∼2 kcal/mol in the dissociation ΔG values as compared with WT. It is clear that mutation on VL4 does not significantly alter the dissociation ΔG, so we checked whether these mutations had any effect on angular stability of the membrane-bound PHDs. To quantify the orientation flexibility of membrane-bound PHD, we looked at the angle that the PHD helix makes with the membrane normal θ (see inset of Figure 7A) and also the angle that it makes when projected to a reference axis on the membrane surface (φ), represented in the 360degree polar coordinate system. We compare the angles for WT data against I533A, F579A, and K583A (θ in Figure 7A and φ in Figure 7B). The distribution in WT is consistent with our cryo-EM reconstructions analysis (Sundborger et al., 2014; Kong et al., 2018) and further shows that the membrane-bound PHD is flexible in its orientation. However, VL4 mutations such as F579A and K583A show a significant increase in the range of angle distribution, while I533A shows hardly any change in the distributions. In our AAMD simulations and free energy calculations, we find that mutants have weaker membrane-binding free energy and many of them increase the orientation fluctuations of the PHD. We hypothesize that the mutation that makes the PHD highly labile (orientationally) adversely affects the collar assembly process, leading to compromised fission behavior. So even if, on the face value, the PHD is not membrane defective with VL4 mutants, the high orientation fluctuations are likely to affect the fission kinetics to a large extent. By attaching to the membrane using VL1 as the primary pivot and VL4 loop as the auxiliary pivot, the PHD stabilizes the dynamin on the membrane—in terms of both anchoring and orientation flexibility—thereby likely facilitating better assembly and collar formation. Incidentally, an artificially higher PIP2 concentration on the membrane has a similar effect on rescuing the kinetics for the I533A mutation to some extent (Dar et al., 2015), which lends further credence to our theory. While a high PIP2 concentration rescues the fission kinetics by increasing the “anchoring” binding energy, it will be interesting to check whether fission kinetics can be rescued for mutations that make the PHD unusually labile on the membrane. We carried out some tests toward that with double mutations as shown in Supplemental Figure S5.
FIGURE 7:
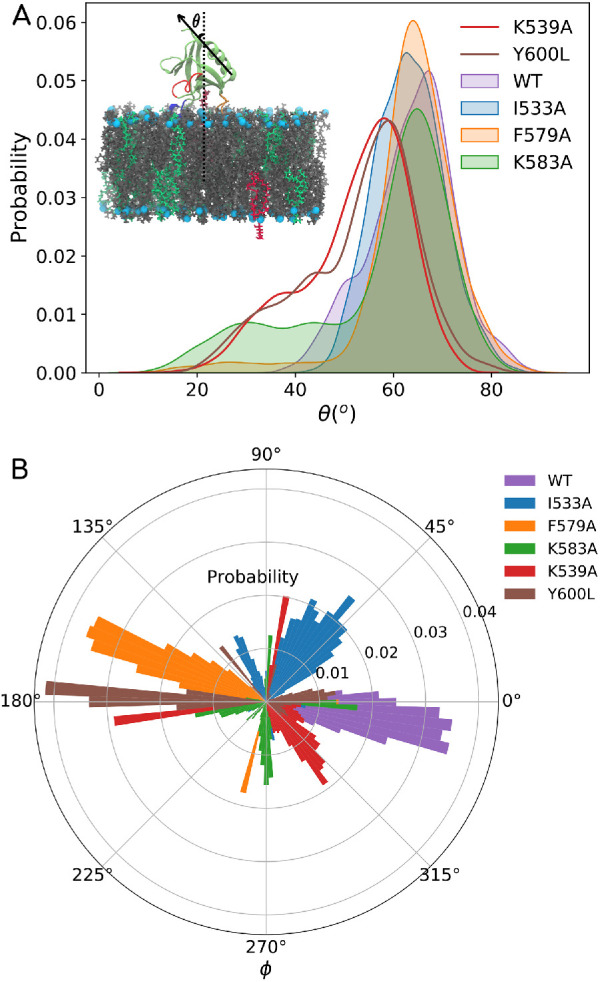
Data showing orientation distribution for PHD for WT and various mutant systems. (A) Inset in the top panel shows a schematic where θ is the angle that the helical axis makes with the membrane normal. θ angle distributions for the WT, I533A, F579A, K583A, Y600L, and K539A systems are shown in the top panel. (B) φ is the angle that the helical axis makes with a reference axis on the plane of the membrane and is shown in the bottom panel for all the WT and mutant systems.
Key residues stabilizing the PIP 2-PHD binding are found in multiple VLs
The role of VL1 in dynamin-catalyzed vesicle scission is undisputed (Ramachandran et al., 2009; Shnyrova et al., 2013; Mehrotra et al., 2014; Dar et al., 2015; Dar and Pucadyil, 2017). Fission is shown to be impaired with VL1 mutants such as I533A and M534C that attenuate its hydrophobic character (Ramachandran and Schmid, 2008; Ramachandran et al., 2009). Several studies have also shown the importance of VL3 in stabilizing the membrane interaction of PHD (Lee et al., 1999; Vallis et al., 1999; Chappie et al., 2011; Liu et al., 2011; Mehrotra et al., 2014). Owing to its polar nature, VL3’s direct engagement with the hydrophobic core is thought to be unlikely and loss in function with mutations such as Y600L is primarily attributed to the overall instability of the dynamin polymer on the membrane surface (Mehrotra et al., 2014). Additionally, our work consistently shows that VL4 engages with the acyl-chain hydrocarbon core and likely acts as a secondary pivot or a “buoy” that stabilizes the protein on the surface. Cryo-EM maps of the dynamin polymer, reconstructed with different modeled orientations of the PHD, further reinforce the possibility of PHD associating with multiple orientation with the membrane (Chappie et al., 2011; Sundborger et al., 2014; Kong et al., 2018).
To test the multiple orientation and binding geometries and also to look more carefully into the molecular-scale features that possibly drive and stabilize these associations, we carried out well-tempered metadynamics (WT-MTD)-based enhanced-sampling simulations (Barducci et al., 2008; Bonomi et al., 2009; Bonomi and Parrinello, 2010; Dama et al., 2014) using a single PHD and head group mimic of the PI(4,5)P2 (IP3: inositol triphosphate). We set up three different systems for WT-MTD simulations: two systems where the distance collective variables (CVs) are chosen such that the variable loops are sampled preferentially and another system with no bias for any loop or pocket. CVs can be thought of as reaction coordinates or order parameters that can distinguish between different conformational states of the PIP2-PHD complex in this work. The schematic of CV selection is shown in the left panel of Figure 8, and the corresponding free energy surface (FES) profiles are shown in the right panel. The details of CV definition are discussed in the Supplemental Information. The convergence plot for the three systems and the time evolution of distance in and out of the ligand from the domain’s pocket are shown in Supplemental Figure S6. To explore whether a variety of binding geometries exist, we considered the geometries in the range of ∼2 kBT of the deepest minima. We observed a range of possible geometries suggesting degeneracy, which implies that PIP2, or rather the IP3 ligand, can bind with different sets of residues in the PHD. Results from IP3-PHD simulations show that pockets formed by VL1-VL3 and VL1-VL4 are both available for binding the inositol head group.
