

Abstract
Patients with transthyretin amyloid polyneuropathy (ATTR‐PN) show decreased motor and sensory nerve amplitudes and conduction. Electrophysiological changes over time may be sensitive indicators of progression. This analysis from the Transthyretin Amyloidosis Outcomes Survey (THAOS) assessed longitudinal changes in nerve conduction as signals of neurologic disease progression in patients with hereditary ATTR (ATTRv) amyloidosis. Patients with ATTRv in THAOS with recorded nerve conduction values were included (data cut‐off: January 6, 2020); changes in nerve amplitude and velocity over time were assessed. Patients (n = 1389) were 45.0% male; 80.4% were the Val30Met (p.Val50Met) genotype. Mean (SD) age at enrollment was 43.6 (14.5) years; duration of symptoms was 9.3 (6.4) years. Median (10th, 90th percentile) sural nerve amplitude and velocity was 18.0 (4.9, 35.0) μV and 50.7 (41.0, 57.9) m/s; peroneal conduction was 13.0 (4.4, 27.0) μV and 51.0 (41.7, 59.7) m/s, respectively. Median (10th, 90th percentile) percentage change from baseline in sural nerve amplitude was variable, but generally decreased over time from −7.4 (−43.2, 52.4) at year 1 to −14.4 (−76.9, 46.7) at year 8. Percent change from baseline in sural nerve velocity declined similarly: −0.1 (−14.5, 15.3) at year 1 and − 6.4 (−21.3, 10.5) at year 8. The decline was more pronounced in patients with greater disability at baseline. Similar patterns were observed for the peroneal nerve. These data show an association between nerve amplitudes and velocities and disease severity, suggesting progressive deterioration in nerve conduction may be an indicator of ATTRv amyloidosis disease progression.
Keywords: ATTR‐PN, nerve amplitude, nerve conduction, transthyretin amyloid polyneuropathy
1. INTRODUCTION
The rare, progressive, life‐threatening disorder transthyretin amyloid polyneuropathy (ATTR‐PN) is characterized by an increasingly incapacitating sensorimotor polyneuropathy and autonomic dysfunction.1 ATTR‐PN is an autosomal dominant inherited form of ATTR amyloidosis, caused by the systemic deposition of transthyretin amyloid fibrils in peripheral nerves and vital organs, leading to advanced polyneuropathy.2, 3, 4, 5 It is caused by mutations in the transthyretin (TTR) gene,4 and diagnosis and monitoring of disease progression is complicated by the considerable phenotypic heterogeneity seen among patients with the disease.
In the early stages of ATTR‐PN, amyloid fibrils usually affect the small nerve fibers,2, 6, 7 as evidenced by altered pain and temperature sensation, but relatively low impact on light touch, and with little effect seen on awareness of the position and movement of the body.2, 7 Muscle strength and tendon reflexes remain normal in the early stages of disease.2 However, as the disease progresses, ATTR‐PN presents with altered motor and sensory nerve conduction; namely, a decrease in nerve amplitude and velocity.8 Larger sensory and motor nerve fibers are affected as ATTR‐PN progresses further, with motor deficits apparent in the distal lower limbs, and weakening of light touch and deep sensations.2 Subsequently, patients experience increased difficulty in walking without assistance, due to imbalance and gait issues. Previous nerve conduction studies have indicated that sensory fibers are more affected than motor fibers: sensory nerve conduction amplitudes can be reduced or even nonexistent, while motor nerve conduction amplitudes can remain normal, or be reduced.7, 8 Conduction velocities have been reported as ranging from normal to only slightly reduced.7 Due to the effects of amyloid fibrils on the peripheral nerves, further characterization of electrophysiological changes over time may be useful in assessing disease progression.
The Transthyretin Amyloidosis Outcomes Survey (THAOS) is the largest ongoing, global, longitudinal, observational survey of patients with ATTR amyloidosis, including both inherited (ATTRv) and wild‐type disease, and asymptomatic patients with TTR mutations (ClinicalTrials.gov: NCT00628745).9 THAOS allows the opportunity to examine the potential of exploratory indicators of ATTR‐PN disease progression.9 The primary objective of this analysis was to assess whether nerve conduction can serve as a marker of disease progression in patients with ATTR‐PN.
