

Abstract
Herein, we describe an extracellular function of the vertebrate high-mobility group box 1 protein (HMGB1) in the proliferation of bacterial biofilms. Within host cells, HMGB1 functions as a DNA architectural protein, similar to the ubiquitous DNABII family of bacterial proteins; despite that, these proteins share no amino acid sequence identity. Extracellularly, HMGB1 induces a proinflammatory immune response, whereas the DNABII proteins stabilize the extracellular DNA-dependent matrix that maintains bacterial biofilms. We showed that when both proteins converged on extracellular DNA within bacterial biofilms, HMGB1, unlike the DNABII proteins, disrupted biofilms both in vitro (including the high-priority ESKAPEE pathogens) and in vivo in 2 distinct animal models, albeit with induction of a strong inflammatory response that we attenuated by a single engineered amino acid change. We propose a model where extracellular HMGB1 balances the degree of induced inflammation and biofilm containment without excessive release of biofilm-resident bacteria.
Keywords: Immunology, Infectious disease
Keywords: Bacterial infections, Drug therapy, Immunotherapy

Introduction
Bacteria within a human host prefer a community architecture, also known as a biofilm. Biofilms form when free-living (planktonic) bacteria adhere to each other (aggregate biofilms) or adhere to a surface (attached biofilm) and initiate a developmental program that includes alterations in gene expression, intercellular communication, and importantly, production of a self-made extracellular matrix (extracellular polymeric substances or EPSs). The resident bacteria within each of these community architectures are recalcitrant to the host immune system and antimicrobials (1, 2), which enables the bacteria to persist and serve as reservoirs to maintain chronic and recurrent infections. Hence, there is a critical need to develop targeted strategies to resolve bacterial biofilms.
Although the constituent molecules of EPSs vary among bacterial species, extracellular DNA (eDNA) serves as a common underlying structural component throughout diverse bacterial biofilms (3). We recently further characterized the eDNA structure and determined that the eDNA lattice is composed of Holliday junction–like (HJ-like) structures that are integral to the stability of the eDNA-dependent bacterial biofilm EPSs (4). The bacterial DNABII family of DNA-binding proteins, which includes integration host factor (IHF) and histone-like protein (HU), bind to these HJ-like structures within the eDNA lattice and serve as linchpin proteins that maintain the structural integrity of eDNA-dependent EPSs (4). Sequestration of free DNABII proteins via exposure to specific antibodies (α-DNABII) directed against the DNA-binding domain of the DNABII proteins shifts the equilibrium from the eDNA-bound state to the unbound state, which subsequently causes bacterial biofilms to collapse (4–14).
DNABII proteins are absent in vertebrates, but eukaryotes possess a partial functional orthologue, high-mobility group box 1 protein (HMGB1), that binds to similar bent DNA structures such as HJ DNA (15). HMGB1 is a ubiquitous protein in eukaryotes and a native part of the chromatin (16, 17). It functions as a monomer and consists of 2 tandem DNA-binding domains and an acidic C-terminus tail (18) and often has posttranslational modifications that dictate its location (nucleus, cytoplasm, or extracellular) and activity (reviewed in ref. 19). HMGB1 serves as an accessory protein in multiple DNA-protein transactions that include recombination, DNA repair, and transcription via its ability to bind to and bend DNA in a sequence-independent manner (20–22). HMGB1 also functions as a damage-associated molecular pattern molecule that induces a proinflammatory cascade upon its release from eukaryotic cells into the extracellular milieu via the NF-κB pathway by binding to TLR2, TLR4, TLR9, and RAGE (23–26), and thus serves as an alarmin with the potential to cause sepsis, which has devastating consequences for the host (27, 28). Extracellularly, HMGB1 also has a wide array of functions that include tissue regeneration and wound healing; senescence; and at very high concentrations (1.75 μM–12 μM), bacterial killing (29–32). Perhaps most importantly, extracellular HMGB1 is an integral part of the eDNA of neutrophil extracellular traps (NETs), the host’s primary means to sequester bacteria for further elimination (33–36) and, as proposed herein, can additionally serve as a tactic to prevent proliferation of bacterial biofilms.
The eukaryotic host innate-immune effector HMGB1 and the bacterial DNABII proteins serve similar roles as accessory proteins in nucleoprotein transactions in the host and bacteria, respectively (reviewed in refs. 19, 37). HMGB1 and DNABII proteins share no discernible sequence identity or secondary structure, but they can nonetheless functionally replace each other in in vitro transactions. As such, HMGB1 was originally thought to be a functional orthologue of DNABII proteins (38, 39). Although both proteins bind to and bend DNA, they do so via distinct mechanisms. Both HMGB1 (as a monomer) and DNABII proteins (as dimers) bind to DNA via its minor groove; HMGB1 stabilizes the DNA bend from the convex surface, whereas DNABII proteins stabilize the DNA bend from the concave surface (40, 41). Given their extraordinary functional similarities intracellularly, and because HMGB1 and DNABII proteins are also found extracellularly, we were interested in determining the interaction of HMGB1 within the eDNA-dependent EPSs of bacterial biofilms.
In the present study, we examined the effects of HMGB1 on bacterial biofilms. Remarkably, we found that, despite their similarities in DNA substrate preference, DNABII proteins stabilized biofilm structural integrity, whereas HMGB1 disrupted bacterial biofilm structure. We first determined the breadth of antibiofilm activity of HMGB1 against multiple pathogenic biofilms in vitro via an assay against multiple high-priority pathogens followed by assessment of HMGB1’s ability to therapeutically resolve biofilm-mediated infections in 2 distinct animal models of human disease. Moreover, we assessed whether we could engineer HMGB1 to eradicate its proinflammatory activity without loss of its antibiofilm activity via modification of a single key amino acid. We then tested a therapeutic cocktail of host-derived modified HMGB1 plus pathogen-directed antibody against DNABII protein to determine the ability of this cocktail to eradicate biofilms formed by a predominant bacterial pathogen of the human respiratory tract via use of an experimental model of otitis media. Finally, we propose a model to describe the native extracellular functions of HMGB1, now inclusive of its antibiofilm activity.
Results
HMGB1 and DNABII proteins localized to distinct regions on the lattice structure of eDNA within an in vivo biofilm.
Since HMGB1 is bound to DNA within NETs (36), the primary host defense against pathogens, and to demonstrate that HMGB1 and DNABII proteins functionally complement each other in several in vitro transactions by virtue of their shared ability to bind to and bend DNA, we first attempted to localize host HMGB1 and bacterial DNABII proteins within a biofilm that had formed in vivo to begin to characterize the potential role of HMGB1 in host defense against bacterial biofilms. To localize HMGB1 and DNABII proteins within the eDNA-rich matrix of biofilms recovered from the chinchilla middle ear during experimental otitis media induced by nontypeable Haemophilus influenzae (NTHI), biofilm specimens were probed with antibodies specific for either HMGB1 or DNABII proteins. We first identified a region with clear delineation of bacterial biofilm from that of neutrophils/NETs, with an approximately 236 μm region of apparent overlap (Figure 1, A and B). We then used immunofluorescence microscopy to label eDNA (white), DNABII proteins (green), or HMGB1 (red) within the regions of exclusive bacterial biofilm, the overlapping interface, and the region exclusively occupied by neutrophil/NETs (Figure 1, C, D, and G). eDNA lattice can be seen in all 3 panels with DNABII proteins bound to the eDNA exclusively at the vertices of crossed strands of eDNA in the biofilm panel (Figure 1C), whereas HMGB1 bound to eDNA exclusively in the neutrophil/NET panel (Figure 1G). In contrast, at the interface there was clear labeling of both DNABII proteins and HMGB1 on the eDNA. We detected HMGB1 (red) in close proximity to DNABII protein (green) at the crossed strands of eDNA; however, these proteins did not colocalize at the vertices (Figure 1D, would be detectable as yellow) despite what might appear to be a small amount of yellow color within the demarked box in the upper left-hand corner of panel D, which is an artifact due to compression of multiple Z-stack images. This is evident within the individual Z-stack images (Figure 1, E and F). This result suggested that host HMGB1 was indeed incorporated into the eDNA-dependent extracellular matrix of bacterial biofilms, similar to bacterial DNABII proteins. However, unlike DNABII proteins, HMGB1 was never observed at the vertices of crossed strands of eDNA within the lattice, which suggested that it did not stabilize the HJ-like structures, yet another function that is dissimilar to that of the DNABII proteins.
Figure 1. Composite of images that depict the zone between the biofilm and the abundance of PMNs elicited to the site of infection.

(A) Low-magnification light micrograph of an H&E-stained frozen section of a 17-day biofilm produced by nontypeable Haemophilus influenzae (NTHI) in the middle ear of the chinchilla during experimental otitis media. Area of dense PMN infiltration primarily located in top right-hand corner region, whereas NTHI-induced biofilm occupies the lower left-hand region of image. Zone where these 2 regions meet is demarked by dashed lines. Scale bar: 100 μm. (B) Image of a serial section of the in situ biofilm shown in A, where the PMN-rich area intersects with the NTHI biofilm, immunolabeled with antibodies directed against elastase to demark the PMNs (shown in violet) and antibodies directed against NTHI outer membrane proteins to demark the NTHI-induced biofilm (shown in green) as well as the area of intersection where both fluorochromes are visible as admixed. Scale bar: 100 μm. High-magnification immunolabeled confocal image of an 11-day-old NTHI biofilm recovered from the chinchilla middle ear: (C) NTHI biofilm nearly exclusively labeled with antibodies to the DNABII protein HU (green) where HU labeling is detected on strands of bacterial eDNA (white); (D) area where NTHI-induced biofilm intersects with PMN-rich region where anti-DNABII (HU) labeling is evident (green) as well as labeling with anti-HMGB1 (red); (E and F) are consecutive 1 μm Z-plane images of the inset designated in D, which demonstrate that there is no physical overlap of DNABII and HMGB1 labeling. (G) PMN-rich area wherein the labeling is exclusively by anti-HMGB1. Scale bars in C–G: 5 μm.
Recombinant HMGB1 disrupted biofilms formed by diverse critical and high-priority human pathogens in vitro.