FIGURE 8:

Well-tempered metadynamics setup with docking region for IP3 (ligand) biased toward the VL1 and VL3 loop region (A1), toward the VL1 and VL4 loop region (B1), and with no bias toward any loop region (C1). The free energy landscapes for the three systems are shown in panels A2, B2, and C2, respectively. The lowest energy configurations are picked from the lowest basin and region free energy difference of <2kBT from the minimum basin.
It should be noted that the WT-MTD runs were carried out with an isolated lipid head group (IP3), and so we wanted to check how well the information from these runs would relate back to membrane interaction modes of the PHD. To address this issue as well as to gain further insights into the configurations of binding geometry of bilayer-bound PHD, we performed the following simulation. We extracted the coordinates of PHD-IP3 from the deepest basins of free energy landscapes obtained from the WT-MTD runs. The first set of coordinates were obtained from WT-MTD data where the IP3 was positioned in the pocket between the VL1 and VL4 loops. The other set of coordinates was taken from simulations where the IP3 was positioned in the pocket between the VL1 and VL3 loops. The coordinates were superimposed onto the membrane bilayer (DOPC:DOPS:PIP2 in 80:19:1 ratio), making sure that the orientation of PHD-IP3 remained unchanged. This was done by aligning the IP3 ligand with the head group of PIP2, the membrane with only rotational and translational modes allowed during superimposition. The prescription is similar to what was done with the electron paramagnetic resonance-based structure of IP3-PHD of GRP1 to simulate a bilayer-bound PHD (Chen et al., 2012; Lai et al., 2013). We then let the system evolve in an all-atom NPT ensemble simulation for 500 ns each. This allowed us to test the stability of the configurations obtained from IP3-PHD WT-MTD runs in the presence of bilayer. We find that the simulation initiated from the orientation taken from minima obtained in the region of loops 1–3 is unstable compared with the orientation observed in the minima obtained from the loop 1–4 system. Please see Figure 9A, where the root-mean-square deviation (RMSD) is plotted keeping the first frame (configuration from converged WT-MTD runs) as the reference indicating that the VL1-VL4 basin configuration is robust. We chose this system for further analysis to look into residues that consistently engage with the acyl-chain hydrocarbon core as well as into residues that have nontransient interactions with PIP2 lipid and explore whether any of the interactions localizes the anionic PS lipids and stabilizes the interactions further.
FIGURE 9:

(A) RMSD plot of AAMD runs for two systems, one started with PIP2 docking derived from a metadynamics run with IP3 ligand in VL1-VL3 and the other with IP3 ligand in the VL1-VL4 pocket. The latter shows stable convergence and was chosen for 500 ns of the MD simulation, where we explored the lipid contacts of the PHD. (B) One of the last snapshots of the MD run (water and ions not shown). PC lipids are shown in gray, PS in green, and PIP2 lipid in red. The PHD is shown in a transparent white isosurf representation, and VL1, VL3, and VL4 are shown in opaque blue, red, and orange, respectively. The full movie file of this trajectory is also reported in Supplemental Movie-AA. (C–F) Normalized contacts that different kinds of lipids make with residues on various loops.
In Figure 9B, we show a snapshot at the end of the 500 ns, which shows the lipid engagements of various loops. Contact analysis of our trajectories shows that lipid tail atoms consistently interact with residues I533 and M534 of VL1 (Figure 9C), indicating hydrophobic association of the VL1 loop. However, what we find very interesting is how the PHD is stabilized further using residues such as K535 and K359 that recruit PS lipids near the VL1. This is quite evident in the Supplemental Movie-AA. VL1 accesses the hydrophobic core in a nonspecific manner, but once the PS lipids and PIP2 are recruited to the location, the I533 and M534 residue stabilize PS and PIP2 in its place by making contact with its tail. It is also amply clear from our HMMM simulations (see Supplemental Movie-HMMM) and AAMD simulations (see Supplemental Movie-AA) that VL3 has vital interactions with PIP2. HMMM runs show that the PIP2 is locked to the binding pocket once it makes contact with the polar VL3. AAMD analyses also highlight how PIP2 is localized without VL3 partitioning into the membrane. A plot of the z-distance of the C-alpha and z-distance of the terminal “hydroxyl oxygen” atom of Y600 as a function of time reveals nominal association with the membrane. While Y600L mutation is tried and tested for fission inhibition behavior, our results indicate R594 and R601 as critical residues on VL3 (Figure 9D) involved in interaction with PIP2. This could be tested experimentally to provide further insights into dynamin’s membrane association mechanisms. While VL1 and VL3 loops have been studied in depth for more than a decade now (and VL2 is not important for membrane association but shows some role in PS recruitment; Figure 9E through K554), no study has so far looked into the role of VL4. A clinical observation of a large family of Charcot–Marie–Tooth patients with the Met580Thr mutation supports the importance of this loop (Haberlová et al., 2011). On the other hand, in a hydrogen/deuterium exchange(HDX)-mass spectroscopy–based study, Srinivasan et al. (2016), explored the accessibility of dynamin residues upon nucleotide and/or membrane binding. Unlike our findings, they did not see any detectable change in protection of VL4 associated peptides, which can be misinterpreted as an absence of membrane association for VL4. It should be noted that the membrane accessibility of a given residue was reported based on the difference in solvent exchange behaviors of the residue in the membrane-bound state of dynamin1 with that of its apo state (existing as a dimer or tetramer in solution). It is possible that VL4 residues may be inaccessible to solvent in the apo state owing to its existence as dimers/tetramers in solution. In that case, the membrane-bound state and the apo state of the residue will show no difference in solvent exchange, which may likely reconcile the inconsistency with our results that clearly shows that VL4 is membrane bound. From our analyses, we show that VL4 consistently partitions into the hydrophobic core of the membrane though the association seems facile given that we see only marginal loss in binding free energies with mutations on VL4 residues. However, mutations such as F579A and K583A have very noticeable effects on the orientation stability of the PHD on the membrane despite no visible loss in membrane partitioning. Figure 9D clearly shows the heavy contacts that F579 and M580 and other residues of VL4 make with the membrane. Our observations can inform experiments to test the role played by VL4 in terms of orientation stability of PHD.