2. MATERIALS AND METHODS
2.1. Trial design and patients
All patients with a pathogenic TTR disease causing mutation in THAOS with any sural or peroneal amplitude or velocity value (left or right) non‐missing and greater than 0 at any time were included in the analysis (data cut‐off: 06 January 2020). If the nerve conduction measurement was taken on both the left and right side, the average of the two values was used for the analysis. Demographic and clinical characteristics were reported using descriptive statistics.
2.2. Nerve conduction studies
Nerve conduction assessments are standardized in THAOS. The amplitude and velocity of sural and peroneal nerves was measured base to peak at annual visits. Data are shown as mean and median values and overall percentage change at each year post baseline, from years 1 to 8. Investigators recorded whether these were measured at left, right, or both sides, along with the amplitude and velocity recorded. Results were also stratified by modified polyneuropathy disability (mPND) score, to examine the relationship with disease severity and nerve conduction. mPND is scored from I to IV (I indicates sensory disturbance in lower limbs but preserved walking capacity; II indicates difficulties in walking but no need of a walking stick; IIIa indicates one stick or one crutch required for walking; IIIb indicates two sticks or two crutches required for walking; IV indicates patient confined to a wheelchair or bed. For those patients who were symptomatic, but for whom no lower limb sensory/motor deficits were recorded, an mPND score of 0 was recorded.10
3. RESULTS
3.1. Patient characteristics
There were 3371 patients with ATTRv amyloidosis in the overall population (55.5% male). The most common genotypes were Val30Met (p.Val50Met; 63.4%), Val122Ile (p.Val142Ile; 8.3%) and Thr60Ala (p.Thr80Ala; 3.4%), and most patients came from Portugal (35.6%), United States (14.1%) and Brazil (6.9%).
A total of 1389/3371 patients with ATTRv amyloidosis represented the subset of those included in the analysis with any sural or peroneal amplitude or velocity value non‐missing and greater than 0 at any time (Table 1). Almost half of the patients were male (45.0%), and the predominant TTR genotype was Val30Met (80.4%). The mean (SD) age at onset of ATTR amyloidosis symptoms was 39.7 (13.7), with a mean (SD) duration of symptoms of 9.3 (6.4) years (Table 2). The majority of patients came from Portugal (65.7%), followed by France (7.1%), Brazil (6.6%), and Spain (6.0%; Table 1).
TABLE 1.
Demographic characteristics of patients with ATTRv amyloidosis and nerve conduction studies at enrollment in THAOS
| All patients (N = 1389) | |
|---|---|
| Age at enrollment, mean (SD), years | 43.6 (14.5) |
| Sex, n (%) | |
| Male | 625 (45.0) |
| Female | 764 (55.0) |
| TTR genotype, n (%) | |
| Val30Met (p.Val50Met) | 1117 (80.4) |
| Glu89Gln (p.Glu109Gln) | 61 (4.4) |
| Val122Ile (p.Val142Ile) | 33 (2.4) |
| Ser77Tyr (p.Ser97Tyr) | 19 (1.4) |
| Asp38Ala (p.Asp58Ala) | 16 (1.2) |
| Ser50Arg (p.Ser70Arg) | 15 (1.1) |
| Phe64Leu (p.Phe84Leu) | 14 (1.0) |
| Thr60Ala (p.Thr80Ala) | 14 (1.0) |
| Ser77Phe (p.Ser97Phe) | 10 (0.7) |
| Othera | 90 (6.5) |
| Country, n (%)b | |
| Argentina | 8 (0.6) |
| Brazil | 92 (6.6) |
| Bulgaria | 57 (4.1) |
| France | 99 (7.1) |
| Germany | 12 (0.9) |
| Italy | 35 (2.5) |
| Japan | 8 (0.6) |
| Mexico | 17 (1.2) |
| Portugal | 913 (65.7) |
| South Korea | 26 (1.9) |
| Spain | 83 (6.0) |
| United Sates | 32 (2.3) |
| Race, n (%) | |
| Caucasian | 357 (25.7) |
| Asian | 34 (2.4) |
| Other | 32 (2.3) |
| Latino American | 28 (2.0) |
| African descentc | 12 (0.9) |
| American Hispanic | 3 (0.2) |
| Missing | 923 (66.5) |
Abbreviations: ATTR‐PN, transthyretin amyloidosis with polyneuropathy; THAOS, Transthyretin Amyloidosis Outcomes Survey; TTR, transthyretin.
