

Abstract
The pathogenesis of Parkinson’s disease (PD) remains elusive, but mitochondrial dysfunction is believed to be one crucial step in its pathogenesis. The mitochondrial unfolded protein response (UPRmt) is an important mitochondrial quality control strategy that maintains mitochondrial function in response to disturbances of mitochondrial protein homeostasis. Activation of the UPRmt and the beneficial effect of rescuing mitochondrial proteostasis have been reported in several genetic models of PD. However, the pathogenic relevance of the UPRmt in idiopathic PD is unknown. The present study examined the link between the UPRmt and mitochondrial dysfunction in 1-methyl-4-phenylpyridinium (MPP+)-treated SH-SY5Y cells. Treatment with MPP+ induced activation of the UPRmt, reflected by an increase in the expression of UPRmt-related chaperones, proteases, and transcription mediators. UPRmt activation that was induced by overexpressing mutant ornithine transcarbamylase significantly reduced the production of mitochondrial reactive oxygen species (ROS) and improved cell survival in SH-SY5Y cells following MPP+ treatment. Moreover, the overexpression of activating transcription factor 5 (mammalian UPRmt transcription factor) conferred protection against MPP+-induced ROS production and against cell death in SH-SY5Y cells. Overall, our results demonstrate the beneficial effect of UPRmt activation in MPP+-treated cells, shedding new light on the mechanism of mitochondrial dysfunction in the pathogenesis of PD.
Keywords: MPP+, UPRmt, ATF5, Parkinson’s disease
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, characterized by the presence of Lewy bodies and degeneration of dopaminergic neurons in the dense part of the substantia nigra and striatum. Parkinson’s disease affects around 10 million people worldwide [1]. Despite the lack of a complete understanding of the molecular mechanisms that underlie PD, efforts have been made to identify risk factors for developing PD. Mutant genes, such as α-synuclein (SNCA), PTEN-induced putative kinase protein 1 (PINK), parkin, and protein deglycase DJ-1, can lead to familial PD. Over 90% of idiopathic cases are believed to result from multiple factors, including mutations of low-risk genes (e.g. Glucosylceramidase Beta), chronic inflammation in the central nervous system, and microbiota dysbiosis in the gut [2, 3].
Although the pathogenesis of PD remains elusive, numerous studies suggest an important role for mitochondrial dysfunction in its pathogenesis. Parkinson’s disease patients exhibit deficits in electron transport chain function [4]. Accumulating evidence from various mouse models of PD indicates the accumulation of mitochondrial reactive oxygen species (ROS), impairments in mitophagy, and the dysregulation of mitochondrial proteostasis [5–8]. Moreover, mitochondrial toxins, including 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and rotenone, have been shown to selectively impair dopaminergic neurons by inhibiting mitochondrial complex I. Thus, these mitochondrial toxins have been widely used to model PD [9–11]. Additionally, variants of genes that are involved in mitochondrial function have been shown to cause familial PD, including parkin and PINK1 [12, 13].
The mitochondrial unfolded protein response (UPRmt) recently emerged as a novel quality control mechanism that regulates mitochondrial proteostasis. Similar to the endoplasmic reticulum unfolded protein response, the UPRmt is generally stimulated and activated when an overload of unfolded or misfolded proteins occurs in the mitochondrial matrix [14]. Conditions that increase mitochondrial proteotoxicity, such as mitochondrial DNA depletion and perturbations of oxidative phosphorylation (OXPHOS), can elicit the UPRmt [15, 16]. The UPRmt is a retrograde signaling that is mediated by b-ZIP (basic leucine zipper) transcription factors, such as stress-activated transcription factor 1 (ATFS-1) in C. elegans and activating transcription factor 5 (ATF5) in mammalian cells [17, 18]. Under physiological conditions, ATFS-1 and ATF5 are imported into mitochondria and degraded by mitochondrial proteases [17, 18]. Upon stimulation, the transcription mediator is translocated to the nucleus, together with the co-factors ubiquitin-like protein 5 (UBL5)/defective proventriculus homolog protein (DVE-1)/C/EBP-homologous protein (CHOP), to induce the expression of mitochondrial chaperones (e.g., heat shock protein 10 [HSP10]/HSP60/HSP70) and proteases (e.g., ClpP/ClpX, Caseinolytic Mitochondrial Matrix Peptidase subunit P/X) which are transported back to mitochondria to rescue protein homeostasis and accommodate the stress conditions [19, 20]. Activation of the UPRmt induces mitochondrial biogenesis through OXPHOS and promotes pathogen resistance and lifespan extension [16, 21, 22]. Thus, the UPRmt can promote metabolic balance and protein homeostasis in response to mitochondrial stressors.
Previous studies suggested a protective effect of the UPRmt, specifically in genetic models of PD. However, whether UPRmt activation is beneficial in idiopathic cases is unknown. In the present study, we observed an increase in the expression of UPRmt-related proteases, chaperones, and transcription factors in dopaminergic SH-SY5Y cells, and this expression decreased over time upon treatment with 1-methyl-4-phenylpyridinium (MPP+), a metabolic product of MPTP in the brain. Activation of the UPRmt by overexpressing mutant ornithine transcarbamylase by the deletion of amino acids 30–114 (Δ-OTC) significantly reduced the production of mitochondrial reactive oxygen species (ROS) and cell death that were induced by the addition of MPP+. Lastly, ATF5 overexpression prevented mitochondrial ROS production and improved cell viability in SH-SY5Y cells that were treated with MPP+. Our results suggest that UPRmt activation can mitigate mitochondrial stress and protect against neuronal damage in PD.
