

Abstract
The inherited childhood blindness caused by mutations in NPHP5, a form of Leber congenital amaurosis, results in abnormal development, dysfunction, and degeneration of photoreceptors. A naturally occurring NPHP5 mutation in dogs leads to a phenotype that very nearly duplicates the human retinopathy in terms of the photoreceptors involved, spatial distribution of degeneration, and the natural history of vision loss. We show that adeno-associated virus (AAV)-mediated NPHP5 gene augmentation of mutant canine retinas at the time of active degeneration and peak cell death stably restores photoreceptor structure, function, and vision with either the canine or human NPHP5 transgenes. Mutant cone photoreceptors, which failed to form outer segments during development, reform this structure after treatment. Degenerating rod photoreceptor outer segments are stabilized and develop normal structure. This process begins within 8 weeks after treatment and remains stable throughout the 6-month posttreatment period. In both photoreceptor cell classes mislocalization of rod and cone opsins is minimized or reversed. Retinal function and functional vision are restored. Efficacy of gene therapy in this large animal ciliopathy model of Leber congenital amaurosis provides a path for translation to human treatment.
Keywords: ciliopathy, gene therapy, inherited retinal disease, Leber congenital amaurosis, photoreceptors, retina, retinal degeneration
Graphical abstract
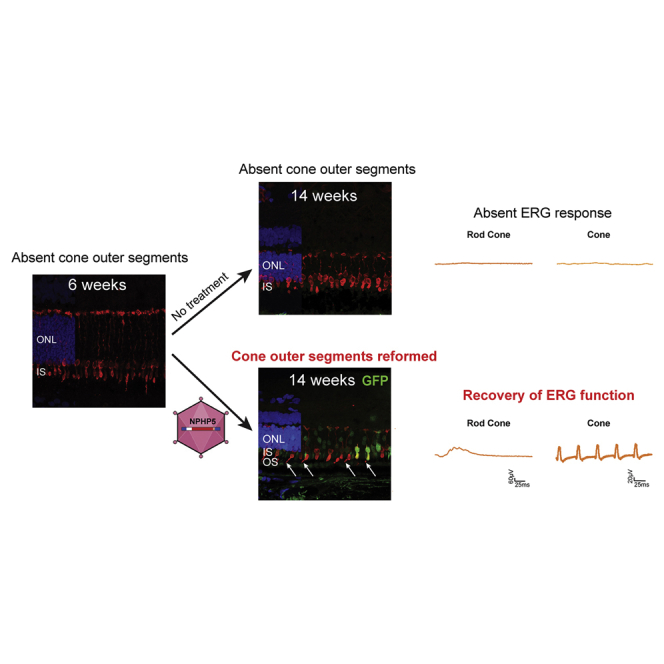
Using the canine NPHP5 model of Leber congenital amaurosis, the authors show that gene therapy is highly effective at the stage of disease when there is rapid cell death and active degeneration. After treatment, cone outer segments reform, retinal and visual function is restored, and degeneration is arrested.
Introduction
The hereditary childhood blindness Leber congenital amaurosis (LCA) is a heterogeneous group of disorders affecting the photoreceptor cells or retinal pigment epithelium (RPE) that are characterized by severe visual impairment or early-onset childhood blindness and currently considered to be mostly untreatable.1 To date, disease-causing mutations have been identified in autosomal recessive (n = 23) or dominant (n = 3) forms of LCA (https://sph.uth.edu/retnet/sum-dis.htm#B-diseases; February 19, 2021). These genes play essential but varied roles in outer retinal function and vision, ranging from vitamin A isomerization in the visual cycle to phototransduction, photoreceptor development and maintenance, and intracellular transport, among others2 (see den Hollander et al.3 for review). Of these, mutations in genes involved in transport through the photoreceptor ciliary transition zone (aka connecting cilium) result in retinal ciliopathies and comprise a major subclass of LCA with at least 8 genes (https://sph.uth.edu/retnet/sum-dis.htm#B-diseases)3, 4, 5, 6 as well as members of the BBSome complex.7, 8, 9 In addition to the retinal phenotype, CNS abnormalities and/or renal cystic disease can occur. One such ciliopathy resulting from NPHP5 (IQCB1) mutations, Senior Loken syndrome (SLSN), includes nephronophthisis (NPHP) and LCA, with the cystic kidney disease being the most frequent cause of end-stage renal failure in the first three decades of life.10
NPHP5 mutations represent a rare form of retinal disease estimated to affect ~5,000 patients worldwide, mainly in South East Asian and Northern European populations.11 However, the NPHP5 ciliopathy shares many common and extensive abnormalities that occur during retinal development or at birth and compromise photoreceptor structure and function. This is evident in at least four of the human ciliopathies—CEP290, NPHP5, RPGRIP1, TULP1—which show central outer nuclear layer (ONL) preservation with more peripheral loss and dramatically reduced cone function that does not reflect the degree of cone cell preservation.12, 13, 14, 15, 16, 17, 18 Such extensive photoreceptor structural abnormalities, which usually involve the outer segments (OSs), may preclude functional and structural correction after gene augmentation therapy, even if the transfer of a therapeutic transgene is successful. However, by using a translationally relevant animal model whose disease closely parallels the human, it is possible to examine and address these questions and facilitate the path to human treatments.
The canine NPHP5 disease homolog recapitulates exactly the human disease, with early-onset disease, central preservation of non-functional cone photoreceptors, and relentless progression to blindness.19 We have used this model to examine questions of photoreceptor targeting and treatment at a stage when photoreceptors are severely diseased and active degeneration is ongoing. Treatment was delivered at a time when there is a lack of cone function because most cone OSs have failed to form, and rod function was severely compromised because of marked OS structural abnormalities.19 Gene augmentation therapy led to formation of normal cone OSs, stabilized the comparable rod structures, and reversed the abnormal immunolocalization of cell class-specific markers as assessed by immunohistochemistry (IHC) and confocal microscopy. In parallel, there was robust and sustained recovery of normal retinal and visual function.
Results
Vectors
Because of the severe and rapidly progressive degenerative changes that occur early in the disease,19 we tested four different AAV vector constructs based on AAV2/5 and AAV2/8 with excellent photoreceptor tropism,20 with the aim of identifying a potential therapeutic candidate vector and promoter for treatment (Figure 1, vectors identified as Vectors A–D). These vectors were AAV based, either single-stranded (ss) or self-complementary (sc); the sc vectors bypass the need for double-stranded DNA synthesis prior to expression, an advantage for treating an aggressive and rapidly progressing disease.21 As well, Vector D had a single capsid mutation, Y733F, that changed a tyrosine for phenylalanine at position 733 to bypass ubiquitination and increase nuclear targeting and transgene expression.22
Figure 1.

Schematics of recombinant AAV vector constructs
Size (in kilobases [kb]) of AAV vector cassettes containing either the human GRK1 (hGRK1) or human IRBP (hIRBP) promoters driving expression of either the canine or human NPHP5 (cNPHP5 or hNPHP5, respectively) cDNA packaged as single-stranded (ss) or self-complementary (sc) constructs in either AAV8(Y377F) or AAV5 capsids. All other elements (SV40 SD/SA and poly A signals) within these constructs are identical. TR, AAV2-inverted terminal repeat; Δtrs, deletion of terminal resolution site in mutant TR; SV40 SD/SA, simian virus 40 splice donor/splice acceptor element; bGH pA, bovine growth hormone polyadenylation signal. The vectors are identified in text as Vectors A–D. A fifth vector, scAAV2/8(Y733F)-IRBP-cNPHP5, is not listed as it was used only once with no efficacy.