Other includes all genotypes contributing <10 patients: Ala109Ser (p.Ala129Ser); Ala120Ser (p.Ala140Ser); Ala19Asp (p.Ala39Asp); Ala19Asp (p.Ala39Asp)/Gly6Ser (p.Gly26Ser); Arg34Ser (p.Arg54Ser); Asp18Asn (p.Asp38Asn); Glu54Gln (p.Glu74Gln); Glu54Leu (p.Glu74Leu); Glu54Ser (p.Glu74Ser); Glu61Lys (p.Glu81Lys); Glu62Lys (p.Glu82Lys); Glu89Lys (p.Glu109Lys); Gly47Ala (p.Gly67Ala); Gly47Glu (p.Gly67Glu); Gly6Ser/Val30Met; His88Srg (p.His108Arg); Ile107Val (p.Ile127Val); Ile68Leu (p.Ile88Leu); Ile73Val (p.Ile93Val); Leu58His (p.Leu78His); Lys35Asn (p.Lys55Asn); Met13dup (p.Met33dup); Phe33Leu (p.Phe53Leu); Ser23Asn (p.Ser43Asn); Ser52Pro (p.Ser72Pro); Thr119Met (p.Thr139Met)/Val30Met; Thr49Ile (p.Thr69Ile)/Gly6Ser (p.Gly26Ser); Thr59Lys (p.Thr79Lys); Thr75Ile (p.Thr95Ile); Tyr116Ser (p.Tyr136Ser); Tyr69His (p.Tyr89His); Val28Met (p.Val48Met); Val30Met (p.Val50Met)/Pmp22Del (p.Pmp42Del); Val71Ala (p.Val91Ala); delVal122 (p.delVal142); p.Met33dup, p.Asp119Asn.
Countries contributing ≤5 patients not shown: Belgium, n = 1; the Netherlands, n = 4; Romania, n = 1; Turkey, n = 1.
African descent includes African American and Afro‐Caribbean.
TABLE 2.
Clinical characteristics of patients with ATTRv amyloidosis and nerve conduction studies at enrollment in THAOS
| All patients (N = 1389) | |
|---|---|
| Age at onset of symptoms, mean (SD), years | 39.7 (13.7) |
| Duration of symptoms, mean (SD), years | 9.3 (6.4) |
| Time from symptom onset to diagnosis, mean (SD), years | 4.0 (5.7) |
| Sural nerve conduction | |
| Mean (SD) | |
| Amplitude, μV | 19.2 (12.4), n = 1225 |
| Velocity, m/s | 50.7 (28.2), n = 1221 |
| Median (10th, 90th percentile) | |
| Amplitude, μV | 18.0 (4.9, 35.0) |
| Velocity, m/s | 50.7 (41.0, 57.9) |
| Peroneal nerve conduction | |
| Mean (SD) | |
| Amplitude, mV | 16.4 (34.3), n = 920 |
| Velocity, m/s | 51.7 (32.1), n = 918 |
| Median (10th, 90th percentile) | |
| Amplitude, mV | 13.0 (4.4, 27.0) |
| Velocity, m/s | 51.0 (41.7, 59.7) |
Abbreviations: ATTR‐PN, transthyretin amyloidosis with polyneuropathy; THAOS, Transthyretin Amyloidosis Outcomes Survey.
3.2. Nerve conduction at enrollment
At enrollment, the mean (SD) recorded sural nerve sensory nerve action potential (SNAP) amplitude was 19.2 (12.4) μV and the velocity was 50.7 (28.2) m/s (Table 2).
Regarding compound muscle action potential (CMAP), the mean (SD) peroneal nerve amplitude was 16.4 (34.3) mV, and the velocity was 51.7 (32.1) m/s.