Conclusions
The dynamin collar undergoes a series of segmental rearrangements while executing membrane fission. Each PHD must be capable of adapting to the progressively remodeled underlying membrane. Previous results from three-dimensional (3D) reconstructions of dynamin mutants trapped in a constricted state (Chappie et al., 2011; Sundborger et al., 2014; Kong et al., 2018) as well as data from biochemical and multiple fluorescence spectroscopic approaches (Mehrotra et al., 2014) suggest that the PHD can associate with the membrane in different orientations. Theoretical analyses of determinants that could lower the energy barrier for fission have invoked tilting of the PHD to conform with the evolving membrane curvature and thereby creating a low-energy pathway for fission (Shnyrova et al., 2013; Pannuzzo et al., 2018; Kadosh et al., 2019). Our multiscale simulations data validate these possibilities and provide molecular insights in terms of effects of PHD on membrane properties and lipid conformations and on the binding geometries and orientation stability of PHD on membrane, all of which may affect processes downstream of membrane binding.
We show that binding of the PHDs lowers the membrane-bending rigidity, indicating that molecular interactions at the membrane interface confer a change in the bending rigidity of the membrane. These changes are brought about by polar and charged residues at the membrane interface that locally reduce membrane thickness and increase chain flexibility. Recent evidence of the existence of nonbilayer configurations of lipids formed at the stalk and hemifission intermediates prompted us to go beyond models that assume the membrane as an elastic sheet capable of undergoing fission through curvature instability (Leibler, 1986; Kozlov, 2001; Kooijman et al., 2003; Liu et al., 2011; Frolov et al., 2015; Fuhrmans and Müller, 2015) and focus on the molecular degrees of freedom of constituent lipids. Thus, higher fluctuations in the lipid tilt and lipid tail splay may have a significant role in facilitating the membrane to stochastically cross-over to the nonbilayer intermediates once a threshold constriction of the lumen is reached. The formation of nonbilayer intermediates dictates that a physical coupling between the two leaflets of the bilayer is reduced (Shchelokovskyy et al., 2011). However, our analysis reveals no apparent change in the interdigitation between the two leaflets, and this could reflect the fact that our simulations are currently not designed to recapitulate dynamics of the constricted tubular intermediate seen during fission.
With growing structural, biochemical, and modeling evidence supporting the role for PHD–lipid interactions, it is important to put our results into the context of the two prevalent models for the dynamin-induced fission mechanism (Antonny et al., 2016). The mechanochemical (constriction/ratchet) model and the catalytic activities (constriction/stochastic–cross-over) model may not be mutually exclusive and could together constitute the underlying fission mechanism. This aspect is very elegantly presented in the contribution from Frolov and Bashkirov in a recently published topical review (Bassereau et al., 2018). In our work, through molecular-scale modeling we also try to clearly bring out the catalytic aspect of PHD, and our work provides molecular insights into how the various variable loops may mediate membrane association, assembly, membrane mechanical properties, and prefission lipid conformations. The role of membrane rigidity in dynamin-mediated fission is established firmly by experiments (Morlot et al., 2012; Pinot et al., 2014), and our data on the role of PHDs in inducing local curvatures and enhanced membrane fluctuations (reducing rigidity) lend further credence to the mechanism that proposes stochastic cross-over to fission once the constriction reaches a reversible hemifission state (Shnyrova et al., 2013; Mehrotra et al., 2014; Frolov et al., 2015; Mattila et al., 2015; Dar and Pucadyil, 2017; Zhang and Müller, 2017). The mechanochemical constrictase/ratchet model for fission (Chappie et al., 2010, 2011; Faelber et al., 2011; Ford et al., 2011; Reubold et al., 2015) treats dynamin as a pure GTP-driven motor protein that triggers sliding of the helical turn, leading to membrane constriction and eventual fission. The stalk domain in dynamin, which mediates dimerization/assembly as well the power-stroke sliding motion, is connected to the activity-providing GTPase on the one end and to the membrane-associating PHD on the other end. The x-ray diffraction–derived structural data on dynamin constructs (Chappie et al., 2011; Faelber et al., 2011; Reubold et al., 2015) provide a strong basis for the existence of the above-mentioned model. However, for the stalk domain in particular, which is central to this mechanism, the structural data do not reconcile with the later cryo-EM data (Sundborger et al., 2014; Kong et al., 2018). This inconsistency could be due to the possible conformational rearrangements in the stalk domain, particularly during the membrane-bound assembly process. If that is the case, for the stalk domain to function effectively in the proposed mechanism, the role of PHDs as a highly regulated flexible pivot further comes to the fore and may be used to further reconcile the two prevalent proposed mechanisms in the literature.
The PHD is a ubiquitous membrane-binding domain for many different proteins. Several studies have explored the molecular mechanism by which it associates with PIP2/PIP3 in the membrane (Lai et al., 2013; Srivastava and Voth, 2014; Jian et al., 2015; Vonkova et al., 2015; Naughton et al., 2016, 2018; Yamamoto et al., 2016; Chan et al., 2017; Pant and Tajkhorshid, 2020; Soubias et al., 2020). Analysis of inositol-bound PHD structures clearly reveals the presence of a highly basic region that favors binding to the highly anionic phosphoinositide lipids (Ferguson et al., 1995; Lemmon et al., 1995). Our results from AAMD, WT-MTD, and HMMM runs extend these models and provide molecular-level insights into dyn-PHD engagement with membrane. Our simulation studies show that membrane binding of dyn-PHD has features that are quite unique. In general, multiple binding site studies suggest a mechanism with canonical and atypical phosphoinositide-binding sites that allows efficient switching between active and inactive forms in proteins (Jian et al., 2015; Yamamoto et al., 2016, 2020; Soubias et al., 2020). There is an important difference between the degeneracy in binding sites in our work as compared with that observed in other systems including ASAP1-PHD. Residues from multiple variable loops engage with the same PIP2 lipid in the case of dyn-PHD. The “catalytic” role of dyn-PHD is evident in our work due to its role in lowering the barrier to fission. Multiple binding loops around a PIP2 may explain how the dyn-PHD is able to “dynamically” keep itself anchored to the membrane while undergoing very rapid shape changes in the midst of the fission process.
