
Figure 1. Overview of the online iNMF algorithm.
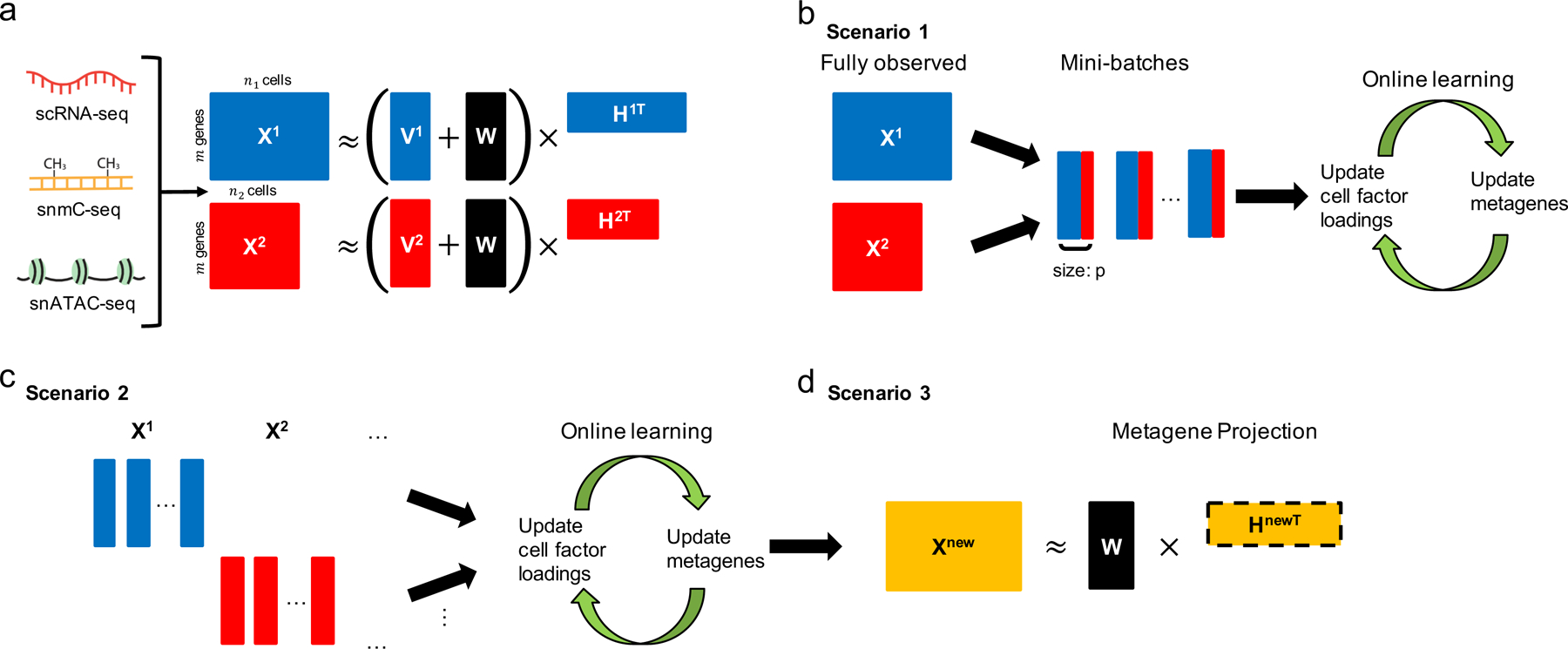
a, Schematic of integrative nonnegative matrix factorization (iNMF): the input single-cell datasets are jointly decomposed into shared and dataset-specific metagenes and corresponding “metagene expression levels” or cell factor loadings . These metagenes and cell factor loadings provide a quantitative definition of cell identity and how it varies across biological settings. b-d, Three different scenarios in which online learning can be used for single-cell data integration. (b) Scenario 1: the single-cell datasets are large but fully observed. Online iNMF processes the data in random mini-batches, enabling memory usage and/or disk storage independent of dataset size. Each cell may be used multiple times in different epochs of training to update the metagenes. (c) Scenario 2: the datasets arrive sequentially, and online iNMF processes the datasets as they arrive, using each cell to update the metagenes exactly once. (d) Scenario 3: online iNMF is performed as in scenario 1 or scenario 2 to learn and . Then cell factor loadings for the newly arriving dataset are calculated using the shared metagenes learned from previously processed datasets. The new dataset is not used to update the metagenes.