When the SNAP and CMAP values were stratified by mPND score, the pattern of an inverse relationship was observed; in general, progressive decreases were seen as disability increased (Table 3).
TABLE 3.
Sural and peroneal nerve conduction at enrollment by latest mPND score in patients with ATTRv amyloidosis and nerve conduction studies in THAOS
| Nerve | Amplitude (μV or mV)a | Velocity (m/s) |
|---|---|---|
| Sural nerve conduction, median (10th, 90th percentile) | ||
| mPND score | ||
| 0 | 22.0 (10.0, 39.0), n = 302 | 52.5 (44.3, 60.0), n = 301 |
| I | 17.5 (5.4, 33.0), n = 682 | 50.9 (41.2, 57.5), n = 682 |
| II | 10.0 (2.5, 32.5), n = 138 | 46.5 (38.0, 53.6), n = 136 |
| IIIa | 8.6 (2.4, 21.0), n = 27 | 43.8 (37.0, 51.6), n = 27 |
| IIIb | 4.1 (0.8, 50.3), n = 10 | 39.7 (4.0, 56.9), n = 9 |
| IV | 2.5 (0.6, 10.0), n = 5 | 43.0 (33.0, 46.0), n = 5 |
| Peroneal nerve conduction, median (10th, 90th percentile) | ||
| mPND score | ||
| 0 | 16.0 (7.6, 31.0), n = 225 | 52.4 (44.3, 59.5), n = 225 |
| I | 13.2 (4.7, 26.0), n = 524 | 51.1 (42.3, 60.0), n = 524 |
| II | 7.5 (2.0, 25.0), n = 102 | 45.6 (37.5, 56.1), n = 101 |
| IIIa | 12.0 (1.7, 20.0), n = 15 | 47.8 (38.0, 52.2), n = 15 |
| IIIb | 5.0 (0.9, 25.0), n = 7 | 54.0 (4.6, 63.9), n = 7 |
| IV | 2.9 (1.4, 8.1), n = 3 | 45.0 (37.5, 45.7), n = 3 |
Note: mPND score: 0, symptomatic, but no lower limb sensory/motor deficits; I, sensory disturbance in lower limbs but preserved walking capacity; II, difficulties in walking but no need of a walking stick; IIIa, one stick or one crutch required for walking; IIIb, two sticks or two crutches required for walking; IV, patient confined to a wheelchair or bed.
Abbreviations: mPND, modified polyneuropathy disability.
μV for sural nerve and mV for peroneal nerve.
3.3. Nerve conduction over the duration of the study
Across annual visits, the median (10th, 90th percentile) percentage change from baseline in sural nerve SNAP amplitude (μV) generally decreased (year 1: −7.4 [−43.2, 52.4], n = 797; year 8: −14.4 [−76.9, 46.7], n = 168). A concomitant decline in velocity (m/s) was also seen (year 1: −0.1 [−14.5, 15.3], n = 796; year 8: −6.4 [−21.3, 10.5], n = 168) (Figure 1); however, in general, changes were relatively small and variable.
FIGURE 1.
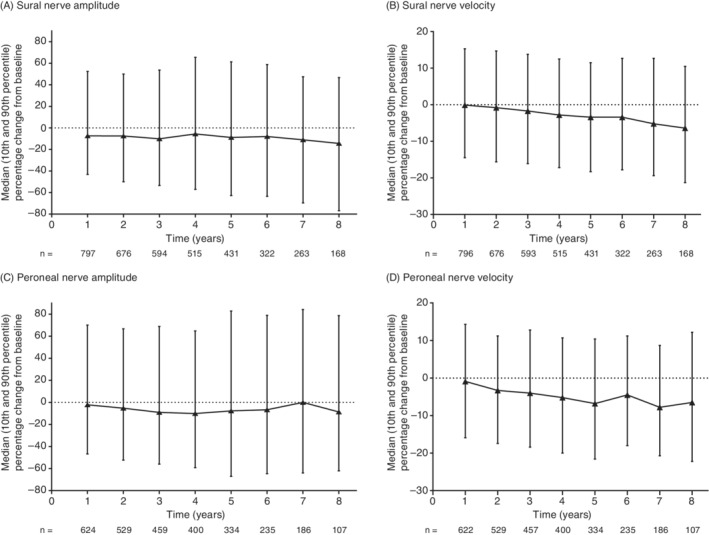
Median (10th and 90th percentile) percentage change from baseline in sural and peroneal nerve amplitude and velocity in patients with ATTRv amyloidosis and nerve conduction studies. Change from baseline (enrollment) is the baseline value subtracted from the follow‐up value, where both are non‐missing. The data presented are cross‐sectional by follow‐up time in THAOS. ATTRv, hereditary transthyretin amyloidosis; THAOS, Transthyretin Amyloidosis Outcomes Survey
Similar patterns were observed for peroneal nerve CMAP amplitude (mV) (year 1: −2.2 [−46.7, 70.0], n = 624; year 8: −8.7 [−62.1, 78.6], n = 107) and velocity (m/s) (year 1: −0.9 [−15.9, 14.3], n = 622; year 8: −6.5 [−22.2, 12.2], n = 107) (Figure 1).