Since HMGB1 could be incorporated within bacterial biofilm EPSs, we attempted to directly determine the effect of recombinant HMGB1 (rHMGB1) on bacterial biofilm architecture. To do so, we examined in vitro multiple human pathogens that in part mediate their virulence through the biofilm state. These included uropathogenic E. coli (UPEC), Burkholderia cenocepacia (Bc), NTHI, Enterobacter spp. (E), Staphylococcus aureus (S), Klebsiella pneunomiae (K), Acinetobacter baumanii (A), Pseudomonas aeruginosa (P), and Enterococcus faecium (E) that are defined as critical and high priority by the WHO (42). Twenty-four-hour biofilms formed by each of these pathogens were incubated with 200 nM rHMGB1 for 16 hours with the exception of E. faecium, wherein the incubation period was shortened to 1 hour to avoid potential degradation of rHMGB1 by its secreted proteases (43). Antibody directed against E. coli IHF (α-IHFEc; recognizes both IHF and HU), which disrupts biofilms formed by multiple bacterial species (4–7, 9, 44, 45), was used as a positive control.
As shown in Figure 2, we found that rHMGB1 disrupted biofilms formed by each of these pathogens, as evidenced by a significant reduction in biofilm average thickness compared with the control, wherein biofilms were incubated in medium only (Figure 2A). Only S. aureus and E. faecium required a greater, albeit nonbactericidal, dose (800 nM) of rHMGB1 to achieve a similar reduction in biofilm average thickness (Figure 2A). Additionally, the antibiofilm activity of native HMGB1 (nHMGB1), purified from calf thymus, on UPEC and B. cenocepacia biofilms was equivalent to that of rHMGB1 (Figure 2A), which indicated that any potential differences in posttranslational modifications between nHMGB1 and rHMGB1 did not significantly affect the antibiofilm activity.
Figure 2. HMGB1 variants disrupted biofilms formed by diverse high-priority human pathogens.

(A) Indicated isoforms of HMGB1 (200 nM unless otherwise indicated) were added to 24-hour biofilms in vitro for 16 hours. Exceptions were 800 nM rHMGB1 or 200 nM mHMGB1 for S. aureus (ESKAPE); 800 nM rHMGB1, 800 nM mHMGB1, and 3.3 mM α-IHFEc IgG for E. faecium (ESKAPE) and for only 1 hour to avoid potential degradation by E. faecium–produced proteases. Biofilms were stained with LIVE/DEAD stain and visualized via confocal laser scanning microscope and analyzed by COMSTAT to calculate average thickness. Percentage change in biofilm thickness compared with control was plotted. Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 via 1-way ANOVA (corrected for multiple comparisons by Dunnett’s test). (B) Representative images of UPEC biofilms incubated with the indicated concentrations of rHMGB1. Data collectively showed that rHMGB1, nHMGB1, and mHMGB1 significantly disrupted bacterial biofilms formed by diverse human pathogens and further showed that rHMGB1 induced dose-dependent disruption of a UPEC biofilm.
The dose-dependent activity of rHMGB1 to disrupt UPEC biofilms is demonstrated in Figure 2B. Next, we enumerated the relative concentrations of the planktonic versus the biofilm-resident UPEC after incubation with rHMGB1 and found that rHMGB1 did not exhibit any bactericidal effect in that there was no statistically significant difference in total CFUs compared with the control (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI140527DS1). However, and interestingly, rHMGB1 appeared to induce a shift of bacteria from biofilm residence into the planktonic state, as evidenced by the statistically significant increase in the planktonic bacteria and the concomitant statistically significant decrease in biofilm-resident bacteria within this culture system (Supplemental Figure 1B).
Finally, to directly test the efficacy of antibiotics in the presence of rHMGB1, we incubated NTHI biofilms with either antibiotics (ampicillin [32 μg/mL] or amoxicillin-clavulanate [1 μg/mL]) alone (8) or in combination with rHMGB1 (200 nM) for 16 hours. We then enumerated the relative concentrations of the planktonic versus the biofilm-resident NTHI and found that while rHMGB1 induced a shift of bacteria from biofilm residence into the planktonic state, rHMGB1 in combination with either ampicillin or amoxicillin-clavulanate killed a statistically significant amount of the total bacteria, likely by the ability of these antibiotics to kill the planktonic bacteria and rHMGB1 to drive the bacteria into the vulnerable planktonic state (Supplemental Figure 2). These data suggested that rHMGB1 synergized with antibiotics in vitro in the clearance of bacterial biofilms.
Oxidation of rHMGB1 negatively affected the antibiofilm activity of rHMGB1.
Several posttranslational modifications are described for HMGB1 that modulate its location, function, and ability to bind DNA (reviewed in ref. 19). Although liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis of rHMGB1 and nHMGB1 revealed that less than 20% of the peptides from either exhibited any posttranslational modifications (data not shown), we nonetheless examined the effect of oxidation on the antibiofilm activity of rHMGB1 given that the redox state of HMGB1 is known to modulate its function (46). The oxidation state of the 3 cysteine (C) residues at positions 23, 45, and 106 influences the inflammatory properties (47). HMGB1 with the C106 thiol group and a C23-C45 disulfide bond triggers inflammation, whereas terminally oxidized or reduced cysteines promote resolution of inflammation (48). To this end, HMGB1 was first oxidized with hydrogen peroxide as described (49). Ox-rHMGB1 was then evaluated to determine its relative ability to disrupt biofilms formed by UPEC. Ox-rHMGB1 lost its antibiofilm effect as compared with rHMGB1 (Figure 3). This result suggested that oxidation of rHMGB1 significantly impaired its biofilm disruption ability.
Figure 3. Oxidation of rHMGB1 negatively affected its antibiofilm activity.
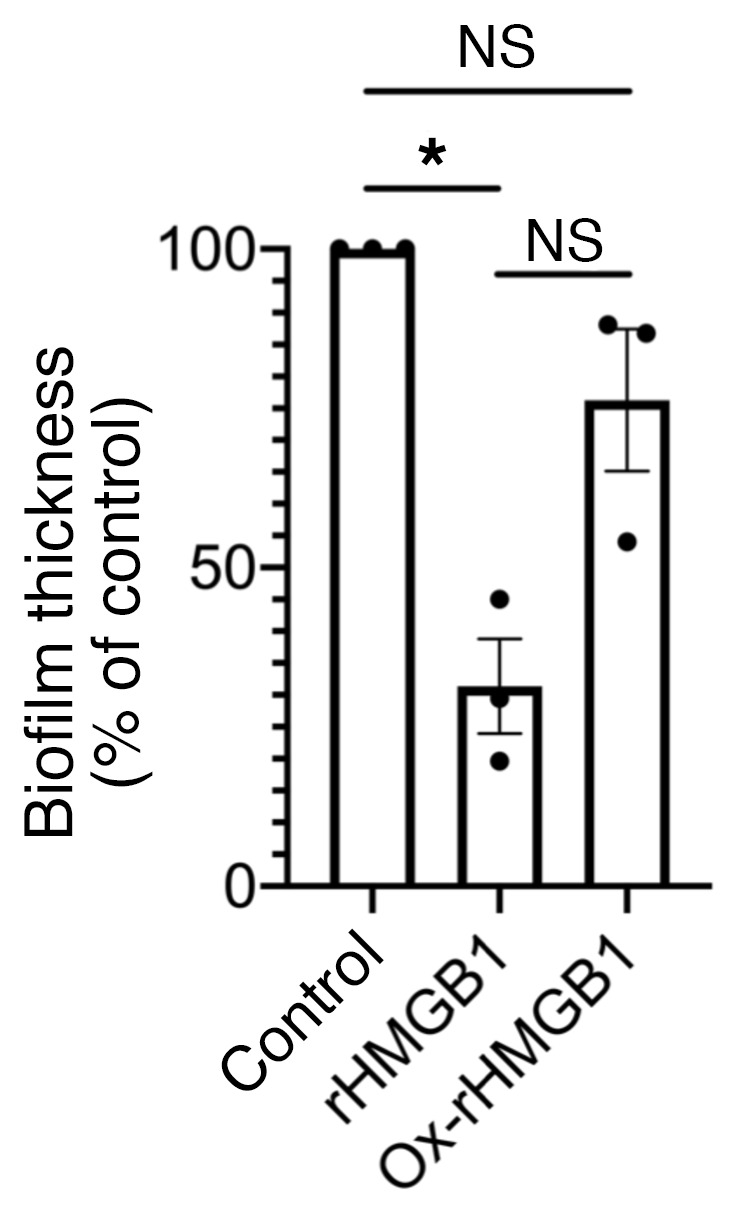
Twenty-four-hour biofilms formed in vitro by UPEC were incubated with ox-rHMGB1 (200 nM) for 16 hours. Biofilms were stained with LIVE/DEAD stain and visualized via confocal laser scanning microscope and analyzed by COMSTAT to calculate average thickness. Data are shown as mean ± SEM. *P < 0.05 as assessed by 1-way ANOVA (corrected for multiple comparisons by Tukey’s test). Note that the antibiofilm function of HMGB1 was significantly reduced upon oxidation of rHMGB1.
rHMGB1 with an engineered single amino acid mutation C45S retained its antibiofilm function in vitro against multiple human pathogens.
To alleviate any undesired proinflammatory effect of rHMGB1, we engineered a C45S point mutation to generate modified HMGB1 (mHMGB1) that abrogates disulfide bond formation with C23. To validate this variant, we first demonstrated that mHMGB1 retained its ability to bind to HJ DNA, a known binding substrate of HMGB1 (Supplemental Figure 3). Next, we assayed the antibiofilm function of mHMGB1 as described above and demonstrated that mHMGB1 fully retained the antibiofilm activity (Figure 2A). Only E. faecium required a higher dose of mHMGB1 to achieve a similar reduction in biofilm average thickness. These data suggested that the engineered C45S variant of HMGB1 retained biofilm disruption capability.