Recovery of rod and cone function after NPHP5 augmentation
Prior to treatment at 5–6 weeks of age, the mutant retina shows marked structural abnormalities (Figure 2). Cones are present, but the majority lack OSs based on high-resolution confocal microscopy and loss of cone-specific molecular markers (Figures 2A–2C). Lack of cone OSs results from failure of these structures to form during development.19 The retina has a normal complement of rods, but the OSs are disorganized and disoriented and show extensive rod opsin (RHO) mislocalization (Figure 2D). Despite these early OS structural abnormalities in both photoreceptor classes, detection of glutamylated RPGRORF15 (Figure 2E), a ciliary protein that interacts with NPHP5, suggests that the integrity of the photoreceptor ciliary transition zone initially is not compromised. At this time, TUNEL labeling shows a 10- to 15-fold increase in ongoing photoreceptor cell death in comparison to age-matched controls.19
Figure 2.

Treatment restores retinal structure and rod and cone ERG function in NPHP5 mutant retina
(A–E) Retinal structure of mutant retina at the 6 weeks of age time point. (A) Human cone arrestin (hCA) antibody shows the normal labeling of the cone IS, perinuclear cytoplasm, and cone axons/pedicles, but only a small number of cone OSs are present (arrows). (B and C) M/L- and S-cone opsins label the few OSs present of the specific cone subclasses and show mislocalization into IS, perinuclear region, and axons (∗, C). (D) Rod OSs are irregular, and rod opsin (RHO) is mislocalized into IS and ONL (∗). (E) GT335, a surrogate marker for glutamylated RPGRORF15 in photoreceptor ciliary transition zone (∗), shows distinct labeling at the tips of the IS. (F–J) Untreated regions of treated eye at 14 weeks of age, 8 weeks post injection (PI), shows progressive degeneration of photoreceptors, loss of ONL, and decreased labeling with GT335 in the ciliary transition zone. (K–O) Eye that received a subretinal injection with Vector D (scAAV2/8(Y733F)-GRK1-cNPHP5) at 1.5E+12 vg/mL; treated region identified by GFP fluorescence (green). Cone-specific molecular markers show in treated areas reformation of cone OSs and reduced M/L and S opsin mislocalization. Cone OSs are short, and some cone ISs lack an OS. The ISs are more bulbous and similar in shape to untreated cone ISs. RHO expression in OSs is intense and mislocalization reduced (∗). Localization of glutamylated RPGRORF15 in the photoreceptor ciliary transition zone (∗) is distinct. (P) Electroretinograms of the same WT control (black tracing) and mutant (WM46-right eye; orange tracing) dogs before and 8 weeks after gene augmentation therapy in the mutant. There is recovery of rod- and cone-mediated ERG responses, with cone responses similar in waveforms and amplitudes to WT control. Light intensities used to elicit the illustrated responses are Rod = −1.7 log cd·s·m−2; Rod Cone = 0.5 log cd·s·m−2; Cone 1Hz = 0.5 log cd·s·m−2; Cone 5Hz = 0.25 log cd·s·m−2; Cone 29Hz = 0.25 log cd·s·m−2. ONL, outer nuclear layer; IS, inner segment layer; OS, outer segment layer. Hoechst nuclear label is used in all sections.
Early in life, NPHP5 mutants show absence of photopic vision and cone electroretinogram (ERG) signals and rod responses that are markedly reduced in amplitude or absent (Figure 2P). Subretinal injection of the therapeutic vectors was directed at the high-cone density temporal retinal quadrant with the visual streak/fovea-like region. After treatment, this dog showed a robust recovery of rod and cone ERG signals under all stimulus conditions, and the ERG cone responses were comparable to the unaffected control (Figure 2P).
We used the 4 therapeutic vectors to treat 9 dogs unilaterally (n = 6) or bilaterally (n = 3) (Table S1). When assessed 7–9 weeks post injection (PI), the 9 dogs recovered rod- and cone-mediated function, and the ERG recovery was stable for >6 months (Figures 2P and 3A). Although the 9 treated dogs recovered ERG rod and cone responses, differences in amplitudes were present and could have resulted from the vector, the dose, or the area of injection (Figure 3). The single dog treated with the vector having the human transgene, Vector C, also had stable recovery of function, and the response amplitudes were broadly in the same range as with the other vectors. The one treatment failure not included among the 9 dogs treated unilaterally or bilaterally, dog AS2-396, received scAAV2/8(Y733F)-IRBP-cNPHP5 vector at low and high doses but did not recover ERG function at any of the 3 PI time points evaluated (Table S1). Common to all the therapeutic vector constructs was the absence of clinical evidence of inflammation or toxicity in the 1.5E+11 to 4.7E+12 vg/mL titer range.
Figure 3.

Robust and stable recovery of ERG rod and cone function following NPHP5 gene augmentation therapy
(A) ERG responses from untreated mutants, 13–14 weeks of age (left column), and one wild-type (WT) control (right column) for comparison. Center panels show the individual responses of dogs treated with 4 different vector combinations at 3 different post injection (PI) intervals. All dogs show recovery of rod and cone ERG responses that remains stable for 6 months. Note that the ERG responses for WM46-right eye at the 8 week time point also are included in Figure 2P to illustrate the functional recovery 8 weeks after treatment. (B) Summary data of the amplitudes recorded for individual treated dogs shown in (A) in response to rod-, rod-cone-, and cone-selective stimuli at 3 different PI intervals. Data for control (n = 4 dogs/8 eyes; 22.2 ± 3.4 weeks of age) and untreated mutants (n = 8; 13–14 weeks of age) is provided for comparison and represent the mean ± 1 SD of the amplitudes. Light intensities used to elicit the illustrated responses are: Rod = -1.7 log cd·s·m−2; Rod Cone = 0.5 log cd·s·m−2; Cone 1 Hz = 0.5 log cd·s·m−2; Cone 29 Hz = 0.25 log cd·s·m−2. Note: all but one vector has the canine NPHP5 transgene; the human transgene is used in Vector C (scAAV2/5-GRK1-hNPHP5). Vector titers are in vg/mL, and eyes received 70-μL subretinal injections. Individual animal identifiers are in parentheses. LE, left eye; RE, right eye.
Suboptimal injections occurred in two of the treated left eyes (dogs AS2-404 LE and WM46 LE [Table S1]), either because of a retinal tear at the retinotomy site with vector escaping into the vitreous or failure to form a large single subretinal bleb (Figure S1A). Despite the actual low vector volumes administered, there was distinct recovery of rod- and cone-mediated ERG responses that were sustained throughout the 25- to 32-week PI study interval (Figures S1B and S1C). Compared to the optimally injected eyes (Figure 3), the responses were lower in amplitude but distinct.
NPHP5 gene augmentation rescues vision
Functional vision was assessed in an obstacle avoidance course (Video S1) at 28 weeks PI in four unilaterally treated mutant dogs under two ambient illumination conditions. Based on ERG functional recovery after treatment, these dogs were considered poor-to-moderate responders; the two high responders, dogs WM46 and AS2-407, were not included in the assessment. All treated eyes showed faster transit time (Figure 4A) and fewer collisions in transit (Figure 4B), on average, under both scotopic (0.003 lux) and photopic (646 lux) conditions. The differences between treated and untreated eyes, regardless of vector, dose, or transgene, were statistically significant (p < 0.05), especially under photopic conditions. Under scotopic conditions, inter-animal variability was greater, and significance was achieved only for 1 dog, AS21-7, that was considered a poor responder based on ERG functional recovery (Figures 3, 4A, and 4B).
Figure 4.

Improved visual function following gene therapy
Visual behavior results in an obstacle avoidance course for 4 NPHP5 mutants treated unilaterally with 3 therapeutic vectors. (A and B) Visual function was assessed at 28 weeks PI under (A) dim scotopic (0.003 lux) and (B) bright photopic (646 lux) conditions. The horizontal gray bar in (A) represents the 95% confidence interval of the transit time for 4 WT control dogs. (B) No collisions were observed in the WT dogs. A paired t test was used to analyze the mean difference in transit time and number of collisions under each ambient illumination condition between treated eyes and the contralateral control eyes, and p < 0.05 was considered to be statistically significant. Tx, treated eye; Untx, untreated eye. Vector titers are in vg/mL, and eyes received 70-μL subretinal injections.