When the percentage change‐from‐baseline values were stratified by mPND score (0, I, II; higher levels not shown due to low sample sizes [n = 0 or 1]), larger decreases in amplitude and velocity relative to baseline were observed as disability increased (Figure 2).
FIGURE 2.
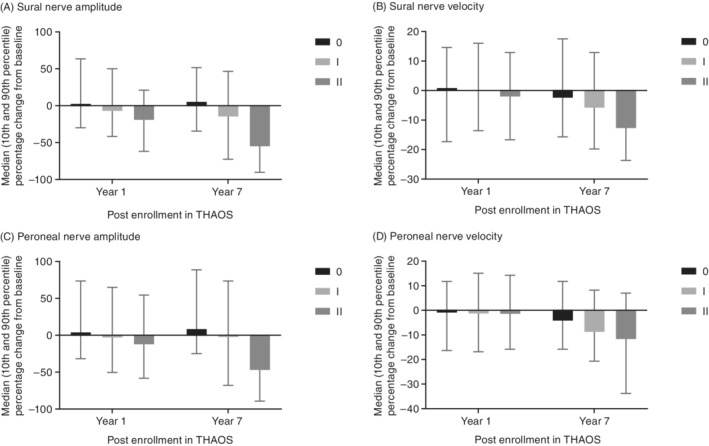
Median (10th and 90th percentile) percentage change from baseline in sural and peroneal nerve conduction by latest mPND score in patients with ATTRv amyloidosis and nerve conduction studies. mPND score IIIa, IIIb, and IV values are either not available (no patients with data) or not shown due to low n (n = 1). Data from year 8 not shown due to small number of patients in certain subgroups. ATTRv, hereditary transthyretin amyloidosis; mPND, modified polyneuropathy disability; THAOS, Transthyretin Amyloidosis Outcomes Survey
4. DISCUSSION
This large, exploratory analysis of patients with ATTR‐PN in THAOS suggests that the amplitude and velocity of sural and peroneal nerve conduction may serve as a sensitive marker of neurologic disease progression. The analysis revealed a decrease over time in these parameters, particularly in patients with more severe disease. Median (10th, 90th percentile) percentage change from baseline in sural nerve amplitude was variable, but generally decreased over time from year 1 to year 8. Velocity broadly declined over time, and similar patterns were observed for the peroneal nerve. The decrease over time to year 7 was more pronounced in patients with higher mPND score at baseline. Progressive deterioration in nerve conduction may be a useful indicator of ATTR‐PN disease progression.