Despite the fact that all three VLs appear to engage with the membrane, albeit to different extents and through different modes, the degeneracy in orientations lends credence to the notion that unlike being rigidly clamped to the membrane, the PHDs are best represented as units that are flexible and “moored” to the membrane. VL1 acts as a primary anchor that dictates the binding affinity with the membrane, a feature driven by both hydrophobic interactions (I533A showing reduced dissociation free energy) and stabilization by recruitment of PS lipids at VL1 by residues such as K535 and K539 on the loop. Interestingly, residue K554 on VL2 seems to also favor PS lipid recruitment to the PHD, though VL2 is quite distant from the membrane surface. VL3, a highly polar loop, never partitions with the bilayer but is instrumental in localizing PIP2. Given that the protruded head group of PIP2 allows such interactions, it would be interesting to see how mutations on VL3 residues behave in membranes with different curvatures and anionic lipid composition. VL4 has a subtler role in dictating the orientation flexibility of dyn-PHD, and we provide some testable hypotheses toward that end. None of the experimental studies has probed the role of VL4 in PHD–membrane interaction, possibly because the center of the loop is rich in hydrophobic residues with distal lysines unlike VL1 and VL3 (Lemmon and Ferguson, 2000), where the positively charged residues are at the tip of the loop. Our study identifies this novel interaction between VL4 and the membrane and highlights its role as an auxiliary pivot providing orientation stability to the PHD—a feature that we believe is important for effective polymerization of the dynamin collar. Our findings with respect to both multiple binding sites (Jian et al., 2015; Soubias et al., 2020; Yamamoto et al., 2020) and multiple orientations (Shnyrova et al., 2013; Mehrotra et al., 2014) have to be reexamined in the light of the auxiliary pivot theory. Put together, our results indicate that the membrane binding and orientation stability is tightly regulated by concerted interactions among three different loops on the dyn-PHD, which acts as a flexible pivot and modulates effective and expedited fission.
MATERIALS AND METHODS
Request a protocol through Bio-protocol.
Complete descriptions of the all-atom simulation parameters, metadynamics simulation parameters, and umbrella sampling simulations as well as the details of Martini and HMMM CG modeling are provided below or are available in the Supplemental Information. Moreover, all input files, including starting coordinates of the various systems, required to run all the simulation jobs for all methods, areavailable on the Github platform (https://github.com/codesrivastavalab/dynPHD).
CG simulations
Two CG bilayer systems with and without dyn1-PHDs (randomly oriented) were generated using the Martini bilayer builder in the CHARMM-GUI online server (Jo et al., 2008). The lipid bilayer was composed of 2048 lipids with a composition of DOPC-DOPS-PIP2 (80:19:1). These systems were simulated in the NPT (constant number of particles, pressure and temperature) ensemble using the MARTINI Polarizable force field (Version 2.2P) in GROMACS (Marrink et al., 2004, 2007; Monticelli et al., 2008). Both these systems were minimized using a steepest descent algorithm followed by an equilibration of 5–10 ns before proceeding to a 4 μs production run at 310 K temperature and 1 atm pressure. The temperature and pressure were maintained at 310 K and 1 atm using the V-rescale thermostat and Berendsen barostat (with semiisotropic coupling scheme), respectively. To understand the influence of dyn1-PHD on the mechanical properties of the lipid bilayer, we estimated the BM of the lipid bilayer with and without dyn1-PHDs. In this regard we adopted the reciprocal space-based method of Brandt et al. (2011) to obtain membrane undulation fluctuation spectra. The spectra so obtained were used to extract the BM using the Helfrich theory. To evaluate the mean and SD on the BM estimate, we performed the spectra evaluation on the full trajectory as well as on blocks of 1 μs of the full trajectory. The reported values were evaluated from the estimates from individual blocks. All the relevant data files, Python script, and c++ codes can be found at the Github location https://github.com/codesrivastavalab/dynPHD/.
Curvature analysis
Curvature analysis was performed on martini CG patches with and without PHDomains using the following protocol. The coordinates of phosphate head groups of lipids corresponding to one of the leaflets were extracted. A 2D Delaunay triangulation algorithm was then employed to generate a polygonal mesh of vertices from the coordinates of phosphate head groups. The surface, so generated, was used for evaluating the mean and gaussian curvature at various vertices of the mesh. These calculations were performed using Python scripts coupled with built-in subroutines from the VTK (Visualization ToolKit) library (Schroeder et al., 2004) which is used in Memsurfer to perform curvature calculations (Bhatia et al., 2019).The results were visualized using Paraview (Ahrens et al., 2005; Bethel et al., 2012) and its plugins.
HMMM simulations
HMMM simulations (Ohkubo et al., 2012; Vermaas and Tajkhorshid, 2014a) were performed using GROMACS with the CHARMM36 protein force field (Huang et al., 2017) and CHARMM36 phospholipid force field (Klauda et al., 2010) with a time step of 2 fs. We ran 12 different replicates with starkly different initial orientations of PHD, and each system was run for a minimum of 500 ns. The membrane was composed of a 80:19:1 mixture of DOPC, DOPS and POPI head groups (100 lipid molecules per leaflet). The core of the bilayer was made of DCLE molecules (∼1200). The bilayer–PHD systems were solvated and ionized with 150 mM NaCl using VMD (Visual Molecular Dynamics) (Humphrey et al., 1996). For each system, the following simulation protocol was used: the system was well equilibrated in both NVT (constant number of particles, volume and temperature) and NPT ensemble for 20 ns prior setting up for production run. Nonbonded forces were calculated with a 12 Å cutoff (10 Å: switching distance). Long-range electrostatic forces were calculated at every other time step using the particle mesh Ewald method (Essmann et al., 1995). The system is maintained at a temperature of 310 K and pressure of 1 atm using a Nose–Hoover thermostat and Parrinello–Rahman (with semi-isotropic coupling) barostat with time constants 1.0 and 5.0 ps–1, respectively.
All-atom simulations
We performed a series of AAMD NPT-simulations on WT and mutant dyn-PHD with membrane using GROMACS (Table 1) to understand the dynamics and to obtain deeper molecular insights into the dyn1-PHD on the membrane surface. Each of these systems was energy minimized and equilibrated for 20 ns. Each system comprises PHD, ∼200 lipids, ∼23,000 water molecules, and 150 mM NaCl. The CHARMM36 force field (Klauda et al., 2010) was used for particle definitions. Analysis of thickness variation across the membrane surface and protein was carried out using Python-based FATSLiM (Buchoux, 2017). The lipid conformational analysis was carried out using an in-house VMD TCL script or the VMD membrane plug-in tool (Guixà-González et al., 2014).