Photoreceptor layer preservation with treatment
The natural history of canine NPHP5 disease shows widespread degeneration of the photoreceptor and ONL by ~6 months of age, with a slight degree of preservation within the visual streak and fovea-like region.19 In comparison to wild-type (WT) controls, BSS-injected eyes showed extensive retinal degeneration that was comparable to untreated eyes (Figures 5A and 5B). In contrast, NPHP5 gene augmentation resulted in a distinct area of ONL preservation within the treated region (Figures 5C–5F and S2A–S2D). Comparable results were obtained with Vectors A–D, and there were no obvious differences that could be attributed to dose or the canine versus human NPHP5 transgene. The distribution of ONL rescue, however, was dependent generally on the bleb size (Figures 5C–5F and S2A–S2D). In the one dog where the focal bleb became widely distributed throughout the superior retinal quadrants because of an entrapped air bubble in the injection fluid, ONL preservation was commensurate with the vector distribution (Figure 5F); the untreated fellow eye showed advanced degeneration (data not shown).
Figure 5.

In vivo retinal imaging shows preservation of photoreceptor outer nuclear layer (ONL) in treated areas
(A and B) Topographical maps of ONL thickness in (A) WT control and (B) untreated or BSS-injected eyes. (C–F) All eyes treated with the therapeutic vectors show preservation of ONL thickness in comparison to untreated areas of the same eye. Retained ONL thickness in some cases extends beyond the bleb border into a penumbral region. Anatomic landmarks added include the fovea-like region (temporal white or black circle), the optic disc (black), and the tapetal boundary (wavy yellow line). In treated eyes, the red line identifies the subretinal injection bleb determined photographically immediately after the injection. The dashed arrow/red line in (F) shows the putative spread of the subretinal injection bleb from a trapped air bubble in the injection fluid. BSS, balanced salt solution; LE, left eye; RE, right eye. Vector titers are in vg/mL, and eyes received 70-μL subretinal injections.
Expression of therapeutic transgene in treated retinas
Neither commercial nor custom-made antibodies directed against human NPHP5 gave specific immunolabeling with/without antigen retrieval when used on retinas treated with the canine NPHP5 transgene (Table S2). To specifically localize NPHP5 transgene expression, we used RNA in situ hybridization (ISH). The treated areas showed intense RNA hybridization signals primarily localized in the ONL, with scattered signals present in the inner retinal layers and RPE, albeit at much reduced intensity (Figures S3A–S3C). In contrast, the untreated areas of the same eye, or the uninjected fellow eye, showed only very slight background signal. Co-injection with an AAV2/5-interphotoreceptor retinoid-binding protein (IRBP)-GFP reporter vector, and detection of GFP fluorescence in similar retinal areas on sequential sections, confirmed that NPHP5 mRNA expression was limited to the bleb region. Similar ISH labeling patterns were found in dogs treated with Vector D and analyzed at 8, 27, and 29 weeks PI. At the longer PI intervals, the treated areas had much better ONL and photoreceptor preservation than untreated areas of the same or fellow eyes (Figures S3B and S3C, lower panels).
Reforming normal photoreceptor structure after gene augmentation
IHC was used to examine the photoreceptor structural correlates that accompanied the recovery of retinal function following treatment. Eight weeks PI, the treatment areas delineated by GFP fluorescence showed that cones began to reform. Cone OSs developed, started to elongate, and expressed M/L- and S-cone opsins. The cone inner segments (ISs) were short and bulbous, similar to those in untreated areas, but cone opsin mislocalization was reduced (compare Figures 2F–2H to 2K–2M). Similarly, RHO mislocalization was minimized, and RHO immunolabeling was more intense (Figures 2I and 2N). At the photoreceptor ciliary transition zone, there was more intense expression of glutamylated RPGRORF15, a protein that interacts with NPHP5 in photoreceptors,4,23 than in untreated areas (Figures 2J and 2O).
Longer PI intervals clearly emphasized the significant positive treatment effects. Cone ISs became slender and longer, and, with few exceptions, all expressed M/L- or S-cone opsins in well-formed elongated OSs (Figures 6A–6C, 6F–6H, S4A–S4C, and S4F–S4H). Similarly, based on high-resolution confocal microscopy and IHC, rod IS and OS structures were normal, and glutamylated RPGRORF15 localization in the photoreceptor ciliary transition zone was also normal (Figures 6D, 6E, 6I, 6J, S4D, S4E, S4I, and S4J). In contrast, untreated areas of the same eye showed marked thinning of the ONL, loss of photoreceptors, and extensive mislocalization of rod and cone opsins into the perinuclear region of the few remaining ONL nuclei. These positive treatment effects emphasized that very abnormally developed photoreceptors can reform normal structure following gene augmentation therapy, and that this reversal was stable.
Figure 6.

Stable recovery of normal rod and cone structure after treatment
(A–J) Sections from (A E) untreated and (F–J) treated areas (Vector D; scAAV2/8(Y733F) GRK1-cNPHP5) of the right eye of dog AS2-404 at 29 weeks after therapy; treated areas identified by GFP fluorescence (green) in photoreceptor and outer nuclear layer nuclei. (A–C and F–H) Cone-specific molecular markers show normal cone structure in treated areas, and all cone ISs have corresponding OSs (white arrows). Untreated areas show absence of cone ISs and OSs and cone opsin mislocalization to ONL (B and C, ∗). (D and I) Rod OSs are structurally normal, and opsin mislocalization is almost completely reversed in treated area, but in untreated area rod degeneration has advanced and remaining rod nuclei have perinuclear RHO localization (D, ∗). (E and J) Localization of glutamylated RPGRORF15 in the ciliary transition zone (∗) is distinct, while the untreated region has none. Hoechst nuclear label is used in all sections. ONL, outer nuclear layer; IS, inner segment; OS, outer segment; RE, right eye; vg/mL, vector genomes/mL.
Discussion
The momentum generated by FDA’s approval of LUXTURNA (voretigene neparvovec-rzyl) for treating patients with biallelic RPE65 mutations24,25 has given hope that successful treatment for previously untreatable blinding diseases is possible. Gene therapy trials for other inherited retinal diseases have been initiated26 and provide further optimism for eventual cures. Among candidate diseases, the ciliopathy disorders provide a compelling therapeutic target, especially those for which there is a dissociation of structure and function. The absence of retinal function measured objectively by ERG, decreased retinal sensitivity, and/or impaired vision performance in a mobility test permits an early assessment of a successful functional outcome in treated eyes in which non-invasive analysis ascertains the presence of a sufficient number of photoreceptors suitable for treatment.27,28 These outcome measures can be identified soon after treatment, as was the case in the RPE65 clinical trials (see Jacobson et al.29,30 for review and study summaries), a form of LCA also characterized by a dissociation of structure and function.27 In contrast, outcomes for therapies that aim to halt disease progression and preserve vision are more difficult to assess quickly and usually require many years to establish positive outcomes.31
The canine NPHP5 model is an excellent platform to develop therapies for the class of ciliopathies characterized by early onset, aggressive clinical course, and dissociation of retinal structure and function. In both dogs and patients there is marked peripheral rod loss, but the degree of cone function loss is much greater than the degree of central cone preservation.19 In patients, cross-sectional in vivo imaging of the cone-only foveal region shows that in most the ONL thickness is normal, and the presence of a distinct ellipsoid region in the foveal cones (EZ line) indicates that cone ISs have formed; similarly, dogs show that cones, both in the cone-enriched fovea-like region and more peripherally, initially have normal numbers, but these lack an OS.19,32
In dogs, the disease is characterized by an overlap of abnormal photoreceptor development and progressive degeneration occurring in two phases, an early peak at ~5 weeks of age and a constant rate of loss thereafter.19 At the initial disease stage, ~85% of cones but not rods fail to form an OS, but rod OSs have irregular contours and extensive RHO mislocalization into the ISs, ONL, and synaptic terminals. Despite the cone structural abnormalities, a subset of cone-specific genes, particularly the cone opsins and the cyclic nucleotide-gated α and β subunits, are unchanged in expression, whereas the expression of rod-specific and rod/cone-enriched genes is downregulated.19 Thus the structural, functional, and molecular abnormalities in this form of LCA result in severely compromised photoreceptors.