Nerve amplitude and velocity are known to be lower, and to deteriorate over time in ATTR‐PN,7, 8 with worsening of nerve conduction in serial measurements reported previously.11 A study examining electrophysical features of patients with ATTR‐PN Val30Met (44 cases of late‐onset compared with 21 cases of early‐onset ATTR‐PN) indicated lower amplitude and velocity regardless of time of onset of ATTR‐PN.8 Mean (SD) sural nerve amplitudes were lower in both patients with late‐onset (0.4 [1.4] μV vs 14.9 [± 9.3] μV in healthy controls) and patients with early‐onset (2.8 μV ± 5.0 vs 18.6 μV ± 9.7 in healthy controls).8 Sural nerve velocity was also lower in patients with ATTR‐PN (although to a lesser degree than amplitude), and similar patterns were seen with motor nerve amplitudes and conduction velocity (measured using the tibial and median nerves).8 The variability observed in this analysis is consistent with earlier reports noting diversity in baseline nerve conduction measurements in ATTR‐PN,11 and with the variability of repeated nerve conduction measurements in general.12 This analysis adds to the available data, showing a direct association between reduced nerve amplitude and conduction velocity and disease severity. The data also suggest that nerve amplitude and velocity worsen further over time.
While evidence suggests that nerve amplitude reductions can be related to aging13; patients with ATTR‐PN have lower nerve amplitude and velocities compared with age‐matched healthy individuals, and nerve conduction parameters reduce steadily over time.7, 8, 11 In general, the slowing of nerve conduction seen in this study was not in the range of classic demyelination criteria14 and more likely is related to fiber loss and amplitude decrease. This analysis is limited by the fact that nerve conduction measures are unable to detect small fiber neuropathy,15 the predominant early sign of neuropathy in patients with ATTR‐PN.15, 16 Various other techniques for assessing small fiber neuropathy have been examined, with sudometers showing promise,17, 18, 19 though requiring further validation in larger cohorts. However, it is important to consider that most patients in this cohort had mild–moderate neurologic disease. Additionally, nerve conduction studies were used in our analysis to allow examination of the development of more severe progression in these patients. Patients were excluded from the analysis if they did not have a SNAP >0 which may have introduced some bias in the assessments. The changes observed in this study were small, and relatively variable; this may be due to the heterogeneity of the disease in general.4 Patients with ATTR‐PN may display patterns of nerve amplitude and conduction that can lead to a misdiagnosis of chronic inflammatory demyelinating polyneuropathy. In particular, this misdiagnosis can occur in patients with ATTR‐PN experiencing electrophysical demyelinating features without reduction in nerve conduction,20, 21 or severe axonal degeneration,22 where no biopsy is performed, or the results of the biopsy are negative.20, 23 While the impact of disease‐modifying treatment on nerve conduction would be interesting to examine, limitations in this THAOS dataset precluded comparative analysis of treated vs untreated patients.
Overall, this large analysis from THAOS provides further evidence that the amplitude and velocity of the sural and peroneal nerve conduction may be a useful indicator for assessing ATTR‐PN disease progression. Monitoring patients closely for the earliest signs of advancing disease may improve the clinical management of patients with ATTR‐PN.
CONFLICT OF INTEREST
Márcia Waddington‐Cruz received research funding, consulting fees, and travel support for advisory boards and meetings from FoldRx Pharmaceuticals and Pfizer. Yukio Ando declares receipt of consulting fee or honorarium, support for travel to meetings, and provision for writing assistance from Pfizer. Yoshiki Sekijima has received royalties from Pfizer related to tafamidis patents and has received speaker honoraria from Pfizer. Leslie Amass, Doug Chapman, and Jan Kiszko are full‐time employees of Pfizer and hold stock and/or stock options with Pfizer.
AUTHOR CONTRIBUTIONS
All authors contributed to the design and conduct of the analysis; interpretation of the data; and preparation, review, and approval of the manuscript.
ACKNOWLEDGEMENTS
The THAOS registry and this analysis were sponsored by Pfizer. We thank all THAOS patients and investigators for their important contributions to this study. We thank Rajiv Mundayat for valuable assistance with statistical analysis and interpretation. Medical writing support was provided by Caitlin Watson, PhD, of Engage Scientific Solutions, and funded by Pfizer.