Unlike DNABII proteins, HMGB1 did not stabilize model HJs and the lattice-like eDNA network within K.
pneumoniae biofilms in vitro. We recently showed the presence of HJ-like structures within the eDNA lattice of bacterial biofilms (4); given its high affinity for such branched DNA structures (15), we hypothesized that HMGB1 likely destabilized HJ DNA upon binding, which was why we never observed it at the vertices of crossed strands of eDNA (see Figure 1). To test our hypothesis, we incubated HJ DNA with either HMGB1; the DNABII protein IHF; or the prototypic HJ DNA binding protein, RuvA at room temperature or 55°C (melting temperature of HJ DNA) and resolved the complexes by nondenaturing PAGE. Although all 3 proteins bound to HJ DNA to form stable complexes at room temperature, only HMGB1 was unable to efficiently stabilize the HJ DNA at 55°C (Supplemental Figure 4), as indicated by the decrease in abundance of the shifted HJ DNA-HMGB1 complex (arrow) and the concomitant increase in the constituent melted oligos (asterisk). These data suggested that despite a similar HJ DNA binding site preference to both IHF and RuvA, HMGB1 was unable to likewise stabilize the HJ structure.
To directly evaluate the effect of HMGB1 isoforms on the eDNA-dependent extracellular matrix, we incubated 24-hour K. pneumoniae biofilms (used here as a representative model bacterial biofilm) with either rHMGB1 or mHMGB1 for 16 hours. Unfixed biofilms were labeled with a monoclonal antibody against dsDNA to visualize the eDNA within the biofilm matrix. Bacteria were labeled with FilmTracer FM 4-64 (gray). eDNA within K. pneumoniae biofilms was organized into a complex web-like structure (yellow), which was notably disrupted upon incubation with rHMGB1 or mHMGB1. This outcome corresponded with a concurrent substantial reduction in biofilm bacteria in the presence of either isoform of HMGB1 (Figure 4). Collectively, these data suggested that both rHMGB1 and mHMGB1 directly destabilized the biofilm extracellular matrix by specifically disrupting the eDNA lattice, which resulted in biofilm disruption in vitro. This result was consistent with what was observed when we labeled a biofilm that had formed in vivo, wherein HMGB1 was not found at the vertices of crossed strands of eDNA within the scaffold. Per our model, HMGB1 would compete with DNABII proteins, wherein instead of stabilization, HMGB1 would destabilize these HJ-like structures and consequently disrupt the biofilm.
Figure 4. HMGB1 isoforms disrupted the lattice-like eDNA network within K.

pneumoniaebiofilms in vitro. Twenty-four-hour K. pneumoniae biofilms were incubated with the indicated protein (200 nM) for 16 hours. Unfixed biofilms were incubated with α-dsDNA monoclonal antibody, then incubated with goat α-mouse IgG conjugated to Alexa Fluor 488 (yellow). K. pneumoniae were stained with FilmTracer FM 4-64 (white). Biofilms were visualized by confocal laser scanning microscope. Note the intertwined web-like structure in the control and the disruption of the web-like structure by rHMGB1 and mHMGB1. Scale bars: 10 μm.
HMGB1 disrupted biofilms via its ability to bind to HJ-like structures within the biofilm extracellular matrix.
We hypothesized that HMGB1 could mediate its antibiofilm effect either by binding directly to HJ DNA within the eDNA matrix or by binding to DNABII proteins such that these proteins could no longer stabilize the bacterial extracellular matrix. First, we tested the possibility that HMGB1 mediated its antibiofilm effect by directly binding to HJ DNA. To this end, we assayed the antibiofilm function of HMGB1 as described above but only in the presence of exogenously added HU (DNABII protein) or RuvA (prototypic HJ DNA-binding protein) that would compete with mHMGB1 for similar binding sites within the eDNA or CbpA, another bacterial nucleoid–associated protein as a negative control; we have previously shown that RuvA can replace DNABII proteins for function in biofilm EPSs (4), whereas CbpA is not required for the maintenance of the eDNA lattice structure within EPSs (12). We observed that mHMGB1 was unable to disrupt biofilms formed by UPEC in the presence of either added DNABII protein or RuvA in a dose-dependent manner but could still disrupt UPEC biofilms in the presence of CbpA (Figure 5A), which suggested that mHMGB1 mediated its antibiofilm effect via its ability to bind to HJ-like structures within the biofilm extracellular matrix. Second, since alkylation of HMGB1 with N-ethylmaleimide (NEM) has been shown to inhibit HMGB1’s ability to bind to DNA (50), we used NEM-treated rHMGB1 to directly test the mechanism of HMGB1-mediated biofilm disruption. We first verified that rHMGB1 was modified by NEM by Triton X-100–acetic acid–urea gel that revealed a shift in NEM-rHMGB1 as compared with rHMGB1 (Supplemental Figure 5A). Next, we demonstrated that NEM-rHMGB1 was unable to bind to HJ DNA (Supplemental Figure 5B). To verify that NEM-HMGB1 was still folded properly and otherwise functional, we assayed for the ability of NEM-HMGB1 to induce neutrophils to form NETs. Human neutrophils were isolated from fresh blood and were incubated in the absence or presence of NEM-rHMGB1 for 3.5 hours. Neutrophils were fixed and then labeled with a monoclonal antibody against dsDNA to visualize the eDNA (NETs; teal) and a polyclonal antibody against neutrophil elastase to demark the NET-derived eDNA (yellow). Neutrophils themselves were labeled with wheat germ agglutinin (WGA) conjugated with Alexa Fluor 350 (blue). As shown in Supplemental Figure 6, NEM-HMGB1 induced neutrophils to form NETs, which suggested that NEM modification of rHMGB1 only specifically interfered with its ability to bind to HJ DNA. Finally, we assayed the antibiofilm function of NEM-rHMGB1 as described above and demonstrated that NEM-rHMGB1 lost its antibiofilm activity (Figure 5B). These results suggested that HMGB1 disrupted biofilms via its ability to directly bind to HJ-like structures within the biofilm extracellular matrix.
Figure 5. HMGB1 disrupted biofilms via its ability to bind to HJ-like structures within the biofilm extracellular matrix.

(A) Twenty-four-hour biofilms formed in vitro by UPEC were incubated with mHMGB1 (200 nM) in the presence or absence of each of the indicated proteins for 16 hours. (B) Twenty-four-hour biofilms formed in vitro by UPEC were incubated with rHMGB1 (200 nM) or NEM-rHMGB1 (200 nM) for 16 hours. Biofilms were stained with LIVE/DEAD stain, visualized via confocal laser scanning microscope, and analyzed by COMSTAT to calculate average thickness. Data are shown as mean ± SEM. *P < 0.05, **P < 0.01, as assessed by 1-way ANOVA (corrected for multiple comparisons by Dunnett’s test for A and Tukey’s test for B). Note that the antibiofilm function of HMGB1 was lost in the presence of competitors that bind directly to eDNA (HU, RuvA) and therefore compete with HMGB1 for binding to eDNA or upon modification of rHMGB1 that directly affects its ability to bind to HJ DNA.
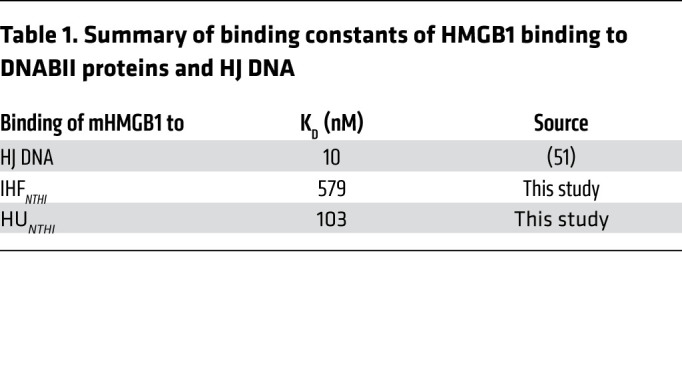
Next, we tested the possibility that HMGB1 mediated its antibiofilm effect by binding to DNABII proteins. To this end, we determined the Kd as a measure of binding affinity of mHMGB1 to IHFNTHI and HUNTHI by Biacore surface plasmon resonance analysis and found that the Kd for mHMGB1 binding to IHFNTHI was 579 nM and to HUNTHI was 104 nM (Table 1). Given that full-length HMGB1 binds to HJ DNA with at least an order of magnitude higher affinity (Kd ~10 nM) (51) as compared with either of the 2 DNABII proteins, these results collectively suggested that HMGB1 antibiofilm effects predominated by directly binding to its high-affinity HJ DNA target and thus destabilized HJs rather than via protein-protein interactions with DNABII proteins.
Table 1. Summary of binding constants of HMGB1 binding to DNABII proteins and HJ DNA.
HMGB1 promoted clearance of B.
cenocepacia aggregates from the mouse lung. Because rHMGB1 and mHMGB1 disrupted biofilms formed by multiple bacteria in vitro, we next evaluated their potential antibiofilm activity in a murine model of lung infection mediated by B. cenocepacia. C57BL/6 mice were challenged intratracheally (i.t.) with 107 CFU of B. cenocepacia, and at either the time of challenge (prevention cohort) or at 24 hours after infection (treatment cohort), mice received 0.2 nmol (i.t.) of either rHMGB1 or mHMGB1. We first immunolabeled B. cenocepacia within the mouse lungs at 72 hours after infection via use of a monoclonal antibody against E. coli elongation factor Tu (α-EF-Tu; cross-reacts with B. cenocepacia) and demonstrated that B. cenocepacia formed aggregates within the lungs (Figure 6A, green). Next, we determined the bacterial burden within the lungs either 18 hours after infection (prevention) or at 72 hours after infection (treatment) and demonstrated that both rHMGB1 and mHMGB1 significantly facilitated the clearance of B. cenocepacia from the lungs, regardless of preventative (Figure 6B) versus treatment strategy used (Figure 6C). We also stained B. cenocepacia within the lung tissue at 72 hours after infection and observed a large number of bacteria within the lungs of the control mice (Supplemental Figure 7A, green). Strikingly, there was a significantly reduced bacterial load in mice treated with either rHMGB1 or mHMGB1 compared with the control (Supplemental Figure 7A). These data suggested that both the preventative and treatment approaches with either rHMGB1 or mHMGB1 facilitated clearance of B. cenocepacia from the mouse lung.