Regardless of the four AAV constructs used, gene augmentation therapy delivered at 5–6 weeks of age resulted in robust recovery of rod- and cone-mediated ERG responses when evaluated 7–9 weeks PI and remained stable to the end of the study. The responses showed normal waveforms, and recovery of cone responses was dramatic, as there was complete absence of cone signals at the time of treatment because most of the OSs had failed to form (present study and Downs et al.19). For functional recovery to occur, the cones had to reform the OSs and express and localize the phototransduction components in the correct subcellular compartment. Given that the subset of cone-specific genes analyzed using a canine-specific qRT-PCR profiling array showed normal expression at the treatment age,19 it is likely that expression of WT NPHP5 in cones restored normal trafficking of proteins through the cone ciliary transition zone, resulting in OS formation and restoration of function.
Rods too have severe functional deficits that result from compromised structure. At the time of treatment rod OSs are present, but their contours are irregular and their parallel orientation is lost, there is extensive RHO mislocalization into the ISs and ONL, and rod cell death is at its peak.19 Rod-isolated ERG responses could be present, but reduced in amplitude by 60%–75%, or absent (present study and Downs et al.19). Impaired trafficking of key phototransduction proteins through the ciliary transition zone explains both opsin mislocalization and abnormal rod ERG function prior to therapy. After gene augmentation therapy, rod function is restored and stabilized during the PI period studied.
In this study, we tested the safety, efficacy, and dose ranges of four therapeutic vectors with the canine or human NPHP5 transgenes regulated by either IRBP or G protein-coupled receptor protein kinase 1 (GRK1) promoters and packaged either as ss or sc constructs (Figure 1). Our initial aim was to identify a superior vector for translational applications. However, we found that a positive therapeutic response is not limited to a specific vector, promoter, or transgene combination, but that the mutant retina appeared quite promiscuous in responding positively to therapeutic intervention. In general, the principal determinants of a positive response appeared to be the dose and the size/distribution of the subretinally injected vector.
A possible concern about the 2 sc vectors used is their more limited cargo-carrying capacity compared with ssAAVs, although some studies have shown that the packaging capacity of scAAV can be expanded beyond previously accepted limits.21,33 Systematic evaluation of scAAV genome size has established that scAAV vectors are capable of packaging up to 3.3 kb (i.e., 6.6 kb ss genome), and the University of Florida co-authors have reliably utilized scAAV vectors over 3 kb in retinal gene replacement studies.34,35 The scAAV vectors utilized in this study fall well below this upper size limit. To this end, the expression cassette of scVector C was sequenced, and this confirmed that the entire 2,800-bp cassette was present, and the size of the packaged scAAV was ~5,600 bp (Figure S5). However, before further development toward human applications, the integrity of any of these scAAV genomes over 5 kb in size will need to be defined.
At the doses used, the four vectors were well tolerated with a good safety profile and resulted in stable recovery of cone and rod function. We found no apparent differences in the functional recovery with either the scAAV2/5 or scAAV2/8(Y733F) vector pseudotypes containing the GRK1 promoter. However, the small sample size and multiple vector/promoter/transgene combinations used precluded definitive assessment of relative efficacy. There appeared to be a trend for higher-amplitude responses with increasing doses; again, the small sample size precluded statistical analysis. Finally, a fifth vector, scAAV2/8(Y733F) IRBP-cNPHP5, was tested in two eyes of a single animal but failed to restore function, and only partially reformed photoreceptor structure, yet stopped ONL loss in the high-dose-treated retina (data not shown). We posit that the IRBP promoter may not provide sufficient expression of the therapeutic transgene in the photoreceptors to fully correct the functional and structural defects. In fact, the only other animal that was treated with a vector containing the IRBP promoter (Vector A, dog AS21-7) had recovery of function and preservation of function but was considered a poor responder based on ERG functional recovery.
There were differences, however, in the degree of recovery of rod and cone function after treatment. Considering that the 70-μL injection volume covered no more than ~25% of the retinal surface, the amplitude of the recovered rod b-wave responses were commensurate with the treatment area, ~20%–30% of normal control values depending on the stimulus conditions used. The one animal where the injection bleb extended into the rod-enriched nasal superior quadrant, and thus covered ~40%–50% of the retinal surface, showed rod ERG responses close to control values.
Cone responses, on the other hand, proportionally were higher in amplitude and at the 1.5 and 4.7E+12 vg/mL titers with the canine transgene were comparable to WT controls; the amplitudes of the recovered cone responses remained unchanged during the 6-month PI period (Figure 3B). As the treatment was directed to the cone-enriched temporal quadrant that included the visual streak and the fovea-like region, it is likely that a higher cone population density was targeted than if treatments had been directed to the nasal or inferior retinal quadrants, where cone densities are lower.32,36 However, the near-normal cone ERG amplitudes when ~25% of the retinal temporal quadrant was treated were unexpected and are not explained by the wider distribution of the vector, as the bleb area corresponded closely to the area of photoreceptor preservation.
Like in humans, functional vision is severely compromised in NPHP5 mutant dogs early in life, particularly under photopic conditions. This was supported by objective vision testing in an obstacle avoidance course. After treatment, transit times were decreased, as were the number of collisions. As it takes several weeks of training after dogs have achieved physical maturity to objectively assess functional vision, the improvement of visual function could not be evaluated at earlier time points after treatment, but only at the end of the current study. At that time, there was a definite improvement in scotopic and photopic vision that was neither AAV capsid, promoter, dose, nor transgene specific, as the three vector combinations tested gave comparable results.
Underpinning the recovery of retinal function and vision were events occurring at the level of the photoreceptor cells that ensured the stability of the therapeutic intervention. Based on ISH results, treated regions showed robust expression of the therapeutic transgene; expression was present at the earliest time point analyzed, 8 weeks PI, and was qualitatively unchanged at the 27–29 weeks PI time points. Within 8 weeks of treatment, and in parallel with recovery of rod and cone ERG function, photoreceptors in the treated area began to reform normal structure. Initially, shortened OSs were associated with many of the cone ISs, which still had the bulbous contours characteristic of the untreated mutant retinas. The newly formed OSs expressed M/L- and S-cone opsins and reversed the protein mislocalization (Figure 2 and Downs et al.19). Rods too showed more robust RHO expression that accompanied a partial reversal of this mislocalized protein. Our prior studies indicated the presence of normal ciliary transition zone structure based on MAP9 expression in mutant retinas;19 now we show that treatment maintains expression of glutamylated RPGRORF15, an NPHP5 interacting protein also localized in the ciliary transition zone.4,23 The reversal of structural abnormalities in the treated retinas was stable. By the end of the study, ~27–29 weeks PI, the rods and cones in treated areas had regained normal structure and expressed key proteins characteristic of each photoreceptor class.