Waddington‐Cruz M, Ando Y, Amass L, et al. Feasibility of assessing progression of transthyretin amyloid polyneuropathy using nerve conduction studies: Findings from the Transthyretin Amyloidosis Outcomes Survey (THAOS). J Peripher Nerv Syst. 2021;26:160–166. 10.1111/jns.12444
Additional THAOS investigators contributing to this analysis are: Sorina Badelita, Institutul De Cardiologie, C. C. Iliescu Spitalului Fundeni, Bucuresti, Romania; Fabio Adrian Barroso, FLENI, Ciudad Autonoma de Buenos Aires, Argentina; Teresa Coelho, Unidade Clinica de Paramiloidose‐Centro Hospitalar Porto, EPE‐Hospital Geral Santo Antonio, Porto, Portugal; Isabel Conceição, Hospital Santa Maria, Lisboa, Portugal; Felix Darstein, Johann‐Gutenberg‐Universität, Mainz, Germany; Angela Dispenzieri, Mayo Clinic, Rochester, United States; Roberto Fernandéz Torrón, Hospital Universitario Donostia, Gipuzkoa ‐ SanSebastian, Spain; Pablo Garcia Pavia, Hospital Universitario Puerta de Hierro, Majadahonda, Spain; Burkhard Gess, University Hospital of RWTH Aachen, Aachen, Germany; Jose Gonzalez Costello, Hospital Universitari de Bellvitge, Barcelona, Spain; Maria Alejandra Gonzalez Duarte Briseno, Instituto Nacional de Ciencia Medicas y Nutricion Salvador Zubiran, Distrito Federal, Mexico; Juan Gonzalez Moreno, Hospital Son Llatzer, Palma de Mallorca, Spain; Stephen Gottlieb, University of Maryland, Baltimore, United States; Eun‐Seok Jeon, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Republic of Korea; Arnt Kristen, Medical University of Heidelberg, Heidelberg, Germany; Samantha, LoRusso, The Ohio University College of Medicine, Columbus, United States; Marco Luigetti, Fondazione Policlinico Gemelli ‐ Universita Cattolica del Sacro Cuore, Roma, Italy; Tessa Marburger, Oregon Health and Science University, Portland, United States; Francisco Munoz Beamud, Hospital Juan Ramon Jimenez, Huelva, Spain; Hans Nienhuis, University Medical Center Groningen, Groningen, the Netherlands; Jeeyoung Oh, Konkuk University Medical Center, Seoul, Republic of Korea; Yesim Parman, Istanbul University, Istanbul Faculty of Medicine, Department of Neurology, Istanbul, Turkey; Violaine Plante‐Bordeneuve, CHU Henri Mondor, Créteil, France; Michael Polydefkis, Johns Hopkins Hospital, Baltimore, United States; Dianna Quan, UC Denver, Aurora, United States; Hartmut Schmidt, Universitatsklinikum Münster ‐ Transplant Hepatology, Münster, Germany; Ivaylo Tarnev, Alexandrovska University Hospital Clinic of Neurology, Sofia, Bulgaria; Johan Van Cleemput, Afdeling Klinische Cardiologie, O&N I, Leuven, Belgium; Giuseppe Vita, AOU Policlinico G. Martino, Messina, Italy.
Funding information Pfizer
DATA AVAILABILITY STATEMENT
Pfizer provides secure access to anonymized patient‐level data to qualified researchers in response to scientifically valid research proposals. Further details can be found at https://www.pfizer.com/science/clinical-trials/trial-data-and-results.