TABLE 1:
WT and mutant PHDs used for all-atom membrane simulations with membrane composition of DOPC-DOPS-PIP2 (80:19:1).
| Membrane + PHD variant | Simulation time (ns) |
|---|---|
| Wild type | 500 |
| I533A | 500 |
| F579A | 500 |
| K539A | 500 |
| K583A | 500 |
| Y600L | 500 |
| I533A-F579A | 500 |
| F579K-K583A | 500 |
| F579A-S581K | 500 |
| K539A-K583A | 500 |
Cryo-EM analysis
The cryo-EM map (Kong et al., 2018) for the constricted collar (K44A mutant) of dynamin (EMD-2701) was obtained from the EM databank (https://www.emdataresource.org/EMD-2701). The map was visualized using chimera (Pettersen et al., 2004) and was colored according to the cylinder radius. An in-house R code (R Core Team, 2020) was used to calculate the distribution of angles taken by PHDs in the cryo-EM map. Our code is available on the aforementioned Github repository.
Protein–ligand metadynamics
The structure of dyn1-PHD (protein) was obtained from the protein data bank (1DYN) (Ferguson et al., 1994). The force-field parameters of inositol triphosphate (ligand) corresponding to the CHARMM force field were generated using the Swissparam server (Zoete et al., 2011). The initial configurations of the protein–ligand system were constructed in cubic box using PYMOL (Schrödinger, 2015). The systems were then solvated with TIP3P water molecules and neutralized using 150 mM NaCl. All the systems were energy minimized and equilibrated to a temperature of 310 K and 1 atm pressure before proceeding to production runs. The velocity rescaling algorithm (Bussi et al., 2007) (time constant of 0.1 ps) and Parrinello–Rahman algorithm (Parrinello and Rahman, 1981) (time constant of 2 ps) were used to maintain temperature and pressure, respectively. Bond constraints were dealt using LINCS algorithm (Hess, 2008). The long-range electrostatic interactions were dealt using a particle mesh Ewald scheme with order 4 and a Fourier spacing of 0.16 while the short-range interactions were dealt using a Verlet scheme with a cutoff of 1.4 nm. All the simulations were performed in GROMACS (Abraham et al., 2015) patched with PLUMED-2.3.5 (Tribello et al., 2014). Short production runs of 10 ns were performed before proceeding for metadynamics runs. The details of different systems and the definitions of their respective CVs can be found in the Supplemental Information as well as in the input files shared on the Github repository.
Umbrella sampling simulations
The initial coordinates for setting up the umbrella sampling simulation were taken from the last frame of the corresponding PHD(WT/mutant)–lipid all-atom trajectory at the end of 500 ns runs. To get the umbrella locations, we placed PHD away from the membrane surface and saved the coordinates of the system at every 1 Å window up to a distance of 25 Å. The systems were then ionized and solvated for AAMD runs. The coordinates thus obtained were used as initial configurations for the 25 windows that constitute umbrella sampling simulations. Here, we chose the distance between the center of masses of the protein and the membrane to be restrained as the window centers (with restraint constant k = 1000 kJ/mol/nm2). Each window was simulated for 50 ns of the production run with the umbrella restraint turned on preceded by an equilibration run. The PMF profile was then calculated using gmx wham tool. The error along the PMF profile was obtained by performing a bootstrap analysis with 100 bootstraps. All profiles were shifted to maintain the PMF value at the membrane plane at 0 kcal/mol value for reference. The same protocol was used for generating the PMF profiles of all the systems of interest.
Supplementary Material
Acknowledgments
A.S. thanks Ryan Bradley from Ravi Radhakrishnan’s lab at the University of Pennsylvania for his help in the analyses and interpretations of the membrane fluctuation spectra. K.J. and A.S.\ thank Shashank Pant for his help in setting up HMMM simulations. A.S. thanks Vikas Dubey, a junior research trainee in our laboratory, for help with CGMD simulation runs. V.D. was supported partially by a grant from Raghavan Varadarajan’s J.C. BOSE project (DSTO/RV-2052). A.S. thanks Varadarajan for timely and much needed support. A.S. also thanks Thomas Pucadyil for carefully reading the manuscript and for his comments. A.S. thanks Himani Khurana from the Pucadyil lab for some very insightful discussions on the subject. Financial support from the Indian Institute of Science–Bangalore and the high-performance computing facility “Arjun” set up from grants by a partnership between the Department of Biotechnology of India and the Indian Institute of Science (IISc-DBT partnership programme) are gratefully acknowledged. A.S. acknowledges a startup grant provided by the Ministry of Human Resource Development of India and an early career grant from the Department of Science and Technology of India. The FIST program sponsored by the Department of Science and Technology and UGC, Centre for Advanced Studies and Ministry of Human Resource Development, India. is gratefully acknowledged by the authors.
Abbreviations used:
- AAMD
all-atom molecular dynamics
- BM
bending modulus
- CG
coarse-grained
- CV
collective variable
- DOPC
1,2-dioleoyl- sn-glycero-3-phosphocholine
- DOPS
1,2-dioleoyl- sn-glycero-3-phospho- l-serine
- HMMM
highly mobile membrane-mimetic model
- PHD
pleckstrin homology domain
- PIP 2
phosphatidylinositol-4,5-bisphosphate
- PMF
potential of mean force
- VL
variable loop
- WT-MTD
well-tempered metadynamics
- WT
wild-type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E20-12-0794) on May 12, 2021.