Figure 6. HMGB1 promotes clearance of B.

cenocepaciaaggregates from the murine lung. (A) Representative immunofluorescence image of a section of lung recovered from a mouse infected with B. cenocepacia (green). C57BL/6 mice were i.t. challenged with 107 CFU and either simultaneously (prevention) or 24 hours later (treatment) received 0.2 nmol of the indicated HMGB1 variant. Bronchoalveolar lavage (BAL) was collected at 18 hours after infection [prevention (B)] or at 72 hours after infection [treatment (C)] then analyzed for CFU. (D) Representative images stained with H&E (10× and 40× magnification). (E) Cells in BAL were stained with α-CD45, CD11b, and Ly-6G and analyzed by flow cytometry to measure relative neutrophil influx (F). Data are shown as mean ± SD. *P < 0.05, **P < 0.01 as assessed by 1-way ANOVA. HMGB1 treatment significantly decreased CFU of B. cenocepacia in the murine lung, and treatment with the engineered C45S mutation within mHMGB1 eliminated the proinflammatory activity.
Although both rHMGB1 and mHMGB1 facilitated clearance of B. cenocepacia from the mouse lung, since HMGB1 is known to reduce the phagocytic capacity of macrophages (52), we evaluated the effect of mHMGB1 on phagocytosis by macrophages. We found that rHMGB1 and mHMGB1 were indistinguishable from the control (Supplemental Figure 7B). Cytochalasin D was used as a positive control. These results suggested that rHMGB1 or mHMGB1 under these conditions did not affect phagocytosis of macrophages.
Since HMGB1 can induce a potentially detrimental proinflammatory response, we assessed the ability of rHMGB1 or mHMGB1 to induce the inflammatory recruitment of neutrophils into the murine peritoneum. Mice were i.p. injected with 0.2 nmol of either rHMGB1 or mHMGB1, and the relative concentration of neutrophils within the peritoneal cavity was determined after 4 hours by flow cytometry with α-CD45, α-CD11b, and α-Ly6G antibodies. Thioglycollate was used as a positive control to induce peritoneal inflammation. Thioglycolate-injected mice showed significant neutrophil migration (2 × 106 neutrophils) to the peritoneal cavity, whereas those that received rHMGB1 showed a moderate (3 × 105 neutrophils) yet significant neutrophil migration to the peritoneal cavity compared with the control (Supplemental Figure 7C). Strikingly, mice injected with mHMGB1 demonstrated a significant reduction in neutrophil migration (<1 × 105 neutrophils) compared with rHMGB1 (Supplemental Figure 7), which suggested that mHMGB1 induced an attenuated inflammatory neutrophil response.
We further evaluated inflammatory cell recruitment into the lungs of mice that had been challenged with B. cenocepacia, then treated with either rHMGB1 or mHMGB1 at 72 hours after infection. Whereas lungs from mice treated with rHMGB1 displayed a significant infiltration of inflammatory cells compared with the control, as evidenced from the H&E stain, lungs from mice treated with mHMGB1 showed no signs of inflammation and instead more closely resembled uninfected lungs (Figure 6D). To identify the inflammatory cells that had migrated into the lungs after treatment with rHMGB1, we performed flow cytometry (using α-CD45, α-Ly6G, and α-CD11b antibodies) on bronchoalveolar lavage (BAL) samples from mice infected with B. cenocepacia and simultaneously treated with either rHMGB1 or mHMGB1 at 18 hours after infection. Although infected mice showed an infiltrate of primarily neutrophils, those similarly infected but also treated with rHMGB1 showed a significantly greater number of neutrophils in BAL compared with the control (Figure 6, E and F). Conversely, mice treated with mHMGB1 showed a significant reduction of neutrophils in BAL compared with those treated with rHMGB1 and were indistinguishable from the control (Figure 6, E and F).
Since excess HMGB1 can mediate dysregulated host response to infection associated with septic shock, we evaluated the ability of rHMGB1 (0.2 nmol; the concentration used to treat in vivo biofilms) to induce septic shock in mice. Mice were i.p. injected with either 0.2 nmol endotoxin-free rHMGB1, LPS (5 mg/kg), or both and then monitored for signs of septic shock for 24 hours. Serum TNF-α, a gold standard for induction of septic shock (53), revealed that although LPS alone induced a significant amount of TNF-α, rHMGB1 did not induce detectable TNF-α and was instead comparable to the control (Supplemental Figure 8). This result suggested that rHMGB1, which we used at the same concentration as a therapeutic in preclinical efficacy studies to resolve biofilms and induce disease resolution, was unlikely to promote systemic inflammation given the lack of evidence for such in these murine studies.
Lastly, since rHMGB1 not only induces neutrophil migration to the site of infection but also induces neutrophils to form NETs, we evaluated the effect of mHMGB1 on induction of NETs by neutrophils. Human neutrophils were isolated from fresh blood and were incubated in the absence or presence of either rHMGB1 or mHMGB1 for 3.5 hours. Neutrophils were fixed and then labeled as described above to visualize the NETs. Although rHMGB1 had a modest effect on induction of NETs, mHMGB1 more strongly induced NET formation of neutrophils in vitro (Supplemental Figure 6). Collectively, these data suggested that although both rHMGB1 and mHMGB1 facilitated clearance of B. cenocepacia aggregates from the mouse lungs, only mHMGB1 did so without the proinflammatory activity of rHMGB1 in a mouse model of lung infection.
rHMGB1 and mHMGB1 promoted resolution of NTHI biofilms within the middle ears in a chinchilla model of experimental otitis media.
We next utilized an established model of experimental otitis media induced by NTHI to further evaluate the ability of rHMGB1 or mHMGB1 to disrupt adherent mucosal biofilms. Four days after transbullar challenge with NTHI, biofilms within both middle ears of chinchillas were treated with either rHMGB1 or mHMGB1 by direct instillation into the middle ear on 2 consecutive days (Figure 7A). As a negative control, an equivalent volume of sterile saline was delivered to a third cohort. One day after receipt of the second therapeutic dose, all animals were euthanized, and NTHI within the middle-ear biofilms was enumerated. The relative amounts of mucosal biofilm and mucosal inflammation were also blindly qualitatively assessed.
Figure 7. mHMGB1 mediated clearance of biofilm-resident NTHI, eradication of established mucosal biofilms, and resolution of experimental disease, an outcome that was enhanced upon codelivery with antibody fragments directed against the immunoprotective domains of a DNABII protein (tip-chimer Fabs).

(A) Study timeline to assess the relative ability of rHMGB1 or mHMGB1 to resolve NTHI biofilms already established in the chinchilla middle ear. (B) Relative quantity of NTHI resident within mucosal biofilms and adherent to the middle-ear mucosa 1 day after completion of therapy. (C) Rubric used to qualitatively assess the amount of middle-ear mucosal biofilm that remained 1 day after completion of treatment. (D) Relative amount of mucosal biofilm within each middle ear per cohort. (E) Rubric used to qualitatively assess the amount of middle-ear mucosal inflammation 1 day after completion of treatment. (F) Relative amount of mucosal inflammation within each middle ear per cohort. (G) Representative image of middle ears from each cohort to demonstrate relative presence or clearance of mucosal biofilm and inflamed or noninflamed state. (H) Study timeline to assess additive potential of mHMGB1 codelivered with tip-chimer Fabs to resolve NTHI biofilms already established in the chinchilla middle ear. (I) Relative quantity of NTHI resident within mucosal biofilms and adherent to the middle-ear mucosa 24 hours after 1 or 2 treatment doses. (J) Relative amount of mucosal biofilm within each middle ear per cohort 24 hours after 1 or 2 treatment doses. When delivered individually, both rHMGB1 and mHMGB1 induced rapid clearance of biofilm-resident NTHI and eradication of established mucosal biofilms, whereas only mHMGB1 induced limited mucosal inflammation. Moreover, codelivery of mHMGB1 with tip-chimer Fab fragments was highly efficacious to eradicate NTHI and associated biofilms from the middle ear. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ANOVA with Kruskal-Wallis multiple comparisons test (B); 1-way ANOVA with Tukey’s multiple comparison test (D, F, J); 1-way ANOVA with Holm-Sidak multiple comparison test (I).
One day after receipt of the second treatment with rHMGB1 or mHMGB1, there was a significantly (more than 3-log) lower amount of NTHI within mucosal biofilms and/or adherent to the middle-ear mucosa compared with the control cohort (Figure 7B) (P ≤ 0.01). No difference in clearance was observed between these 2 treatments. Additionally, 6 blinded evaluators qualitatively ranked the relative amount of mucosal biofilm that remained within the chinchilla middle ears using an established rubric wherein a score of 0 indicated no biofilm present and 4+ indicated a middle ear filled with biofilm (Figure 7C) (54). Middle ears in the negative control cohort scored a mean value of 3.1, i.e., 75% or more of the middle ear remained filled with mucosal biofilm (Figure 7D). In contrast, those treated with rHMGB1 or mHMGB1 received a mean score of 1.0, i.e., less than 25% of the middle ear was occupied by biofilm (P ≤ 0.0001). Again, no difference was observed between treatments for the relative amount of remaining biofilm. Next, middle ears were blindly qualitatively ranked for relative inflammation. A rubric wherein a score of 0 indicated no inflammation and 3+ indicated extensive capillary dilatation and presence of multiple hemorrhagic foci within the middle-ear mucosa was used (Figure 7E). The rHMGB1 cohort scored just slightly lower than the sterile saline control cohort (Figure 7F; 1.7 vs. 2.2, respectively). Conversely, minimal inflammation was seen in the mHMGB1 cohort, significantly less than both the control and rHMGB1 cohorts (score, 0.7; P ≤ 0.0001). Images of representative middle ears are provided in Figure 7G. Importantly, we also found a statistically significant increase in proinflammatory cytokines (IL-1β and IL-17A) in the middle-ear fluids recovered from chinchillas that had been treated with rHMGB1 as compared with treatment with either mHMGB1 or diluent alone. Notably, there was also a statistically significant increase in antiinflammatory cytokine (IL-10) in the middle-ear fluids recovered from chinchillas that had been treated with mHMGB1 as compared with treatment with either rHMGB1 or diluent alone (Supplemental Figure 9). Therefore, although both rHMGB1 and mHMGB1 promoted clearance of mucosal biofilms, only mHMGB1 did so without induction of overt inflammation (Figure 7, F and G).