Along with the stable recovery of photoreceptor structure was the preservation of the ONL. At the ~5–6 weeks of age when treatment was initiated, photoreceptor apoptosis is at its peak and subsequently declines to more constant rates.19 However, by the end of the study it is clear that treatment arrested disease progression, and ONL thickness stabilized. From the en face topographic maps of ONL thickness, it was evident that the region of treatment efficacy extended beyond the bleb borders that were photographically documented at the time of subretinal injection and formed what is termed a penumbral region.37 The en face maps showed a variability in the extent of the penumbral region. In some, preservation was restricted to a narrow region surrounding the treatment bleb, while in others the preservation was more widespread (compare Figure 5E [AS2-391] with Figure S2A [WM51 LE]). The rescue effect outside of the treatment area has been observed in other studies, e.g., Guziewicz et al.37and Beltran et al.38, and is partly attributed to expansion of the subretinal bleb prior to reattachment.39
Like the treated NPHP5 mutant dogs, gene augmentation of knockout (KO) mice that have an artificial retinal all-S-cone environment by being double homozygous for Nphp5−/−;Nrl−/− also shows rescued photopic function, the treated cones formed axonemes, and the phototransduction proteins relocated to the OSs.40 While it is not clear that reforming cone OSs in an artificial all-S-cone environment is relevant for human applications, it provides further support that cones have the ability to reform OSs that never developed, or were lost through disease,41 and emphasizes the plasticity of the photoreceptor sensory cilia that continually renew their OS disc membranes.42 Here we showed that cone OS recovery occurs in a rod and cone environment that, although affected by disease, still models closely the canine and human retina and its disease phenotype.19,32
From the perspective of translational applications, such a model that is a disease homolog, and recapitulates the molecular and cellular defects along with phenotypic identity, will facilitate progress in the clinical development continuum. Furthermore, it will inform on therapeutic strategies for the other ciliopathies that show similar central ONL preservation with more peripheral loss and dramatically reduced cone function that does not reflect the degree of preservation of cone nuclei.12, 13, 14, 15, 16, 17 Obviously, as the treatment stage in this first proof-of concept study is less severe than the potentially more advanced disease stages that will be treated in patients, subsequent studies will need to demonstrate that treatments at later disease stages, as already shown for RPGR-associated X-linked retinal degeneration,43 are equally effective and stable before clinical trials in patients are initiated. These studies are ongoing.
Materials and methods
Canine NPHP5 model
The NPHP5 model originates from a research colony used for mutation identification44 and disease characterization; affected dogs have a severe early-onset retinal disease but no associated renal abnormalities.19 Additional details of the experimental and control dogs, breeding strategies, and number of animals used are provided in Supplemental methods and Table S1. Both mutant and control dogs were bred and maintained at the Retinal Disease Studies Facility, Kennett Square, PA, and housed under identical conditions of diet, medications, vaccinations, and ambient illumination, with cyclic 12 h ON-12 h OFF. For terminal procedures, the dogs were euthanatized with an overdose of Euthasol euthanasia solution (Virbac, Westlake, TX), and the eyes were immediately enucleated. The research was conducted in full compliance with University of Pennsylvania Institutional Animal Care and Use Committee (IACUC) approval, adhered to the Association for Research in Vision and Ophthalmology (ARVO) Resolution for the Use of Animals in Ophthalmic and Vision Research, and followed the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Viral vectors
The objective of this study was to determine whether this aggressive and severe ciliopathy would show a positive response to AAV-mediated gene augmentation therapy and to identify the potential therapeutic candidate vector and promoter for translational development. To this end, four different therapeutic AAV vector constructs were developed; these are identified as Vectors A–D (Figure 1). They carried either the canine (c) or human (h) full-length NPHP5 cDNAs that were synthesized based on the WT canine44 or human (GenBank: AY714228.1) sequences and incorporated a canonical Kozak sequence at the translational initiation site (GenScript). NPHP5 transgenes were placed under control of one of two photoreceptor-specific promoters: a 292-nt segment (positions 1793–2087) of the human G protein-coupled receptor protein kinase 1 promoter (hGRK1, GenBank: AY327580) or a 235-nt segment of the human IRBP promoter that includes the cis-acting element identified in the mouse proximal promoter.45, 46, 47 ss or sc cassettes were packaged into one of two capsids: AAV8(Y733F) or AAV5. A fifth vector, scAAV2/8(Y733F)-IRBP-cNPHP5, is not included in Figure 1 and was only tested in both eyes of one dog that showed no recovery of retinal function or preservation of photoreceptor structure.
To confirm the integrity of scVector C (scAAV2/5-GRK1-hNPHP5), 180 μL of the vector was mixed with 2× benzoate buffer and then incubated with 10 μL of benzoate (3,250 units) to digest possible external DNA. After incubation, 5× proteinase buffer and 10 μL of proteinase K (190 μg) were mixed and incubated to break the proteins and release the DNA. The enzymes are removed by extraction with phenol/chloroform followed by chloroform, and the scAAV-DNA is precipitated with 3 M sodium acetate, 100% ETOH and pellet painted with 2 μL of GlycoBlue. The pellets are cleaned with 70% ETOH and air-dried for sequencing. The expression cassette was sequenced using overlapping primers (Figure S5), and sequences were aligned to the reference plasmid sequence with Sequencher analysis software. This showed that the expression cassette was ~2,800 bp, and the size of the packaged scAAV was ~5,600 bp.
AAV vector preparations were produced by plasmid transfection using polyethylenimine (PEI).48 Vector purification involved discontinuous iodixanol step gradient followed by ion-exchange column chromatography (HiTrap Q Sepharose column) as previously described.49 The vector-containing fractions eluted from the columns were concentrated and titered for DNase-resistant vector genomes by RT-PCR relative to the bovine growth hormone (bGH) PolyA addition sequence of the scAAV viral vector standard. The standard was titered by dot blot and subsequently the vector samples by qPCR, methods that may underreport the vector copy number.50 Finally, the purity of the vector was validated by silver-stained SDS–PAGE and the vector preparation confirmed to have endotoxin levels < 5.0 EU/mL.
Vector administration and post injection treatment
Subretinal injections of recombinant AAV vectors were performed under general anesthesia in mutant dogs between 5.1 and 5.7 weeks of age as previously described (Table S1 details the animals, vectors, and promoters used).43,47,51 Seventy microliters of viral vector diluted in balanced salt solution (BSS) or vehicle (BSS) was delivered with a transvitreal approach without vitrectomy with a custom-modified RetinaJect subretinal injector (SurModics, Eden Prairie, MN, USA)52 under direct visualization with an operating microscope; anterior chamber paracentesis was performed immediately PI to prevent increase in intraocular pressure. Directly after injection, location of the subretinal bleb was documented by RetCam Shuttle fundus photography (Clarity Medical Systems, Pleasanton, CA, USA). The injections were directed to the temporal quadrant and were considered optimal when a large uniform bleb formed that included the area centralis and fovea-like region of the retina even when the injection was slightly off-center.32 Two eyes had suboptimal injections where the bleb was small, flat, or multiloculated or there was a visible retinal tear at the retinotomy site (Figure S1). Details of the clinical management and PI treatment plan are provided in Supplemental methods.
Electroretinography
Full-field flash ERGs were recorded with an Espion ERG system (Diagnosys, Lowell, MA, USA) under general anesthesia (induction with intravenous [i.v.] propofol; maintenance with isoflurane) using a custom-built Ganzfeld dome fitted with the LED stimuli of a ColorDome stimulator (Diagnosys) by methods previously described.43 Waveforms were processed with a digital low-pass 50-Hz filter to reduce recording noise if necessary. After 20 min of dark adaptation, rod- and mixed rod-cone-mediated responses (averaged 4 times) to single 4-ms white flash stimuli of increasing intensities (from −3.74 to 0.5 log cd·s·m−2) were recorded. After 5 min of white light adaptation (1.025 log cd·s·m−2), cone-mediated signals (averaged 10 times) to a series of single flashes (from −2.74 to 0.5 log cd·s·m−2) and to 5-Hz (averaged 20 times; from −2.74 to 0.25 log cd·s·m−2) and 29.4-Hz flicker (averaged 20 times; from −2.74 to 0.25 log cd·s·m−2) stimuli were recorded. These protocols separately assessed rod- and cone-mediated responses.