REFERENCES
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindah E (2015). Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25. [Google Scholar]
- Achiriloaie M, Barylko B, Albanesi JP (1999). Essential role of the dynamin pleckstrin homology domain in receptor-mediated endocytosis. Mol Cell Biol 19, 1410–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal A, Ramachandran R (2019). Exploring the links between lipid geometry and mitochondrial fission: emerging concepts. Mitochondrion 49, 305–313. [DOI] [PubMed] [Google Scholar]
- Agrawal H, Zelisko M, Liu L, Sharma P (2016). Rigid proteins and softening of biological membranes—with application to HIV-induced cell membrane softening. Sci Rep 6, 25412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonny B, Burd C, De Camilli P, Chen E, Daumke O, Faelber K, Ford M, Frolov VA, Frost A, Hinshaw JE, et al. (2016). Membrane fission by dynamin: what we know and what we need to know. EMBO J 35, 2270–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahrens J, Geveci B, Law C (2005). Paraview: An end-user tool for large data visualization. Visua Handbook 717, 8. [Google Scholar]
- Barducci A, Bussi G, Parrinello M (2008). Well-tempered metadynamics: a smoothly converging and tunable free-energy method. Phys Rev Lett 100, 20603. [DOI] [PubMed] [Google Scholar]
- Bassereau P, Jin R, Baumgart T, Deserno M, Dimova R, Frolov VA, Bashkirov PV, Grubmüller H, Jahn R, Risselada HJ, et al. (2018). The 2018 biomembrane curvature and remodeling roadmap. J Phys D Appl Phys 51, 343001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylon JL, Lenov IL, Sligar SG, Tajkhorshid E (2013). Characterizing the membrane-bound state of cytochrome P450 3A4: structure, depth of insertion, and orientation. J Am Chem Soc 135, 8542–8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylon JL, Vermaas JV, Muller MP, Arcario MJ, Pogorelov TV, Tajkhorshid E (2016). Atomic-level description of protein–lipid interactions using an accelerated membrane model. Biochim Biophys Acta 1858, 1573–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethel EW, Childs H, Hansen C (eds) (2012). High performance visualization: Enabling extreme-scale scientific insight. Boca Raton, FL: CRC Press. [Google Scholar]
- Bethoney KA, King MC, Hinshaw JE, Ostap EM, Lemmon MA (2009). A possible effector role for the pleckstrin homology (PH) domain of dynamin. Proc Natl Acad Sci 106, 13359–13364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia H, Ingólfsson HI, Carpenter TS, Lightstone FC, Bremer PT (2019). MemSurfer: a tool for robust computation and characterization of curved membranes. J Chem Theory Comput 15, 6411–6421. [DOI] [PubMed] [Google Scholar]
- Blanchard AE, Arcario MJ, Schulten K, Tajkhorshid E (2014). A highly tilted membrane configuration for the prefusion state of synaptobrevin. Biophys J 107, 2112–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonomi M, Barducci A, Parrinello M (2009). Reconstructing the equilibrium Boltzmann distribution from well-tempered metadynamics. J Comput Chem 30, 1615–1621. [DOI] [PubMed] [Google Scholar]
- Bonomi M, Parrinello M (2010). Enhanced sampling in the well-tempered ensemble. Phys Rev Lett 104, 190601. [DOI] [PubMed] [Google Scholar]
- Bradley RP, Radhakrishnan R (2016). Curvature–undulation coupling as a basis for curvature sensing and generation in bilayer membranes. Proc Natl Acad Sci USA 113, E5117–E5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt EG, Braun AR, Sachs JN, Nagle JF, Edholm O (2011). Interpretation of fluctuation spectra in lipid bilayer simulations. Biophys J 100, 2104–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchoux S (2017). FATSLiM: a fast and robust software to analyze MD simulations of membranes. Bioinformatics 33, 133–134. [DOI] [PubMed] [Google Scholar]
- Bussi G, Donadio D, Parrinello M (2007). Canonical sampling through velocity rescaling. J Chem Phys 126, 14101. [DOI] [PubMed] [Google Scholar]
- Chan KC, Lu L, Sun F, Fan J (2017). Molecular details of the PH domain of ACAP1BAR-PH protein binding to PIP-containing membrane. J Phys Chem B 121, 3586–3596. [DOI] [PubMed] [Google Scholar]
- Chappie JS, Acharya S, Leonard M, Schmid SL, Dyda F (2010). G domain dimerization controls dynamin’s assembly-stimulated GTPase activity. Nature 465, 435–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappie JS, Dyda F (2013). Building a fission machine–structural insights into dynamin assembly and activation. J Cell Sci 126, 2773–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappie JS, Mears JA, Fang S, Leonard M, Schmid SL, Milligan RA, Hinshaw JE, Dyda F (2011). A pseudoatomic model of the dynamin polymer identifies a hydrolysis-dependent powerstroke. Cell 147, 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H-C, Ziemba BP, Landgraf KE, Corbin JA, Falke JJ (2012). Membrane docking geometry of GRP1 PH domain bound to a target lipid bilayer: an EPR site-directed spin-labeling and relaxation study. PLoS One 7, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik LV, Kozlov MM (2003). Protein-lipid interplay in fusion and fission of biological membranes. Annu Rev Biochem 72, 175–207. [DOI] [PubMed] [Google Scholar]
- Dama JF, Parrinello M, Voth GA (2014). Well-tempered metadynamics converges asymptotically. Phys Rev Lett 112, 240602. [DOI] [PubMed] [Google Scholar]
- Dandey VP, Budell WC, Wei H, Bobe D, Maruthi K, Kopylov M, Eng ET, Kahn PA, Hinshaw JE, Kundu N, et al. (2020). Time-resolved cryo-EM using Spotiton. Nat Methods 17, 897–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar S, Kamerkar S, Pucadyil T (2015). A high-throughput platform for real-time analysis of membrane fission reactions reveals dynamin function. Nat Cell Biol 17, 1588–1596. [DOI] [PubMed] [Google Scholar]
- Dar S, Pucadyil TJ (2017). The pleckstrin-homology domain of dynamin is dispensable for membrane constriction and fission. Mol Biol Cell 28, 152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deserno M (2007). Fluid Lipid Membranes—A Primer. http://www.cmu.edu/biolphys/deserno/pdf/membrane_theory. pdf. 2007. [Google Scholar]
- Durieux A-C, Prudhon B, Guicheney P, Bitoun M (2010). Dynamin 2 and human diseases. J Mol Med 88, 339–350. [DOI] [PubMed] [Google Scholar]
- Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG (1995). A smooth particle mesh Ewald method. J Chem Phys 103, 8577–8593. [Google Scholar]
- Faelber K, Posor Y, Gao S, Held M, Roske Y, Schulze D, Haucke V, Noé F, Daumke O (2011). Crystal structure of nucleotide-free dynamin. Nature 477, 556–560. [DOI] [PubMed] [Google Scholar]
- Ferguson SM, De Camilli P (2012). Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol 13, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson KM, Lemmon MA, Schlessinger J, Sigler PB (1994). Crystal structure at 2.2 Å resolution of the pleckstrin homology domain from human dynamin. Cell 79, 199–209. [DOI] [PubMed] [Google Scholar]