REFERENCES
- 1.Yamashita T, Ando Y, Okamoto S, et al. Long‐term survival after liver transplantation in patients with familial amyloid polyneuropathy. Neurology. 2012;78:637‐643. [DOI] [PubMed] [Google Scholar]
- 2.Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin‐related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med. 1997;337:898‐909. [DOI] [PubMed] [Google Scholar]
- 4.Planté‐Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011;10:1086‐1097. [DOI] [PubMed] [Google Scholar]
- 5.Planté‐Bordeneuve V. Transthyretin familial amyloid polyneuropathy: an update. J Neurol. 2018;265:976‐983. [DOI] [PubMed] [Google Scholar]
- 6.Koike H, Ikeda S, Takahashi M, et al. Schwann cell and endothelial cell damage in transthyretin familial amyloid polyneuropathy. Neurology. 2016;87:2220‐2229. [DOI] [PubMed] [Google Scholar]
- 7.Shin SC, Robinson‐Papp J. Amyloid neuropathies. Mt Sinai J Med. 2012;79:733‐748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koike H, Kawagashira Y, Iijima M, et al. Electrophysiological features of late‐onset transthyretin Met30 familial amyloid polyneuropathy unrelated to endemic foci. J Neurol. 2008;255:1526‐1533. [DOI] [PubMed] [Google Scholar]
- 9.Planté‐Bordeneuve V, Suhr OB, Maurer MS, White B, Grogan DR, Coelho T. The Transthyretin Amyloidosis Outcomes Survey (THAOS) registry: design and methodology. Curr Med Res Opin. 2013;29:77‐84. [DOI] [PubMed] [Google Scholar]
- 10.Suhr OB, Holmgren G, Steen L, et al. Liver transplantation in familial amyloidotic polyneuropathy. Follow‐up of the first 20 Swedish patients. Transplantation. 1995;60:933‐938. [PubMed] [Google Scholar]
- 11.Koike H, Tanaka F, Hashimoto R, et al. Natural history of transthyretin Val30Met familial amyloid polyneuropathy: analysis of late‐onset cases from non‐endemic areas. J Neurol Neurosurg Psychiatry. 2012;83:152‐158. [DOI] [PubMed] [Google Scholar]
- 12.Kimura J. Long and short of nerve conduction measures: reproducibility for sequential assessments. J Neurol Neurosurg Psychiatry. 2001;71:427‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palve SS, Palve SB. Impact of aging on nerve conduction velocities and late responses in healthy individuals. J Neurosci Rural Pract. 2018;9:112‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chung T, Prasad K, Lloyd TE. Peripheral neuropathy: clinical and electrophysiological considerations. Neuroimaging Clin N Am. 2014;24:49‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gwathmey KG, Pearson KT. Diagnosis and management of sensory polyneuropathy. BMJ. 2019;365:l1108. [DOI] [PubMed] [Google Scholar]
- 16.Koike H, Misu K, Sugiura M, et al. Pathology of early‐ vs late‐onset TTR Met30 familial amyloid polyneuropathy. Neurology. 2004;63:129‐138. [DOI] [PubMed] [Google Scholar]
- 17.Casellini CM, Parson HK, Richardson MS, Nevoret ML, Vinik AI. Sudoscan, a noninvasive tool for detecting diabetic small fiber neuropathy and autonomic dysfunction. Diabetes Technol Ther. 2013;15:948‐953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luigetti M, Bisogni G, Romano A, et al. Sudoscan in the evaluation and follow‐up of patients and carriers with TTR mutations: experience from an Italian Centre. Amyloid. 2018;25:242‐246. [DOI] [PubMed] [Google Scholar]
- 19.Zouari HG, Ng Wing Tin S, Wahab A, Damy T, Lefaucheur JP. Assessment of autonomic innervation of the foot in familial amyloid polyneuropathy. Eur J Neurol. 2019;26:94‐e10. [DOI] [PubMed] [Google Scholar]
- 20.Cortese A, Vegezzi E, Lozza A, et al. Diagnostic challenges in hereditary transthyretin amyloidosis with polyneuropathy: avoiding misdiagnosis of a treatable hereditary neuropathy. J Neurol Neurosurg Psychiatry. 2017;88:457‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rajabally YA, Adams D, Latour P, Attarian S. Hereditary and inflammatory neuropathies: a review of reported associations, mimics and misdiagnoses. J Neurol Neurosurg Psychiatry. 2016;87:1051‐1060. [DOI] [PubMed] [Google Scholar]
- 22.Ohashi N, Kodaira M, Morita H, Sekijima Y. Electrophysiological demyelinating features in hereditary ATTR amyloidosis. Amyloid. 2019;26:15‐23. [DOI] [PubMed] [Google Scholar]
- 23.Planté‐Bordeneuve V, Ferreira A, Lalu T, et al. Diagnostic pitfalls in sporadic transthyretin familial amyloid polyneuropathy (TTR‐FAP). Neurology. 2007;69:693‐698. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Pfizer provides secure access to anonymized patient‐level data to qualified researchers in response to scientifically valid research proposals. Further details can be found at https://www.pfizer.com/science/clinical-trials/trial-data-and-results.