Combinatorial treatment with host-derived rHMGB1 and pathogen-directed α-DNABII antibody eradicated NTHI biofilms within the chinchilla middle ears.
To now determine whether there might be added value in treating biofilms that had formed in vivo with a combination of mHMGB1 plus antibody (in the form of an antigen-binding fragment, Fab) against a DNABII protein to thereby develop a combinatorial host-derived plus pathogen-directed therapeutic strategy, we again utilized our chinchilla model of experimental NTHI-induced otitis media. Four days after challenge, with NTHI biofilms now present within the middle ears of chinchillas, cohorts were treated with mHMGB1 (host-derived) and/or antibody against a chimeric peptide designed to mimic the immunoprotective domains of IHF (tip chimer, pathogen directed), either alone or in combination (Figure 7H). First, we evaluated the effectiveness of a single treatment. To this end, a subset of animals in each cohort was euthanized 1 day after receipt of a single therapeutic dose, whereas the remaining animals in each cohort were euthanized 24 hours after receipt of a second treatment (Figure 7H). Treatments included 200 nM mHMGB1 alone or admixed with 342 nM Fab fragments derived from rabbit anti–tip-chimer IgG (tip-chimer Fabs), or tip-chimer Fabs alone. As a negative control, Fab fragments derived from antibody directed against nonprotective domains of IHF were used (tail-chimer Fabs) (14) either alone or combined with mHMGB1.
We then treated infected middle ears with either of the aforementioned HMGB1 proteins alone or in combination with Fab fragments against either the tail or tips of IHF. Tail-chimer Fabs failed to alter the extant biofilms regardless of the number of doses, as expected (Figure 7I, red symbols). Receipt of mHMGB1 plus tail-chimer Fabs promoted a significant reduction in NTHI compared with receipt of tail-chimer Fab alone after 1 or 2 doses (P ≤ 0.01 and P ≤ 0.0001, respectively), and 50% (3 of 6) of middle ears cleared NTHI from this niche (purple symbols). This latter clearance was, however, due to the action of mHMGB1, which alone mediated significant clearance of NTHI from 50% (3 of 6) of middle ears after 1 dose (green symbols), and NTHI was further reduced after receipt of 2 doses. A single therapeutic dose of the DNABII-derived tip-chimer Fabs resulted in a significant 5-log reduction in biofilm-resident NTHI, and 67% (4 of 6) of middle ears were culture-negative (yellow symbols). This was significant compared with delivery of tail-chimer Fab alone after 1 or 2 doses (P ≤ 0.01 and P ≤ 0.0001, respectively), which was further reduced by receipt of a second dose as we reported previously (55). Combining mHMGB1 with tip-chimer Fabs was the most effective therapy tested: 86% (5 of 6) of middle ears cleared NTHI from the middle ear (orange symbols; P ≤ 0.001 and P ≤ 0.0001 compared with tail-chimer Fab alone after 1 or 2 doses, respectively), which demonstrated at least an additive effect. Strikingly, treatment with 2 doses of mHMGB1 plus tip-chimer Fab eradicated NTHI from the middle ears of all animals in this cohort below the level of detection, indicating at least an additive effect.
We also qualitatively evaluated the middle ears of animals in these cohorts for the relative amount of remaining NTHI biofilm. Receipt of 1 or 2 doses of tail-chimer Fabs was not effective, and mean mucosal biofilm scores were more than 3.0 at each time point, which again indicated that 75% or more of the middle ear remained filled with NTHI biofilm (Figure 7J, red bars). Admixing mHMGB1 with tail-chimer Fabs significantly reduced the amount of biofilm present by half; however, biofilms still filled between 25% and 50% of the middle ears in this cohort (purple bars; P ≤ 0.0001 vs. tail-chimer Fabs). Receipt of mHMGB1 alone was highly effective (green bars), as was receipt of 1 or 2 treatments with tip-chimer Fabs (yellow bars; compared with 1 and 2 doses of tail-chimer Fab alone, P ≤ 0.001). Overall, however, the most effective therapeutic treatment was mHMGB1 admixed with tip-chimer Fabs (orange bars). A single dose cleared preformed NTHI biofilms from 67% (4 of 6) of middle ears, with a mucosal biofilm score of 0.1. A second dose further enhanced this response, and no NTHI biofilms were seen in 5 of 6 of middle ears (83%). Further, after receipt of 1 dose of the combined treatment, significantly less mucosal biofilm was observed compared with a single dose of tail-chimer Fab plus mHMGB1 (P ≤ 0.0001) or a single dose of mHBGB1 (P ≤ 0.05); whereas after 2 doses of the combined treatment, this difference was significant compared with 2 doses of tail-chimer Fab plus mHMGB1 (P ≤ 0.001). Collectively, these data demonstrated significant reduction in bacterial load and eradication of mucosal NTHI biofilms after receipt of mHMGB1, an outcome that was further augmented when codelivered with tip-chimer Fabs. These results suggested that a combinatorial host-derived plus pathogen-directed therapeutic strategy was highly effective, likely because α-DNABII tip Fabs and mHMGB1 disrupted biofilms by distinct mechanisms that, when used together, accounted for the observed enhanced outcome, as evidenced by virtually complete eradication of preexisting pathogenic biofilms in this animal model.
Discussion
Host HMGB1 and bacterial DNABII proteins are found in the extracellular milieu wherein HMGB1 is involved in several processes that include inflammation, cell migration, invasion and proliferation, tissue regeneration, and antimicrobial defense (reviewed in ref. 19). In contrast, we discovered that extracellularly, DNABII proteins serve as part of an underlying structural component of the eDNA-dependent EPSs of diverse bacterial biofilms (4–9, 10–14, 56–58). Our results showing HMGB1 to be present within the bacterial biofilm EPSs were in line with the localization of other NET components, such as neutrophil elastase and histone H3 during in vivo infection (59).
Given that HMGB1 was localized within the bacterial biofilm EPSs, we hypothesized that biofilm disruption by HMGB1 was mediated by its ability to bind to DNA. The vertices of the crossed strands of eDNA that form the lattice structure of the bacterial biofilm are composed of HJ-like structures wherein DNABII proteins bind and stabilize the biofilm EPSs (4). Although HMGB1 has a high affinity for HJ DNA, heretofore it was never observed at the vertices of the eDNA lattice in vivo. This result suggested to us that when HMGB1 bound to these HJ-like structures of the eDNA lattice, it was unable to stabilize them and as a consequence collapsed the structures, the opposite effect to that of DNABII proteins, which stabilize this structure. This would argue that the antibiofilm activity of HMGB1 was mediated via a competitive inhibition mechanism wherein HMGB1 bound directly to free HJs. Hints to these biological differences between HMGB1 and DNABII proteins can be derived from biochemical analyses. Although DNABII and HMGB1 proteins both bind in the minor groove of DNA, their contacts with HJs occur in different manners (60). DNABII proteins have a binding preference to HJs that adopt an X-like structure (61–63), whereas HJs in a square planar structure is the preferred conformation for HMGB1 (64). We have previously shown that RuvA, a prototypic HJ DNA-binding protein that specifically binds to HJs in the square planar conformation (65), compensates for the loss of DNABII proteins within the biofilm EPSs (4). This result implied that the eDNA lattice within the bacterial biofilm EPSs was composed of HJs that are sufficiently similar to a conformation that is compatible for RuvA and thus HMGB1 to bind, and yet RuvA stabilizes the eDNA lattice and HMGB1 does not. While this is corroborated by our electrophoretic mobility shift assay (EMSA) stabilization studies with HJ substrates, there are clearly other factors in DNA binding as yet to be determined to explain HMGB1’s inability to stabilize the eDNA lattice. Finally, either exogenous addition of DNA-binding proteins that competed with HMGB1 for HJs within the eDNA matrix or modification of HMGB1 with NEM that directly abrogated the ability of HMGB1 to bind to HJ DNA provided direct evidence that supported competitive inhibition of HJ sites for DNABII proteins as the mechanism of action for HMGB1. To verify that this was the sole mechanism of action, we also tested the possibility that HMGB1 could bind directly to free DNABII proteins as a means to mediate the observed biofilm disruption. Surface plasmon resonance analysis demonstrated that HMGB1 bound poorly to DNABII proteins and preferentially bound with high affinity directly to HJ DNA. Thus, within the eDNA, HMGB1 likely destabilized HJs rather than via any limited binding to DNABII proteins.
The capacity for HMGB1 to act as an antibiofilm therapeutic rests with its modular structure with their individual functions. HMGB1 has 2 tandem DNA-binding domains: an A box, a B box, and a C-terminal tail that consists of 30 consecutive acidic amino acid residues (18). Extracellularly, A box alone exhibits antiinflammatory activity and acts as an antagonist for HMGB1, B box alone fully retains the proinflammatory activity of HMGB1, and the C-terminal tail is involved in bacterial killing. When in excess, HMGB1 also functions as late mediator of sepsis, a form of systemic nflammation in response to microbial infection (27, 28). Hence, in the development of HMGB1 as a therapeutic targeted against bacterial biofilms, it is essential to attenuate its proinflammatory activity. We demonstrated that use of rHMGB1 at concentrations that effectively eradicated biofilms did not induce a dysregulated host response to infection associated with septic shock, and we were successful at separation of the antibiofilm activity from the proinflammatory functions of HMGB1 via generation of an engineered derivative of rHMGB1 with a single C45S amino acid mutation (mHMGB1) that exhibited potent antibiofilm function as well as highly attenuated proinflammatory function. Further characterization of each of the individual domains of mHMGB1 in an effort to generate a minimal mHMGB1 molecule that retains its ability to disrupt biofilms with minimal proinflammatory function is currently underway.
