
Figure 3. Composition of FDA-approved ADCs.
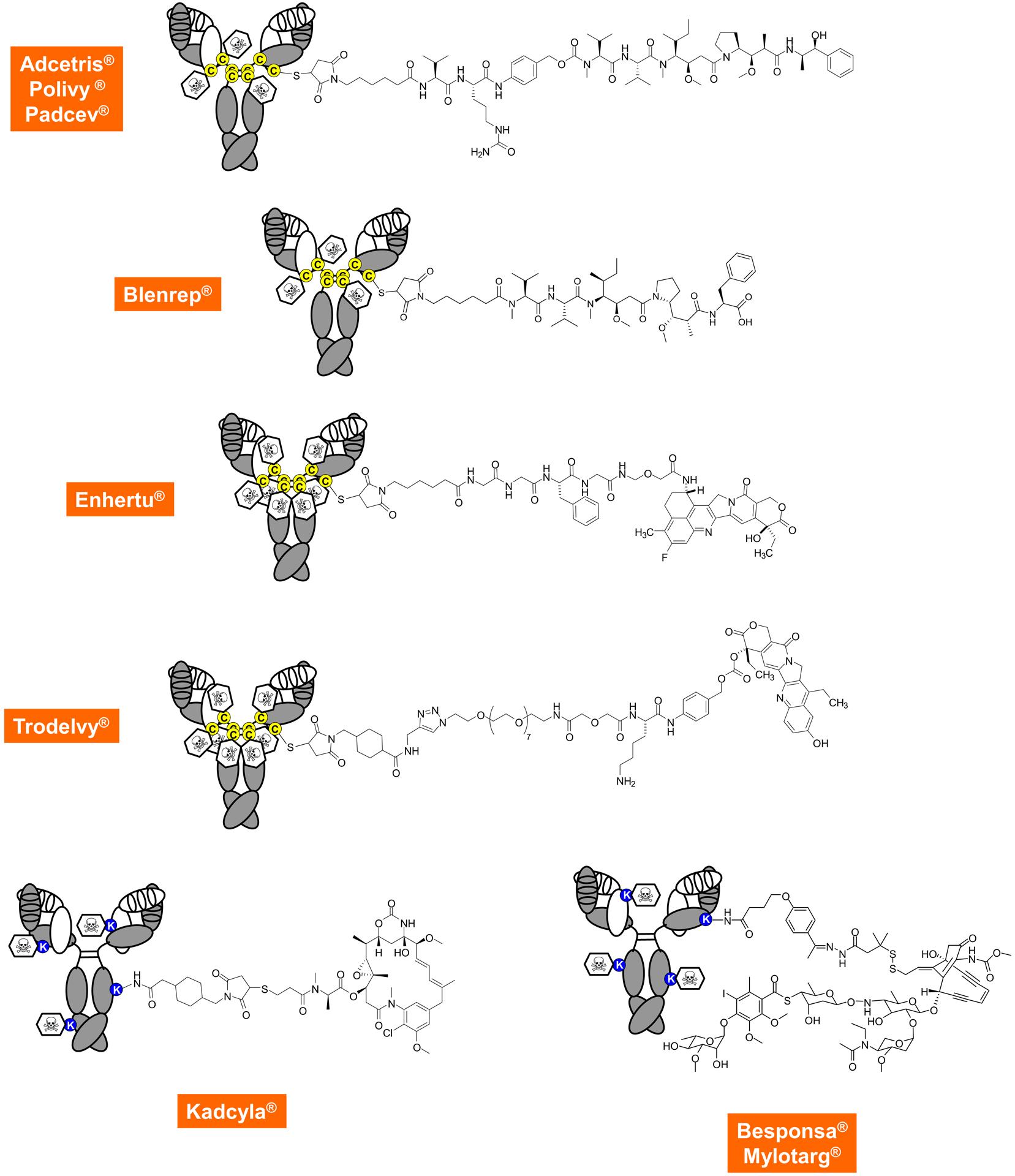
The top row shows first-generation heterogeneous ADCs targeting CD30, CD79B, and NECTIN4, respectively, in which the drug is stochastically linked to reduced cysteine residues in the hinge region using a thiosuccinimide, a caproyl spacer, an enzymatically cleavable unit (a valine-citrulline dipeptide fused to a para-aminobenzylcarbamate), followed by the cytotoxic payload which is monomethylauristatin E (MMAE), a subnanomolar inhibitor of tubulin polymerization. The mean DAR is approximately 4, ranging from 0 to 8. The second row shows another auristatin-based and stochastically assembled ADC that targets BCMA and links monomethylauristatin F (MMAF) through a non-cleavable linker to hinge cysteine residues. Recently, the first homogeneous ADCs shown in the third and fourth row received FDA approval. They target HER2 and TROP2, respectively, and use the same thiosuccinimide connector to hinge cysteine residues but rather than doing this stochastically, all eight cysteines are involved. This requires the use of more hydrophilic linkers but, due to their high DAR, also affords slightly less cytotoxic payloads, which are two different camptothecin derivatives that inhibit topoisomerase I. The bottom row shows the three first-generation ADCs that target HER2, CD22, and CD33, respectively, and use surface lysine residues for drug attachment. With 80–90 of these in the average antibody molecule, the location and distribution is even more stochastic. Cytotoxic payloads include tubulin polymerization inhibitor maytansine linked through a non-cleavable linker (left) and the DNA damaging calicheamicin linked through a hydrazone-disulfide linker that gets cleaved in acidic or reducing conditions (right).