- Ferguson KM, Lemmon MA, Schlessinger J, Sigler PB (1995). Structure of the high affinity complex of inositol trisphosphate with a phospholipase C pleckstrin homology domain. Cell 83, 1037–1046. [DOI] [PubMed] [Google Scholar]
- Ford MGJ, Jenni S, Nunnari J (2011). The crystal structure of dynamin. Nature 477, 561–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francy CA, Alvarez FJD, Zhou L, Ramachandran R, Mears JA (2015). The mechanoenzymatic core of dynamin-related protein 1 comprises the minimal machinery required for membrane constriction. J Biol Chem 290, 11692–11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolov VA, Escalada A, Akimov SA, Shnyrova AV (2015). Geometry of membrane fission. Chem Phys Lipids 185, 129–140. [DOI] [PubMed] [Google Scholar]
- Fuhrmans M, Müller M (2015). Coarse-grained simulation of dynamin-mediated fission. Soft Matter 11, 1464–1480. [DOI] [PubMed] [Google Scholar]
- Guixà-González R, Rodriguez-Espigares I, Ramírez-Anguita JM, Carrió-Gaspar P, Martinez-Seara H, Giorgino T, Selent J (2014). MEMBPLUGIN: studying membrane complexity in VMD. Bioinformatics 30, 1478–1480. [DOI] [PubMed] [Google Scholar]
- Haberlová J, Mazanec R, Ridzo\vn P, Baránková L, Nürnberg G, Nürnberg P, Sticht H, Huehne K, Seeman P, Rautenstrauss B (2011). Phenotypic variability in a large Czech family with a dynamin 2–associated Charcot-Marie-Tooth neuropathy. J Neurogenet 25, 182–188. [DOI] [PubMed] [Google Scholar]
- Hess B (2008). P-LINCS: a parallel linear constraint solver for molecular simulation. J Chem Theory Comput 4, 116–122. [DOI] [PubMed] [Google Scholar]
- Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmüller H, MacKerell AD (2017). CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K (1996). VMD: visual molecular dynamics. J Mol Graph 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Jian X, Tang WK, Zhai P, Roy NS, Luo R, Gruschus JM, Yohe ME, Chen PW, Li Y, Byrd RA, et al. (2015). Molecular basis for cooperative binding of anionic phospholipids to the PH domain of the Arf GAP ASAP1. Structure 23, 1977–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Iyer VG, Im W (2008). CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- Kadosh A, Colom A, Yellin B, Roux A, Shemesh T (2019). The tilted helix model of dynamin oligomers. Proc Natl Acad Sci USA 116, 12845–12850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenniston JA, Lemmon MA (2010). Dynamin GTPase regulation is altered by PH domain mutations found in centronuclear myopathy patients. EMBO J 29, 3054–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD Jr, Pastor RW (2010). Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein DE, Lee A, Frank DW, Marks MS, Lemmon MA (1998). The pleckstrin homology domains of dynamin isoforms require oligomerization for high affinity phosphoinositide binding. J Biol Chem 273, 27725–27733. [DOI] [PubMed] [Google Scholar]
- Koenig JH, Saito K, Ikeda K (1983). Reversible control of synaptic transmission in a single gene mutant of Drosophila melanogaster. J Cell Biol 96, 1517–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Sochacki KA, Wang H, Fang S, Canagarajah B, Kehr AD, Rice WJ, Strub M-P, Taraska JW, Hinshaw JE (2018). Cryo-EM of the dynamin polymer assembled on lipid membrane. Nature 560, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooijman EE, Chupin V, de Kruijff B, Burger KNJ (2003). Modulation of membrane curvature by phosphatidic acid and lysophosphatidic acid. Traffic 4, 162–174. [DOI] [PubMed] [Google Scholar]
- Kozlov MM (2001). Fission of biological membranes: interplay between dynamin and lipids. Traffic 2, 51–65. [DOI] [PubMed] [Google Scholar]
- Lai C-L, Srivastava A, Pilling C, Chase AR, Falke JJ, Voth GA (2013). Molecular mechanism of membrane binding of the GRP1 PH domain. J Mol Biol 425, 3073–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Frank DW, Marks MS, Lemmon MA (1999). Dominant-negative inhibition of receptor-mediated endocytosis by a dynamin-1 mutant with a defective pleckstrin homology domain. Curr Biol 9, 261–265. [DOI] [PubMed] [Google Scholar]
- Leibler S (1986). Curvature instability in membranes. J Phys 47, 507–516. [Google Scholar]
- Lemmon MA, Ferguson KM (2000). Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem J 350, 1–18. [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA, Ferguson KM, O’Brien R, Sigler PB, Schlessinger J (1995). Specific and high-affinity binding of inositol phosphates to an isolated pleckstrin homology domain. Proc Natl Acad Sci USA 92, 10472–10476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y-W, Neumann S, Ramachandran R, Ferguson SM, Pucadyil TJ, Schmid SL (2011). Differential curvature sensing and generating activities of dynamin isoforms provide opportunities for tissue-specific regulation. Proc Natl Acad Sci USA 108, E234–E242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrink SJ, De Vries AH, Mark AE (2004). Coarse grained model for semiquantitative lipid simulations. J Phys Chem B 108, 750–760. [Google Scholar]
- Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, De Vries AH (2007). The MARTINI force field: coarse grained model for biomolecular simulations. J Phys Chem B 111, 7812–7824. [DOI] [PubMed] [Google Scholar]
- Mattila JP, Shnyrova AV, Sundborger AC, Hortelano ER, Fuhrmans M, Neumann S, Müller M, Hinshaw JE, Schmid SL, Frolov VA (2015). A hemi-fission intermediate links two mechanistically distinct stages of membrane fission. Nature 524, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDargh ZA, Deserno M (2018). Dynamin’s helical geometry does not destabilize membranes during fission. Traffic 19, 328–335. [DOI] [PubMed] [Google Scholar]
- Mehrotra N, Nichols J, Ramachandran R (2014). Alternate pleckstrin homology domain orientations regulate dynamin-catalyzed membrane fission. Mol Biol Cell 25, 879–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monticelli L, Kandasamy SK, Periole X, Larson RG, Tieleman DP, Marrink S-J (2008). The MARTINI coarse-grained force field: extension to proteins. J Chem Theory Comput 4, 819–834. [DOI] [PubMed] [Google Scholar]
- Morlot S, Galli V, Klein M, Chiaruttini N, Manzi J, Humbert F, Dinis L, Lenz M, Cappello G, Roux A (2012). Membrane shape at the edge of the dynamin helix sets location and duration of the fission reaction. Cell 151, 619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlot S, Roux A (2013). Mechanics of dynamin-mediated membrane fission. Annu Rev Biophys 42, 629–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle JF (2017). Experimentally determined tilt and bending moduli of single-component lipid bilayers. Chem Phys Lipids 205, 18–24. [DOI] [PubMed] [Google Scholar]