Optical coherence tomography
En face and retinal cross-sectional imaging was performed with the dogs under general anesthesia (induction with i.v. propofol; maintenance with isoflurane) as previously reported.43 Overlapping en face images of reflectivity with near-infrared illumination (820 nm) were obtained (Spectralis HRA+OCT, Heidelberg, Germany) with 30°- and 55°-diameter lenses to delineate fundus features such as optic nerve, retinal blood vessels, and boundaries of injection blebs. Custom MATLAB 7.5 programs (MathWorks, Natick, MA, USA) were used to digitally stitch individual photos into a retina-wide panorama. Spectral domain-optical coherence tomography (SD-OCT) was performed with linear and raster scans (Spectralis HRA+OCT). Overlapping 30° × 20° raster scans were recorded covering large regions of the retina. Post-acquisition processing of OCT data was performed with custom programs (MATLAB 7.5). For retina-wide topographic analysis, integrated backscatter intensity of each raster scan was used to locate its precise location and orientation relative to retinal features visible on the retina-wide mosaic formed by near infrared (NIR) reflectance images. Individual longitudinal reflectivity profiles (LRPs) forming all registered raster scans were allotted to regularly spaced bins (1° × 1°) in a rectangular coordinate system centered at the optic nerve; LRPs in each bin were aligned and averaged. Intraretinal peaks and boundaries corresponding to the ONL were segmented using both intensity and slope information of backscatter signal along each LRP. For all topographic results, locations of blood vessels, optic nerve head, and bleb boundaries were overlaid for reference.
Visual behavior-obstacle avoidance course
The methods to evaluate vision in dogs in an obstacle avoidance course have been previously reported.43,53 For this study we used an abbreviated protocol that evaluated visual function under two ambient light conditions: very dim scotopic (0.003 lux) and bright photopic (646 lux) illumination. The 0.003 lux illumination is ~2 log units lower than the intensity (0.2 lux) where dogs with a selective rod phototransduction defect from a rod PDE6B nonsense mutation show normal visual behavior in the testing environment.54 Eyes were tested individually by placing an AESTEK opaque corneo-scleral shield (Oculo-Plastik, Montreal, QC, Canada) over each cornea after topical anesthesia (proparacaine 0.5%). Both eyes were tested 3 times under each light intensity, and the position of the 5 panels was randomly changed between each of the 3 trials/eye/illumination. The contralateral eye was tested with the same set of panel positions. Random selection of the eye to be tested was made before the session. Dogs were first adapted for 20 min to the lowest ambient illumination (0.003 lux) before running through the course; subsequently, the room illumination was increased to the photopic level of brightness (646 lux), and dogs were adapted for 10 min and tested as previously described. Two digital Sony Handycam DCR-DVD108 cameras (Sony, San Diego, CA, USA) located above the obstacle course recorded the navigation performance of the dogs. The infrared imaging function of the cameras enabled recording under the dimmest light conditions. An experienced observer who was masked to the experimental design reviewed all the videos to measure for each trial the transit time in seconds between the first forward motion at the entrance of the course and the moment the animal completely passed through the exit gate and the total number of collisions. A paired t test was used to analyze the mean difference in transit time and number of collisions under each ambient illumination between treated eyes and the contralateral control eyes, and p <0.05 was considered to be statistically significant.
Retinal morphology, IHC, and in situ hybridization
Preparation and processing of tissues for retinal morphology, IHC, and ISH is detailed in Supplemental methods.
Acknowledgments
The authors thank Dr. John E. Dowling of Harvard University for critical review of the manuscript and helpful comments, Vince Chiodo of the University of Florida for vector production and vector sequencing, Dr. Leonardo Murgiano of the University of Pennsylvania for sequence analysis, Svetlana Savina, Natalia Dolgova, and Evelyn Santana for technical support, Sommer Ainsworth for vision behavior testing, Dr. Karolina Roszak for assisting with some of the in vivo studies-OCT, ERG, and preparations for the subretinal injections, Terry Jordan and the staff of RDSF for excellent animal care and support of in vivo studies, and Lydia Melnyk for research coordination. The study was supported in part by grants R01 EY006855, EY017549, and P30-EY001583 from the National Eye Institute; the content is solely the responsibility of the authors and does not necessarily represent the official views of the National Eye Institute or the NIH. Additional support from the Foundation Fighting Blindness, the Van Sloun Fund for Canine Genetic Research, Hope For Vision, Research to Prevent Blindness Foundation, and the Sanford and Susan Greenberg End Blindness Outstanding Achievement Prize (G.D.A. and W.W.H.).
Author contributions
G.D.A. designed and implemented the studies, analyzed the data, wrote original and final drafts, and supervised the research activity. A.V.C. analyzed and interpreted the noninvasive imaging results, provided critical review and advice at different stages of the study, and was involved in the review and editing of the manuscript. V.L.D. carried out different aspects on the in vivo studies, reviewed and wrote parts of the manuscript, and produced most of the figures. A.R.G. and S.I. carried out eye examinations, medical assessments, ERG, and OCT studies. R.S. carried out the RNA in situ hybridization studies with the assistance of R.N. M.S. used OCT imaging data to create the en face maps of ONL thickness. S.L.B. and W.W.H. designed and produced the vectors. S.G.J. provided critical review and advice at different stages of the study and was involved in the review and editing of the original draft of the manuscript. W.A.B. designed and implemented the studies, carried out the subretinal injections, assessed retinal structural outcomes by histology and immunohistochemistry, analyzed the data, wrote parts of the original and final drafts, and supervised the research activity.
Declaration of interests
G.D.A., W.A.B., S.L.B., A.V.C., W.W.H., and S.G.J. have filed a patent application for NPHP5 gene therapy on behalf of The University of Pennsylvania and the University of Florida. In addition, S.L.B. owns stock in, and is a paid consultant to, the company Atsena Therapeutics, and W.W.H. owns stock in the companies AGTC and BionicSight and is a paid consultant and a member of the scientific board of AGTC.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.03.021.
Contributor Information
Gustavo D. Aguirre, Email: gda@vet.upenn.edu.
William A. Beltran, Email: wbeltran@vet.upenn.edu.