Given our results with HMGB1 and its antibiofilm activity, we suggest an augmented paradigm of NET function. Neutrophils, the first line of host defense against pathogens, release their DNA as NETs in response to proinflammatory stimuli that include LPS, IL-8, TNF-α, and HMGB1 to entrap pathogens in an effort to prevent dissemination. Additionally, NETs are also decorated with HMGB1, histones, elastase, and myeloperoxidase that facilitate eradication of microbes (reviewed in ref. 66). Given that HMGB1 exhibits antibacterial activity and also acts as a proinflammatory cytokine to induce NET formation, we propose the following model. For bacteria to persist and resist clearance in a host, they adopt either an aggregated or attached biofilm state that protects them from host immune effectors. Further, partial sloughing of these communities permits propagation of the pathogens without risk of clearance of the core resident biofilm bacteria. To prevent release of bacteria and subsequent propagation, however, the host releases sufficient concentrations of HMGB1 during NETosis to confine bacterial pathogens, without induction of an uncontrolled inflammatory response. Thus, HMGB1 acts to cordon off and limit biofilm proliferation and to alert the innate-immune system. This model is biologically relevant, as bacteria actively build biofilms to evade the host immune response, whereas the host actively attempts to prevent pathogenic bacterial biofilm propagation via secretion of a well-balanced level of HMGB1; i.e., too little HMGB1 leads to proliferation of biofilms, whereas too much HMGB1 leads to excessive proinflammatory effects that cause a dysregulated host response to infection associated with septic shock. In this study, we demonstrated that a combinatorial host-directed plus pathogen-derived therapeutic approach achieved via delivery of mHMGB1 with antibodies against a DNABII protein facilitated rapid clearance of biofilms without induction of an inflammatory response in a preclinical model and thereby tipped the balance in favor of the host in the eradication of highly recalcitrant bacterial biofilms.
Methods
Bacterial strains.
UPEC strain UTI89 was isolated from a patient with cystitis (67). B. cenocepacia strain K56-2 was isolated from a patient with cystic fibrosis (68). NTHI strain 86-028NP was isolated from the nasopharynx of a child with chronic otitis media at Nationwide Children’s Hospital (69). Enterobacter spp. and K. pneumoniae were isolated from a patient with a urinary tract infection. S. aureus strain 29213, A. baumanii strain 17978, and P. aeruginosa strain 27853 were obtained from ATCC. E. faecium Com12 strain was isolated from feces of healthy human volunteers (70). Each of these strains was maintained at low passage number in liquid nitrogen.
Disruption of bacterial biofilms by various isoforms of HMGB1.
UPEC strain UTI89, B. cenocepacia, K. pneumoniae, Enterobacter spp, and A. baumanii were cultured on Lysogeny broth (LB) agar for 18–20 hours at 37°C in a humidified atmosphere that contained 5% CO2, and then suspended in LB broth to an OD of 0.65 at 490 nm. Cultures were diluted 1:12 in LB broth, and then incubated statically at 37°C 5% CO2 until an OD of 0.6 was reached at 490 nm. The cultures were then diluted in LB broth to contain 2 × 105 CFU/mL, and 200 μL of this suspension was inoculated into each well of an 8-well chambered cover-glass slide (Thermo Fisher Scientific). NTHI and S. aureus were cultured on chocolate agar for 18–20 hours at 37°C in a humidified atmosphere that contained 5% CO2, and then NTHI was resuspended in sBHI (brain heart infusion broth supplemented with heme [2 μg/mL] and b-NAD [2 μg/mL]) broth to an OD of 0.65 at 490 nm. S. aureus was resuspended in tryptic soy broth (TSB) to an OD of 0.65 at 490 nm. NTHI and S. aureus were diluted 1:6 in sBHI and TSB, respectively, and then incubated statically at 37°C 5% CO2 for 3 hours. The cultures were then diluted in their respective media to contain 2 × 105 CFU/mL, and 200 μL of this suspension was inoculated into each well of an 8-well chambered cover-glass slide. P. aeruginosa and E. faecium were cultured on TSB agar and BHI agar, respectively, for 18–20 hours at 37°C in a humidified atmosphere that contained 5% CO2, and then suspended in TSB broth or BHI broth, respectively, to an OD of 0.65 at 490 nm. Cultures were diluted 1:12 in the respective broth, and then incubated statically at 37°C 5% CO2 until an OD of 0.6 was reached at 490 nm. The cultures were then diluted in their respective media to contain 2 × 105 CFU/mL, and 200 μL of this suspension was inoculated into each well of an 8-well chambered cover-glass slide. After 16 hours of incubation of each of the bacterial species at 37°C 5% CO2, the medium was replaced with the respective fresh medium and incubated at 37°C 5% CO2 for another 8 hours. At 24 hours, the medium was replaced with the respective fresh medium (control) or fresh medium that contained one of the HMGB1 (200 nM) isoforms (rHMGB1, mHMGB1, nHMGB1, or Ox-rHMGB1) or α-IHFEc (1000 nM) (5) or rHMGB1 in combination with ampicillin (32 μg/mL) or amoxicillin-clavulanate (1 μg/mL) and incubated at 37°C 5% CO2 for 16 hours. S. aureus was incubated with 800 nM rHMGB1 for 16 hours and E. faecium was incubated with 800 nM rHMGB1 or 800 nM mHMGB1 for 1 hour at 37°C 5% CO2. nHMGB1 was purchased from Chondrex Inc. All biofilms except UPEC and K. pneumoniae were washed twice with sterile saline (0.9%) and stained with LIVE/DEAD stain (Molecular Probes) in accordance with the manufacturer’s instructions. UPEC and K. pneumoniae were washed once with sterile saline and then stained with LIVE/DEAD stain. Biofilms were then washed once with sterile saline and fixed with 1.6% paraformaldehyde, 0.025% glutaraldehyde, and 4% acetic acid in 0.1 M phosphate buffer at pH 7.4. The biofilms were imaged and analyzed as described (4). All in vitro biofilm assays were repeated a minimum of 3 times on separate days. Data are presented as mean ± SEM. Planktonic and biofilm-resident bacteria were enumerated as described (56).
I.t. challenge of the murine lung with B.
cenocepacia and treatment with HMGB1 isoforms. WT male and female C57BL/6 mice were originally purchased from The Jackson Laboratory. Mice were weight and age matched for endotoxicity experiments. All animals were bred and maintained in the Nationwide Children’s Hospital vivarium. B. cenocepacia strain K56-2 was cultured in LB broth (Difco) at 37°C overnight at 200 RPM. Bacterial concentration was adjusted before each experiment based on OD at 600 nm. C57BL/6 mice were infected (i.t.) with 107 CFU of B. cenocepacia. Mice received 0.2 nmol of rHMGB1 or mHMGB1 (i.t.) at the same time of the infection (prevention) or 24 hours after infection (treatment). Animals were euthanized 24 or 48 hours after infection, and BAL was collected with 1 mL of PBS plus 1 mM EDTA. Cells from BAL were stained with α-CD45 brilliant violet 650 (BioLegend, catalog 103151), α-CD11b Alexa Fluor 700 (BioLegend, catalog 101222), and α-Ly6G PerCP-Cy5.5 (BioLegend, catalog 127616) antibodies, and LIVE/DEAD blue discriminator (Invitrogen). Cells were acquired with a LSR II flow cytometer (BD Biosciences). The number of cells were calculated based on the number of neutrophils in BAL (live, CD45+, CD11bhi, Ly6Ghi) and the total count of cells performed by hemocytometer. Two lungs per cohort were fixed and embedded in paraffin; slides were stained with H&E stain. To visualize B. cenocepacia, paraffin section from lungs infected with 107 CFU B. cenocepacia and treated with 0.2 nmol of rHMGB1 or mHMGB1 at 72 hours after infection were deparaffinated, and antigen retrieval was performed in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) for 15 minutes at 121°C. The slides were permeabilized with 0.5% Triton X-100 for 10 minutes, blocked with 10% normal goat serum (Abcam) for 30 minutes, and stained with a monoclonal antibody against E. coli Elongation Factor-Tu (α-EF-Tu; cross reacts with B. cenocepacia) (LSBio, catalog LS-C128699-100) overnight to label B. cenocepacia. The samples were then stained with goat α-mouse IgG Alexa Fluor 488 (Abcam, catalog ab150113) for 1 hour at room temperature, mounted with FluoroShield media with DAPI (Abcam), visualized via the Nikon Eclipse Ti at 40× magnification, and analyzed by ImageJ (NIH). To enumerate bacterial burden, the animals were euthanized 18 or 72 hours after infection, BAL was collected with 1 mL of PBS plus 1 mM EDTA, and an aliquot of BAL was serially diluted and plated on LB agar.
Generation of polyclonal rabbit anti–tip-chimer or anti–tail-chimer Fabs.
Polyclonal rabbit anti–tip-chimer peptide and anti–tail-chimer peptide were generated at Rockland Immunochemical, Inc. IgG was enriched from each rabbit serum by passage through rProtein A and Protein G GraviTrap columns (GE Healthcare) according to the manufacturer’s instructions. Fabs were then generated via Pierce Fab Preparation kit. Digestion of rabbit IgG to Fabs was confirmed by SDS-PAGE with Coomassie Fluor Orange Protein Gel stain (Thermo Fisher Scientific). Bacterial endotoxin test via ToxinSensor Chromogenic LAL endotoxin kit (Genscript) was performed prior to use.
Chinchilla model of experimental otitis media.
Adult chinchillas (Chinchilla lanigera) were obtained from Rauscher’s Chinchilla Ranch, LLC. These outbred, nonspecific pathogen–free animals were randomly divided into cohorts based on body weight and both male and female animals were enrolled. To test biofilm disruption induced by 200 nM rHMHB1 compared with 200 nM mHMGB1 or equivalent volume of sterile saline, 4–6 animals were enrolled in each cohort. Efficacy of 200 nM mHMGB1 versus 342 nM tip-chimer Fabs + 200 nM mHMGB1, 342 nM tip-chimer Fabs alone, 342 nM tail-chimer Fabs + 200 nM mHMGB1, or 342 nM tail-chimer Fabs alone were evaluated with 3 animals per cohort. A concentration of 342 nM was based on prior studies wherein 5 μg intact IgG per 0.1 mL volume was injected into the middle ears of chinchillas (5, 11, 14).