- Naughton FB, Kalli AC, Sansom MSP (2016). Association of peripheral membrane proteins with membranes: free energy of binding of GRP1 PH domain with phosphatidylinositol phosphate-containing model bilayers. J Phys Chem Lett 7, 1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naughton FB, Kalli AC, Sansom MSP (2018). Modes of interaction of pleckstrin homology domains with membranes: toward a computational biochemistry of membrane recognition. J Mol Biol 430, 372–388. [DOI] [PubMed] [Google Scholar]
- Ohkubo YZ, Pogorelov TV, Arcario MJ, Christensen GA, Tajkhorshid E (2012). Accelerating membrane insertion of peripheral proteins with a novel membrane mimetic model. Biophys J 102, 2130–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto PM, Herskovits JS, Vallee RB (1997). Membranes and bioenergetics: role of the basic, proline-rich region of dynamin in Src homology 3 domain binding and endocytosis. 272, 11629–11635. [DOI] [PubMed] [Google Scholar]
- Pannuzzo M, McDargh ZA, Deserno M (2018). The role of scaffold reshaping and disassembly in dynamin driven membrane fission. eLife 7, e39441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant S, Tajkhorshid E (2020). Microscopic characterization of GRP1 PH domain interaction with anionic membranes. J Comput Chem 41, 489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello M, Rahman A (1981). Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys 52, 7182–7190. [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004). UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pinot M, Vanni S, Pagnotta S, Lacas-Gervais S, Payet L-A, Ferreira T, Gautier R, Goud B, Antonny B, Barelli H (2014). Polyunsaturated phospholipids facilitate membrane deformation and fission by endocytic proteins. Science 345, 693–697. [DOI] [PubMed] [Google Scholar]
- R Core Team (2020). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, https://www.R-project.org/. [Google Scholar]
- Ramachandran R, Pucadyil TJ, Liu Y-W, Acharya S, Leonard M, Lukiyanchuk V, Schmid SL (2009). Membrane insertion of the pleckstrin homology domain variable loop 1 is critical for dynamin-catalyzed vesicle scission. Mol Biol Cell 20, 4630–4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Schmid SL (2008). Real-time detection reveals that effectors couple dynamin’s GTP-dependent conformational changes to the membrane. EMBO J 27, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reubold TF, Faelber K, Plattner N, Posor Y, Ketel K, Curth U, Schlegel J, Anand R, Manstein DJ, Noé F, et al. (2015). Crystal structure of the dynamin tetramer. Nature 525, 404–408. [DOI] [PubMed] [Google Scholar]
- Schneck E, Sedlmeier F, Netz RR (2012). Hydration repulsion between biomembranes results from an interplay of dehydration and depolarization. Proc Natl Acad Sci USA 109, 14405–14409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger LLC (2015). The PyMOL molecular graphics system. Version 2.0, https://pymol.org/. [Google Scholar]
- Schroeder W, Lorensen B, Martin K (2004). The visualization toolkit: an object-oriented approach to 3D graphics. New York, NY: Kitware. https://vtk.org/. [Google Scholar]
- Shchelokovskyy P, Tristram-Nagle S, Dimova R (2011). Effect of the HIV-1 fusion peptide on the mechanical properties and leaflet coupling of lipid bilayers. New J Phys 13, 25004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomovitz R, Gov NS, Roux A (2011). Membrane-mediated interactions and the dynamics of dynamin oligomers on membrane tubes. New J Phys 13, 65008. [Google Scholar]
- Shnyrova AV, Bashkirov PV, Akimov SA, Pucadyil TJ, Zimmerberg J, Schmid SL, Frolov VA (2013). Geometric catalysis of membrane fission driven by flexible dynamin rings. Science 339, 1433–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubias O, Pant S, Heinrich F, Zhang Y, Roy NS, Li J, Jian X, Yohe ME, Randazzo PA, Lösche M, et al. (2020). Membrane surface recognition by the ASAP1 PH domain and consequences for interactions with the small GTPase Arf1. Sci Adv 6, eabd1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S, Dharmarajan V, Reed DK, Griffin PR, Schmid SL (2016). Identification and function of conformational dynamics in the multidomain GTPase dynamin. EMBO J 35, 443–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A, Voth GA (2014). Solvent-free, highly coarse-grained models for charged lipid systems. J Chem Theory Comput 10, 4730–4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundborger AC, Fang S, Heymann JA, Ray P, Chappie JS, Hinshaw JE (2014). A dynamin mutant defines a superconstricted prefission state. Cell Rep 8, 734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzi MM, Deserno M (2016). Revisiting tilt in classical curvature elastic theories for membranes. Biophys J 110, 581a. [Google Scholar]
- Tribello GA, Bonomi M, Branduardi D, Camilloni C, Bussi G (2014). PLUMED 2: new feathers for an old bird. Comput Phys Commun 185, 604–613. [Google Scholar]
- Vallis Y, Wigge P, Marks B, Evans PR, McMahon HT (1999). Importance of the pleckstrin homology domain of dynamin in clathrin-mediated endocytosis. Curr Biol 9, 257–263. [DOI] [PubMed] [Google Scholar]
- Van der Bliek AM, Redelmeier TE, Damke H, Tisdale EJ, Meyerowitz EM, Schmid SL (1993). Mutations in human dynamin block an intermediate stage in coated vesicle formation. J Cell Biol 122, 553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermaas JV, Tajkhorshid E (2014a). A microscopic view of phospholipid insertion into biological membranes. J Phys Chem B 118, 1754–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermaas JV, Tajkhorshid E (2014b). Conformational heterogeneity of α-synuclein in membrane. Biochim Biophys Acta 1838, 3107–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonkova I, Saliba AE, Deghou S, Anand K, Ceschia S, Doerks T, Galih A, Kugler KG, Maeda K, Rybin V, et al. (2015). Lipid cooperativity as a general membrane-recruitment principle for PH domains. Cell Rep 12, 1519–1530. [DOI] [PubMed] [Google Scholar]
- Yamamoto E, Doman´ski J, Naughton FB, Best RB, Kalli AC, Stansfeld PJ, Sansom MSP (2020). Multiple lipid binding sites determine the affinity of PH domains for phosphoinositide-containing membranes. Sci Adv 6, eaay5736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto E, Kalli AC, Yasuoka K, Sansom MSP (2016). Interactions of pleckstrin homology domains with membranes: adding back the bilayer via high-throughput molecular dynamics. Structure 24, 1421–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Müller M (2017). Rupturing the hemi-fission intermediate in membrane fission under tension: reaction coordinates, kinetic pathways, free-energy barriers. J Chem Phys 147, 64906. [DOI] [PubMed] [Google Scholar]
- Zoete V, Cuendet MA, Grosdidier A, Michielin O (2011). SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem 32, 2359–2368. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.