Supplemental information
References
- 1.Scholl H.P., Strauss R.W., Singh M.S., Dalkara D., Roska B., Picaud S., Sahel J.A. Emerging therapies for inherited retinal degeneration. Sci. Transl. Med. 2016;8:368rv6. doi: 10.1126/scitranslmed.aaf2838. [DOI] [PubMed] [Google Scholar]
- 2.Kumaran N., Moore A.T., Weleber R.G., Michaelides M. Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. Br. J. Ophthalmol. 2017;101:1147–1154. doi: 10.1136/bjophthalmol-2016-309975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.den Hollander A.I., Roepman R., Koenekoop R.K., Cremers F.P. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 2008;27:391–419. doi: 10.1016/j.preteyeres.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 4.Otto E.A., Loeys B., Khanna H., Hellemans J., Sudbrak R., Fan S., Muerb U., O’Toole J.F., Helou J., Attanasio M. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat. Genet. 2005;37:282–288. doi: 10.1038/ng1520. [DOI] [PubMed] [Google Scholar]
- 5.Perrault I., Saunier S., Hanein S., Filhol E., Bizet A.A., Collins F., Salih M.A., Gerber S., Delphin N., Bigot K. Mainzer-Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am. J. Hum. Genet. 2012;90:864–870. doi: 10.1016/j.ajhg.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eblimit A., Nguyen T.M., Chen Y., Esteve-Rudd J., Zhong H., Letteboer S., Van Reeuwijk J., Simons D.L., Ding Q., Wu K.M. Spata7 is a retinal ciliopathy gene critical for correct RPGRIP1 localization and protein trafficking in the retina. Hum. Mol. Genet. 2015;24:1584–1601. doi: 10.1093/hmg/ddu573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katoh Y., Nozaki S., Hartanto D., Miyano R., Nakayama K. Architectures of multisubunit complexes revealed by a visible immunoprecipitation assay using fluorescent fusion proteins. J. Cell Sci. 2015;128:2351–2362. doi: 10.1242/jcs.168740. [DOI] [PubMed] [Google Scholar]
- 8.Wright A.F., Chakarova C.F., Abd El-Aziz M.M., Bhattacharya S.S. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat. Rev. Genet. 2010;11:273–284. doi: 10.1038/nrg2717. [DOI] [PubMed] [Google Scholar]
- 9.Bramall A.N., Wright A.F., Jacobson S.G., McInnes R.R. The genomic, biochemical, and cellular responses of the retina in inherited photoreceptor degenerations and prospects for the treatment of these disorders. Annu. Rev. Neurosci. 2010;33:441–472. doi: 10.1146/annurev-neuro-060909-153227. [DOI] [PubMed] [Google Scholar]
- 10.Hildebrandt F., Otto E. Cilia and centrosomes: a unifying pathogenic concept for cystic kidney disease? Nat. Rev. Genet. 2005;6:928–940. doi: 10.1038/nrg1727. [DOI] [PubMed] [Google Scholar]
- 11.Hanany M., Rivolta C., Sharon D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. USA. 2020;117:2710–2716. doi: 10.1073/pnas.1913179117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cideciyan A.V., Aleman T.S., Jacobson S.G., Khanna H., Sumaroka A., Aguirre G.K., Schwartz S.B., Windsor E.A., He S., Chang B. Centrosomal-ciliary gene CEP290/NPHP6 mutations result in blindness with unexpected sparing of photoreceptors and visual brain: implications for therapy of Leber congenital amaurosis. Hum. Mutat. 2007;28:1074–1083. doi: 10.1002/humu.20565. [DOI] [PubMed] [Google Scholar]
- 13.Cideciyan A.V., Rachel R.A., Aleman T.S., Swider M., Schwartz S.B., Sumaroka A., Roman A.J., Stone E.M., Jacobson S.G., Swaroop A. Cone photoreceptors are the main targets for gene therapy of NPHP5 (IQCB1) or NPHP6 (CEP290) blindness: generation of an all-cone Nphp6 hypomorph mouse that mimics the human retinal ciliopathy. Hum. Mol. Genet. 2011;20:1411–1423. doi: 10.1093/hmg/ddr022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stone E.M., Cideciyan A.V., Aleman T.S., Scheetz T.E., Sumaroka A., Ehlinger M.A., Schwartz S.B., Fishman G.A., Traboulsi E.I., Lam B.L. Variations in NPHP5 in patients with nonsyndromic leber congenital amaurosis and Senior-Loken syndrome. Arch. Ophthalmol. 2011;129:81–87. doi: 10.1001/archophthalmol.2010.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacobson S.G., Cideciyan A.V., Aleman T.S., Sumaroka A., Schwartz S.B., Roman A.J., Stone E.M. Leber congenital amaurosis caused by an RPGRIP1 mutation shows treatment potential. Ophthalmology. 2007;114:895–898. doi: 10.1016/j.ophtha.2006.10.028. [DOI] [PubMed] [Google Scholar]
- 16.Jacobson S.G., Cideciyan A.V., Huang W.C., Sumaroka A., Roman A.J., Schwartz S.B., Luo X., Sheplock R., Dauber J.M., Swider M., Stone E.M. TULP1 mutations causing early-onset retinal degeneration: preserved but insensitive macular cones. Invest. Ophthalmol. Vis. Sci. 2014;55:5354–5364. doi: 10.1167/iovs.14-14570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacobson S.G., Cideciyan A.V., Huang W.C., Sumaroka A., Nam H.J., Sheplock R., Schwartz S.B. Leber congenital amaurosis: genotypes and retinal structure phenotypes. Adv. Exp. Med. Biol. 2016;854:169–175. doi: 10.1007/978-3-319-17121-0_23. [DOI] [PubMed] [Google Scholar]
- 18.Estrada-Cuzcano A., Koenekoop R.K., Coppieters F., Kohl S., Lopez I., Collin R.W., De Baere E.B., Roeleveld D., Marek J., Bernd A. IQCB1 mutations in patients with leber congenital amaurosis. Invest. Ophthalmol. Vis. Sci. 2011;52:834–839. doi: 10.1167/iovs.10-5221. [DOI] [PubMed] [Google Scholar]
- 19.Downs L.M., Scott E.M., Cideciyan A.V., Iwabe S., Dufour V., Gardiner K.L., Genini S., Marinho L.F., Sumaroka A., Kosyk M.S. Overlap of abnormal photoreceptor development and progressive degeneration in Leber congenital amaurosis caused by NPHP5 mutation. Hum. Mol. Genet. 2016;25:4211–4226. doi: 10.1093/hmg/ddw254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trapani I., Puppo A., Auricchio A. Vector platforms for gene therapy of inherited retinopathies. Prog. Retin. Eye Res. 2014;43:108–128. doi: 10.1016/j.preteyeres.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCarty D.M. Self-complementary AAV vectors; advances and applications. Mol. Ther. 2008;16:1648–1656. doi: 10.1038/mt.2008.171. [DOI] [PubMed] [Google Scholar]
- 22.Zhong L., Zhao W., Wu J., Li B., Zolotukhin S., Govindasamy L., Agbandje-McKenna M., Srivastava A. A dual role of EGFR protein tyrosine kinase signaling in ubiquitination of AAV2 capsids and viral second-strand DNA synthesis. Mol. Ther. 2007;15:1323–1330. doi: 10.1038/sj.mt.6300170. [DOI] [PubMed] [Google Scholar]
- 23.Rao K.N., Anand M., Khanna H. The carboxyl terminal mutational hotspot of the ciliary disease protein RPGRORF15 (retinitis pigmentosa GTPase regulator) is glutamylated in vivo. Biol. Open. 2016;5:424–428. doi: 10.1242/bio.016816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russell S., Bennett J., Wellman J.A., Chung D.C., Yu Z.F., Tillman A., Wittes J., Pappas J., Elci O., McCague S. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390:849–860. doi: 10.1016/S0140-6736(17)31868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maguire A.M., Russell S., Wellman J.A., Chung D.C., Yu Z.-F., Tillman A., Wittes J., Pappas J., Elci O., Marshall K.A. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology. 2019;126:1273–1285. doi: 10.1016/j.ophtha.2019.06.017. [DOI] [PubMed] [Google Scholar]