Disruption of NTHI biofilms resident within the middle ears of chinchillas with experimental otitis media.
Both middle ears of each animal were challenged with 1000 CFU NTHI strain 86-028NP by direct injection to induce experimental otitis media. Four days later, NTHI mucosal biofilms filled more than 50% of the middle ear (54). At this time, treatments were injected into each middle ear (0.1 mL delivered per bulla). Animals were either euthanized 24 hours later or received an identical treatment as described. Animals that received a second treatment were euthanized after an additional 24 hours. To determine the outcome of treatment, animals were euthanized 1 day after completion of antibody therapy, images of mucosal biofilms captured with a Zeiss SV6 dissecting microscope, and then mucosal biofilms and middle ear mucosa were collected, homogenized, and plated on to chocolate agar to semiquantitate the non-planktonic bacterial load within the middle ear (5). Mucosal biofilms were collected and processed as described (5). As an additional assessment, the amount of biofilm in each middle ear was qualitatively determined. Images of each middle ear were captured, randomized, and ranked by 6 blinded reviewers using an established rubric wherein 0 = no mucosal biofilm visible; 1 = less than 25% of middle ear occluded by mucosal biofilm; 2 = 25% to less than 50% occluded; 3 = 50% to less than 75% occluded; 4 = 75%–100% occluded (54). To evaluate relative mucosal inflammation in each middle ear, a second rubric was developed: 0 = no inflammation, mucosa has white hue; 1 = thin capillary dilatation, mucosa has white hue; 2 = thin and thick capillary dilatation, mucosa has red hue, 5 or fewer small hemorrhagic foci; 3 = thick capillary dilatation, mucosa has red hue, more than 5 small and large hemorrhagic foci. For both quantitation of bacterial load and qualitative assessment of mucosal biofilm, each middle ear was considered independent.
Statistics.
Graphical results and statistical tests were performed with GraphPad Prism 8 for all in vitro and in vivo assays. Statistical significance for in vitro assays was assessed by 1-way ANOVA with multiple comparisons. Statistical significance for the mouse model was assessed by 1-way ANOVA with multiple comparisons. Differences in bacterial load, mean mucosal biomass score, and relative mucosal inflammation in the chinchilla model were determined by 1-way ANOVA with multiple comparisons. P values of less than 0.05 were considered significant.
Study approval.
Chinchilla work was performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and under protocol 01304AR approved by the Abigail Wexner Research Institute at Nationwide Children’s Hospital Animal Care and Use Committee. Neither animals nor samples were excluded from evaluation in any study. Mouse work was performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and under protocol AR1300020 approved by the Abigail Wexner Research Institute at Nationwide Children’s Hospital Animal Care and Use Committee. Neither animals nor samples were excluded from evaluation in any study.
Author contributions
AD, LOB, SPS, and SDG designed the study. AD, LAN, FRA, and SDG wrote the paper. AD, LAN, FRA, JRB, LMW, JAJ, and NOT performed experiments and analyzed data.
Supplementary Material
Acknowledgments
We thank Woojin An at University of Southern California for his generous gift of the tagged-HMGB1 expression plasmid. We thank Jennifer S. Downey for cloning human HMGB1 into the pTXB1 vector. We thank Jason Navarro for help with uploading figures. This work was supported by NIH grant R01DC011818 to SDG and LOB; NIH grant R01AI155501 to SDG, LOB, and SPS; and Cystic Fibrosis Foundation grant PARTID18P0 to SPS.
Version 1. 08/16/2021
Electronic publication
Funding Statement
To SDG and LOB
To SPS
Footnotes
Conflict of interest: SDG and LOB are founders, shareholders, and members of the scientific advisory board of Clarametyx Biosciences Inc., to which rights to technologies used herein have been licensed.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(16):e140527.https://doi.org/10.1172/JCI140527.
Contributor Information
Aishwarya Devaraj, Email: aishwarya.devaraj@nationwidechildrens.org.
Laura A. Novotny, Email: novotnyl@musc.edu.
Lauren Mashburn-Warren, Email: Lauren.Warren@nationwidechildrens.org.
Joseph A. Jurcisek, Email: joseph.jurcisek@nationwidechildrens.org.
Natalia O. Tjokro, Email: tjokro@usc.edu.
Santiago Partida-Sanchez, Email: Santiago.Partida-Sanchez@nationwidechildrens.org.
Lauren O. Bakaletz, Email: Lauren.Bakaletz@nationwidechildrens.org.
Steven D. Goodman, Email: Steven.Goodman@nationwidechildrens.org.
References
- 1.Slinger R, et al. Multiple combination antibiotic susceptibility testing of nontypeable Haemophilus influenzae biofilms. Diagn Microbiol Infect Dis. 2006;56(3):247–253. doi: 10.1016/j.diagmicrobio.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 2.Starner TD, et al. Subinhibitory concentrations of azithromycin decrease nontypeable Haemophilus influenzae biofilm formation and diminish established biofilms. Antimicrob Agents Chemother. 2008;52(1):137–145. doi: 10.1128/AAC.00607-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flemming HC, Wingender J. The biofilm matrix. Nat Rev Microbiol. 2010;8(9):623–633. doi: 10.1038/nrmicro2415. [DOI] [PubMed] [Google Scholar]
- 4.Devaraj A, et al. The extracellular DNA lattice of bacterial biofilms is structurally related to Holliday junction recombination intermediates. Proc Nat Acad Sci U S A. 2019;116(50):25068–25077. doi: 10.1073/pnas.1909017116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goodman SD, et al. Biofilms can be dispersed by focusing the immune system on a common family of bacterial nucleoid-associated proteins. Mucosal Immunol. 2011;4(6):625–637. doi: 10.1038/mi.2011.27. [DOI] [PubMed] [Google Scholar]
- 6.Gustave JE, et al. Targeting bacterial integration host factor to disrupt biofilms associated with cystic fibrosis. J Cyst Fibros. 2013;12(4):384–389. doi: 10.1016/j.jcf.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Novotny LA, et al. Structural stability of Burkholderia cenocepacia biofilms is reliant on eDNA structure and presence of a bacterial nucleic acid binding protein. PLoS One. 2013;8(6):e67629. doi: 10.1371/journal.pone.0067629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brockson ME, et al. Evaluation of the kinetics and mechanism of action of anti-integration host factor-mediated disruption of bacterial biofilms. Mol Microbiol. 2014;93(6):1246–1258. doi: 10.1111/mmi.12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brandstetter KA, et al. Antibodies directed against integration host factor mediate biofilm clearance from Nasopore. Laryngoscope. 2013;123(11):2626–2632. doi: 10.1002/lary.24183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rocco CJ, et al. Natural antigenic differences in the functionally equivalent extracellular DNABII proteins of bacterial biofilms provide a means for targeted biofilm therapeutics. Mol Oral Microbiol. 2016;32(2):118–130. doi: 10.1111/omi.12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Novotny LA, et al. Monoclonal antibodies against DNA-binding tips of DNABII proteins disrupt biofilms in vitro and induce bacterial clearance in vivo. EBioMedicine. 2016;10:33–44. doi: 10.1016/j.ebiom.2016.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Devaraj A, et al. The DNABII family of proteins is comprised of the only nucleoid associated proteins required for nontypeable Haemophilus influenzae biofilm structure. Microbiologyopen. 2017;7(3):e00563. doi: 10.1002/mbo3.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freire MO, et al. A bacterial-biofilm-induced oral osteolytic infection can be successfully treated by immuno-targeting an extracellular nucleoid-associated protein. Mol Oral Microbiol. 2017;32(1):74–88. doi: 10.1111/omi.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Novotny LA, et al. Redirecting the immune response towards immunoprotective domains of a DNABII protein resolves experimental otitis media. NPJ Vaccines. 2019;4:43. doi: 10.1038/s41541-019-0137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bianchi ME, et al. Specific recognition of cruciform DNA by nuclear protein HMG1. Science. 1989;243(4894 pt 1):1056–1059. doi: 10.1126/science.2922595. [DOI] [PubMed] [Google Scholar]
- 16.Bianchi ME, Beltrame M. Flexing DNA: HMG-box proteins and their partners. Am J Hum Genet. 1998;63(6):1573–1577. doi: 10.1086/302170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agresti A, et al. GR and HMGB1 interact only within chromatin and influence each other’s residence time. Mol Cell. 2005;18(1):109–121. doi: 10.1016/j.molcel.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 18.Bianchi ME, et al. The DNA binding site of HMG1 protein is composed of two similar segments (HMG boxes), both of which have counterparts in other eukaryotic regulatory proteins. EMBO J. 1992;11(3):1055–1063. doi: 10.1002/j.1460-2075.1992.tb05144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang R, et al. HMGB1 in health and disease. Mol Aspects Med. 2014;40:1–116. doi: 10.1016/j.mam.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Little AJ, et al. Cooperative recruitment of HMGB1 during V(D)J recombination through interactions with RAG1 and DNA. Nucleic Acids Res. 2013;41(5):3289–3301. doi: 10.1093/nar/gks1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sutrias-Grau M, et al. High mobility group protein 1 interacts specifically with the core domain of human TATA box-binding protein and interferes with transcription factor IIB within the pre-initiation complex. J Biol Chem. 1999;274(3):1628–1634. doi: 10.1074/jbc.274.3.1628. [DOI] [PubMed] [Google Scholar]
- 22.Yuan F, et al. Evidence for involvement of HMGB1 protein in human DNA mismatch repair. J Biol Chem. 2004;279(20):20935–20940. doi: 10.1074/jbc.M401931200. [DOI] [PubMed] [Google Scholar]
- 23.Klune JR, et al. HMGB1: endogenous danger signaling. Mol Med. 2008;14(7-8):476–484. doi: 10.2119/2008-00034.Klune. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park JS, et al. Activation of gene expression in human neutrophils by high mobility group box 1 protein. Am J Physiol Cell Physiol. 2003;284(4):C870–C879. doi: 10.1152/ajpcell.00322.2002. [DOI] [PubMed] [Google Scholar]