- 26.Thompson D.A., Iannaccone A., Ali R.R., Arshavsky V.Y., Audo I., Bainbridge J.W.B., Besirli C.G., Birch D.G., Branham K.E., Cideciyan A.V., Monaciano Consortium Advancing Clinical Trials for Inherited Retinal Diseases: Recommendations from the Second Monaciano Symposium. Transl. Vis. Sci. Technol. 2020;9:2. doi: 10.1167/tvst.9.7.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacobson S.G., Aleman T.S., Cideciyan A.V., Sumaroka A., Schwartz S.B., Windsor E.A., Traboulsi E.I., Heon E., Pittler S.J., Milam A.H. Identifying photoreceptors in blind eyes caused by RPE65 mutations: Prerequisite for human gene therapy success. Proc. Natl. Acad. Sci. USA. 2005;102:6177–6182. doi: 10.1073/pnas.0500646102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gardiner K.L., Cideciyan A.V., Swider M., Dufour V.L., Sumaroka A., Komáromy A.M., Hauswirth W.W., Iwabe S., Jacobson S.G., Beltran W.A., Aguirre G.D. Long-Term Structural Outcomes of Late-Stage RPE65 Gene Therapy. Mol. Ther. 2020;28:266–278. doi: 10.1016/j.ymthe.2019.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jacobson S.G., Cideciyan A.V., Ratnakaram R., Heon E., Schwartz S.B., Roman A.J., Peden M.C., Aleman T.S., Boye S.L., Sumaroka A. Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch. Ophthalmol. 2012;130:9–24. doi: 10.1001/archophthalmol.2011.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacobson S.G., Cideciyan A.V., Roman A.J., Sumaroka A., Schwartz S.B., Heon E., Hauswirth W.W. Improvement and decline in vision with gene therapy in childhood blindness. N. Engl. J. Med. 2015;372:1920–1926. doi: 10.1056/NEJMoa1412965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garafalo A.V., Cideciyan A.V., Héon E., Sheplock R., Pearson A., WeiYang Yu C., Sumaroka A., Aguirre G.D., Jacobson S.G. Progress in treating inherited retinal diseases: Early subretinal gene therapy clinical trials and candidates for future initiatives. Prog. Retin. Eye Res. 2020;77:100827. doi: 10.1016/j.preteyeres.2019.100827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beltran W.A., Cideciyan A.V., Guziewicz K.E., Iwabe S., Swider M., Scott E.M., Savina S.V., Ruthel G., Stefano F., Zhang L. Canine retina has a primate fovea-like bouquet of cone photoreceptors which is affected by inherited macular degenerations. PLoS ONE. 2014;9:e90390. doi: 10.1371/journal.pone.0090390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hermonat P.L., Quirk J.G., Bishop B.M., Han L. The packaging capacity of adeno-associated virus (AAV) and the potential for wild-type-plus AAV gene therapy vectors. FEBS Lett. 1997;407:78–84. doi: 10.1016/s0014-5793(97)00311-6. [DOI] [PubMed] [Google Scholar]
- 34.Pang J., Boye S.E., Lei B., Boye S.L., Everhart D., Ryals R., Umino Y., Rohrer B., Alexander J., Li J. Self-complementary AAV-mediated gene therapy restores cone function and prevents cone degeneration in two models of Rpe65 deficiency. Gene Ther. 2010;17:815–826. doi: 10.1038/gt.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koilkonda R.D., Yu H., Chou T.H., Feuer W.J., Ruggeri M., Porciatti V., Tse D., Hauswirth W.W., Chiodo V., Boye S.L. Safety and effects of the vector for the Leber hereditary optic neuropathy gene therapy clinical trial. JAMA Ophthalmol. 2014;132:409–420. doi: 10.1001/jamaophthalmol.2013.7630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Komáromy A.M., Rowlan J.S., Corr A.T., Reinstein S.L., Boye S.L., Cooper A.E., Gonzalez A., Levy B., Wen R., Hauswirth W.W. Transient photoreceptor deconstruction by CNTF enhances rAAV-mediated cone functional rescue in late stage CNGB3-achromatopsia. Mol. Ther. 2013;21:1131–1141. doi: 10.1038/mt.2013.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guziewicz K.E., Cideciyan A.V., Beltran W.A., Komáromy A.M., Dufour V.L., Swider M., Iwabe S., Sumaroka A., Kendrick B.T., Ruthel G. BEST1 gene therapy corrects a diffuse retina-wide microdetachment modulated by light exposure. Proc. Natl. Acad. Sci. USA. 2018;115:E2839–E2848. doi: 10.1073/pnas.1720662115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beltran W.A., Cideciyan A.V., Boye S.E., Ye G.J., Iwabe S., Dufour V.L., Marinho L.F., Swider M., Kosyk M.S., Sha J. Optimization of Retinal Gene Therapy for X-Linked Retinitis Pigmentosa Due to RPGR Mutations. Mol. Ther. 2017;25:1866–1880. doi: 10.1016/j.ymthe.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bruewer A.R., Mowat F.M., Bartoe J.T., Boye S.L., Hauswirth W.W., Petersen-Jones S.M. Evaluation of lateral spread of transgene expression following subretinal AAV-mediated gene delivery in dogs. PLoS ONE. 2013;8:e60218. doi: 10.1371/journal.pone.0060218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanke-Gogokhia C., Chiodo V.A., Hauswirth W.W., Frederick J.M., Baehr W. Rescue of cone function in cone-only Nphp5 knockout mouse model with Leber congenital amaurosis phenotype. Mol. Vis. 2018;24:834–846. [PMC free article] [PubMed] [Google Scholar]
- 41.Horton J.C., Parker A.B., Botelho J.V., Duncan J.L. Spontaneous Regeneration of Human Photoreceptor Outer Segments. Sci. Rep. 2015;5:12364. doi: 10.1038/srep12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Young R.W. A difference between rods and cones in the renewal of outer segment protein. Invest. Ophthalmol. 1969;8:222–231. [PubMed] [Google Scholar]
- 43.Beltran W.A., Cideciyan A.V., Iwabe S., Swider M., Kosyk M.S., McDaid K., Martynyuk I., Ying G.S., Shaffer J., Deng W.T. Successful arrest of photoreceptor and vision loss expands the therapeutic window of retinal gene therapy to later stages of disease. Proc. Natl. Acad. Sci. USA. 2015;112:E5844–E5853. doi: 10.1073/pnas.1509914112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goldstein O., Mezey J.G., Schweitzer P.A., Boyko A.R., Gao C., Bustamante C.D., Jordan J.A., Aguirre G.D., Acland G.M. IQCB1 and PDE6B mutations cause similar early onset retinal degenerations in two closely related terrier dog breeds. Invest. Ophthalmol. Vis. Sci. 2013;54:7005–7019. doi: 10.1167/iovs.13-12915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.al-Ubaidi M.R., Font R.L., Quiambao A.B., Keener M.J., Liou G.I., Overbeek P.A., Baehr W. Bilateral retinal and brain tumors in transgenic mice expressing simian virus 40 large T antigen under control of the human interphotoreceptor retinoid-binding protein promoter. J. Cell Biol. 1992;119:1681–1687. doi: 10.1083/jcb.119.6.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boatright J.H., Borst D.E., Peoples J.W., Bruno J., Edwards C.L., Si J.S., Nickerson J.M. A major cis activator of the IRBP gene contains CRX-binding and Ret-1/PCE-I elements. Mol. Vis. 1997;3:15. [PubMed] [Google Scholar]
- 47.Beltran W.A., Cideciyan A.V., Lewin A.S., Iwabe S., Khanna H., Sumaroka A., Chiodo V.A., Fajardo D.S., Román A.J., Deng W.T. Gene therapy rescues photoreceptor blindness in dogs and paves the way for treating human X-linked retinitis pigmentosa. Proc. Natl. Acad. Sci. USA. 2012;109:2132–2137. doi: 10.1073/pnas.1118847109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zolotukhin S., Byrne B.J., Mason E., Zolotukhin I., Potter M., Chesnut K., Summerford C., Samulski R.J., Muzyczka N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
- 49.Zolotukhin S., Potter M., Zolotukhin I., Sakai Y., Loiler S., Fraites T.J., Jr., Chiodo V.A., Phillipsberg T., Muzyczka N., Hauswirth W.W. Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods. 2002;28:158–167. doi: 10.1016/s1046-2023(02)00220-7. [DOI] [PubMed] [Google Scholar]
- 50.Fagone P., Wright J.F., Nathwani A.C., Nienhuis A.W., Davidoff A.M., Gray J.T. Systemic errors in quantitative polymerase chain reaction titration of self-complementary adeno-associated viral vectors and improved alternative methods. Hum. Gene Ther. Methods. 2012;23:1–7. doi: 10.1089/hgtb.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Komáromy A.M., Alexander J.J., Rowlan J.S., Garcia M.M., Chiodo V.A., Kaya A., Tanaka J.C., Acland G.M., Hauswirth W.W., Aguirre G.D. Gene therapy rescues cone function in congenital achromatopsia. Hum. Mol. Genet. 2010;19:2581–2593. doi: 10.1093/hmg/ddq136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Komáromy A.M., Varner S.E., de Juan E., Acland G.M., Aguirre G.D. Application of a new subretinal injection device in the dog. Cell Transplant. 2006;15:511–519. doi: 10.3727/000000006783981701. [DOI] [PubMed] [Google Scholar]
- 53.Garcia M.M., Ying G.S., Cocores C.A., Tanaka J.C., Komáromy A.M. Evaluation of a behavioral method for objective vision testing and identification of achromatopsia in dogs. Am. J. Vet. Res. 2010;71:97–102. doi: 10.2460/ajvr.71.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ray K., Baldwin V.J., Acland G.M., Blanton S.H., Aguirre G.D. Cosegregation of codon 807 mutation of the canine rod cGMP phosphodiesterase β gene and rcd1. Invest. Ophthalmol. Vis. Sci. 1994;35:4291–4299. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.