- 25.Silva E, et al. HMGB1 and LPS induce distinct patterns of gene expression and activation in neutrophils from patients with sepsis-induced acute lung injury. Intensive Care Med. 2007;33(10):1829–1839. doi: 10.1007/s00134-007-0748-2. [DOI] [PubMed] [Google Scholar]
- 26.He Q, et al. HMGB1 promotes the synthesis of pro-IL-1β and pro-IL-18 by activation of p38 MAPK and NF-κB through receptors for advanced glycation end-products in macrophages. Asian Pac J Cancer Prev. 2012;13(4):1365–1370. doi: 10.7314/APJCP.2012.13.4.1365. [DOI] [PubMed] [Google Scholar]
- 27.Qin S, et al. Role of HMGB1 in apoptosis-mediated sepsis lethality. J Exp Med. 2006;203(7):1637–1642. doi: 10.1084/jem.20052203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diener KR, et al. The multifunctional alarmin HMGB1 with roles in the pathophysiology of sepsis and cancer. Immunol Cell Biol. 2013;91(7):443–450. doi: 10.1038/icb.2013.25. [DOI] [PubMed] [Google Scholar]
- 29.Ranzato E, et al. HMGb1 promotes scratch wound closure of HaCaT keratinocytes via ERK1/2 activation. Mol Cell Biochem. 2009;332(1–2):199–205. doi: 10.1007/s11010-009-0192-4. [DOI] [PubMed] [Google Scholar]
- 30.Gong W, et al. Amino acid residues 201-205 in C-terminal acidic tail region plays a crucial role in antibacterial activity of HMGB1. J Biomed Sci. 2009;16:83. doi: 10.1186/1423-0127-16-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davalos AR, et al. p53-dependent release of alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol. 2013;201(4):613–629. doi: 10.1083/jcb.201206006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zetterstrom CK, et al. High mobility group box chromosomal protein 1 (HMGB1) is an antibacterial factor produced by the human adenoid. Pediatr Res. 2002;52(2):148–154. doi: 10.1203/00006450-200208000-00004. [DOI] [PubMed] [Google Scholar]
- 33.Tadie JM, et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am J Physiol Lung Cell Mol Physiol. 2013;304(5):L342–L349. doi: 10.1152/ajplung.00151.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Remijsen Q, et al. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. 2011;18(4):581–588. doi: 10.1038/cdd.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brinkmann V, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 36.Peng HH, et al. Mineral particles stimulate innate immunity through neutrophil extracellular traps containing HMGB1. Sci Rep. 2017;7(1):16628. doi: 10.1038/s41598-017-16778-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Browning DF, et al. Effects of nucleoid-associated proteins on bacterial chromosome structure and gene expression. Curr Opin Microbiol. 2010;13(6):773–780. doi: 10.1016/j.mib.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 38.Paull TT, et al. The nonspecific DNA-binding and -bending proteins HMG1 and HMG2 promote the assembly of complex nucleoprotein structures. Genes Dev. 1993;7(8):1521–1534. doi: 10.1101/gad.7.8.1521. [DOI] [PubMed] [Google Scholar]
- 39.Segall AM, et al. Architectural elements in nucleoprotein complexes: interchangeability of specific and non-specific DNA binding proteins. EMBO J. 1994;13(19):4536–4548. doi: 10.1002/j.1460-2075.1994.tb06775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanchez-Giraldo R, et al. Two high-mobility group box domains act together to underwind and kink DNA. Acta Crystallogr D Biol Crystallogr. 2015;71(pt 7):1423–1432. doi: 10.1107/S1399004715007452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rice PA, et al. Crystal structure of an IHF-DNA complex: a protein-induced DNA U-turn. Cell. 1996;87(7):1295–1306. doi: 10.1016/S0092-8674(00)81824-3. [DOI] [PubMed] [Google Scholar]
- 42. World Health Organization. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. https://www.who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf Accessed July 6, 2021.
- 43.Ch’ng JH, et al. Biofilm-associated infection by enterococci. Nat Rev Microbiol. 2019;17(2):82–94. doi: 10.1038/s41579-018-0107-z. [DOI] [PubMed] [Google Scholar]
- 44.Brockson ME, et al. Evaluation of the kinetics and mechanism of action of anti-integration host factor mediated disruption of bacterial biofilms. Mol Microbiol. 2014;93(6):1246–1258. doi: 10.1111/mmi.12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Freire MO, et al. A bacterial biofilm induced oral osteolytic infection can be successfully treated by immuno-targeting an extracellular nucleoid associated protein. Mol Oral Microbiol. 2016;32(1):74–88. doi: 10.1111/omi.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Venereau E, et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med. 2012;209(9):1519–1528. doi: 10.1084/jem.20120189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Antoine DJ, et al. A systematic nomenclature for the redox states of high mobility group box (HMGB) proteins. Mol Med. 2014;20:135–137. doi: 10.2119/molmed.2014.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang H, et al. Redox modifications of cysteine residues regulate the cytokine activity of HMGB1. Mol Med. 2021;27(1):58. doi: 10.1186/s10020-021-00307-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu A, et al. Oxidation of HMGB1 causes attenuation of its pro-inflammatory activity and occurs during liver ischemia and reperfusion. PLoS One. 2012;7(4):e35379. doi: 10.1371/journal.pone.0035379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sheflin LG, et al. The specific interactions of HMG 1 and 2 with negatively supercoiled DNA are modulated by their acidic C-terminal domains and involve cysteine residues in their HMG 1/2 boxes. Biochemistry. 1993;32(13):3238–3248. doi: 10.1021/bi00064a005. [DOI] [PubMed] [Google Scholar]
- 51.Xin H, et al. DNA binding by single HMG box model proteins. Nucleic Acids Res. 2000;28(20):4044–4050. doi: 10.1093/nar/28.20.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Banerjee S, et al. Intracellular HMGB1 negatively regulates efferocytosis. J Immunol. 2011;187(9):4686–4694. doi: 10.4049/jimmunol.1101500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stevens NE, et al. Therapeutic targeting of HMGB1 during experimental sepsis modulates the inflammatory cytokine profile to one associated with improved clinical outcomes. Sci Rep. 2017;7(1):5850. doi: 10.1038/s41598-017-06205-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Novotny LA, et al. Transcutaneous immunization as preventative and therapeutic regimens to protect against experimental otitis media due to nontypeable Haemophilus influenzae. Mucosal Immunol. 2011;4(4):456–467. doi: 10.1038/mi.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Novotny LA, et al. Targeting a bacterial DNABII protein with a chimeric peptide immunogen or humanised monoclonal antibody to prevent or treat recalcitrant biofilm-mediated infections. EBioMedicine. 2020;59:102867. doi: 10.1016/j.ebiom.2020.102867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Devaraj A, et al. DNABII proteins play a central role in UPEC biofilm structure. Mol Microbiol. 2015;96(6):1119–1135. doi: 10.1111/mmi.12994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Idicula WA, et al. Identification of biofilms in post-tympanostomy tube otorrhea. Laryngoscope. 2016;126(8):1946–1951. doi: 10.1002/lary.25826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jurcisek JA, et al. Nontypeable Haemophilus influenzae releases DNA and DNABII proteins via a T4SS-like complex and ComE of the type IV pilus machinery. Proc Natl Acad Sci U S A. 2017;114(32):E6632–E6641. doi: 10.1073/pnas.1705508114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alhede M, et al. The origin of extracellular DNA in bacterial biofilm infections in vivo. Pathog Dis. 2020;78(2):ftaa018. doi: 10.1093/femspd/ftaa018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pontiggia A, et al. Protein HU binds specifically to kinked DNA. Mol Microbiol. 1993;7(3):343–350. doi: 10.1111/j.1365-2958.1993.tb01126.x. [DOI] [PubMed] [Google Scholar]
- 61.Bonnefoy E, et al. DNA-binding parameters of the HU protein of Escherichia coli to cruciform DNA. J Mol Biol. 1994;242(2):116–129. doi: 10.1006/jmbi.1994.1563. [DOI] [PubMed] [Google Scholar]
- 62.Vitoc CI, Mukerji I. HU binding to a DNA four-way junction probed by Förster resonance energy transfer. Biochemistry. 2011;50(9):1432–1441. doi: 10.1021/bi1007589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Deng V.Y., Mukerji I. Stability of DNA four-way junctions and characterization of binding to integration host factor. Wesleyan University; 2016. [Google Scholar]
- 64.JR Po, et al. HMG box proteins bind to four-way DNA junctions in their open conformation. EMBO J. 1998;17(3):817–826. doi: 10.1093/emboj/17.3.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hargreaves D, et al. Crystal structure of E.coli RuvA with bound DNA Holliday junction at 6 A resolution. Nat Struct Biol. 1998;5(6):441–446. doi: 10.1038/nsb0698-441. [DOI] [PubMed] [Google Scholar]
- 66.Delgado-Rizo V, et al. Neutrophil extracellular traps and its implications in inflammation: an overview. Front Immunol. 2017;8:81. doi: 10.3389/fimmu.2017.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mulvey MA, et al. Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infect Immun. 2001;69(7):4572–4579. doi: 10.1128/IAI.69.7.4572-4579.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Baldwin A, et al. Multilocus sequence typing scheme that provides both species and strain differentiation for the Burkholderia cepacia complex. J Clin Microbiol. 2005;43(9):4665–4673. doi: 10.1128/JCM.43.9.4665-4673.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Harrison A, et al. Genomic sequence of an otitis media isolate of nontypeable Haemophilus influenzae: comparative study with H. influenzae serotype d, strain KW20. J Bacteriol. 2005;187(13):4627–4636. doi: 10.1128/JB.187.13.4627-4636.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Palmer KL, et al. Comparative genomics of enterococci: variation in Enterococcus faecalis, clade structure in E. faecium, and defining characteristics of E. gallinarum and E. casseliflavus. mBio. 2012;3(1):e00318–11. doi: 10.1128/mBio.00318-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.