Materials and Methods
Antibodies and reagents
Antibodies against ATF5 (catalog no. ab184923, 1:3000), ClpP (catalog no. ab124822, 1:3000), and ClpX (catalog no. ab168338, 1:3000) were purchased from Abcam. Antibodies against HSP60 (catalog no. sc-13115, 1:3000) and c-Myc (9E10) (catalog no. sc-40, 1:4000) were purchased from Santa Cruz Biotechnology. The antibody against Lon protease-like protein (LONP; catalog no. 15440–1-AP, 1:3000) was purchased from Proteintech. The antibody against β-actin (catalog no. A1978, 1:10000) was purchased from Sigma. The antibody against UBL5 (catalog no. LS-C160393–0.1, 1:1000) was purchased from LSBio. MPP+ iodide (catalog no. D048) was purchased from Sigma.
Cell culture and treatment
SH-SY5Y cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM)/F12 (1:1) supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) antibiotics (100 μg/ml penicillin and 100 μg/ml streptomycin) at 37°C in a 5% CO2 incubator. When the cell density of SH-SY5Y cells was higher than 80%, SH-SY5Y cells were incubated with 2 mM MPP+.
Preparation of total cell lysates
SH-SY5Y cells were washed with 1X phosphate-buffered saline (PBS) and then incubated in total lysis buffer (10 mM HEPE-NaOH [pH 7.8], 150 mM NaCl, 1 mM ethylene glycol-bis[β-aminoethyl ether]-N,N,N’,N’-tetraacetic acid [EGTA], 1% Triton X-100, protease inhibitors, and phosphatase inhibitors) for 30 min on ice. Samples were collected and centrifuged at 12,000 rotations per minute for 10 min at 4°C. The supernatants were saved as total lysates.
Western blot
Protein concentrations were measured using the Bradford assay. Proteins (30 μg) were then resuspended in Laemmni buffer, loaded on sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels, and transferred to nitrocellulose membranes. The membranes were probed with the indicated antibodies and visualized by electrochemiluminescence.
Real-time quantitative polymerase chain reaction
Cells were washed with 1X PBS once. Total RNA was extracted using the RNeasy plus mini kit (catalog no. 74034, Qiagen). For each sample, cDNA was generated using the Quantitect reverse transcription kit (catalog no. 205311, Qiagen). The relative amount of cDNA was determined using SYBR Green Polymerase Chain Reaction (PCR) Master Mix (catalog no. 4309155, ThermoFisher Scientific) on the QuantStudio 3 Real-time PCR platform (ThermoFisher Scientific) according to the manufacturer’s instructions. The primer sequences were the following: ClpP (forward: TTGCCAGCCTTGTTATCGCA; reverse: GGTTGAGGATGTACTGCATCG), UBL5 (forward: GGGAAGAAGGTCCGCGTTAAA; reverse: ACGTGGTCCTTAAAAATCGTGT), HSP60 (forward: GTGTAGACCTTTTAGCCGATG; reverse: GTGCCAGTACAGTAGCAGTGG).
Measurement of cell viability
SH-SY5Y cells were plated in 96-well plates with 15,000 cells in each well. After treatment with MPP+, cell viability was determined using the MTT assay kit (catalog no. 11465007001, Roche) according to the manufacturer’s instructions.
Mitochondrial ROS measurement
Cells were cultured on coverslips, washed with PBS, and incubated with 5 μM MitoSOX Red (catalog no. M36008, Invitrogen), a mitochondrial superoxide indicator, and 5 μg/ml Hoechst (catalog no. 33342, Invitrogen) for 10 min at 37°C for SH-SY5Y cells. The images were then visualized under a microscope (Olympus, Fluoview FV100), and mitochondrial ROS were quantified using ImageJ software (National Institutes of Health). At least 100 cells per group were counted for the analysis. The MitoSOX fluorescence density was normalized to the total number of cells that were labeled with 4’,6-diamidino-2-phenylindole (DAPI).
Constructs and transfection
Δ-OTC plasmid was obtained from Addgene (catalog no. 71878). Myc-tagged full-length human ATF5 was purchased from Origene (catalog no. RC200081). Cells were transfected with TransIT-2020 (Mirus Bio) according to the manufacturer’s instructions.
Results
Protein levels of UPRmt-related components were altered in SH-SY5Y cells upon treatment with MPP+
Mitochondrial proteostasis is mainly surveilled and regulated by UPRmt-related chaperones and proteases, including HSP60, ClpP, ClpX, and LONP [22]. Activation of the UPRmt results in the expression of selective mitochondrial proteases and chaperones that rescue mitochondrial protein homeostasis and relieve mitochondrial stress. To determine whether the UPRmt is affected by MPP+ treatment, we first examined protein levels of several mitochondrial proteases that are involved in UPRmt signaling in SHSY5Y cells that were treated with MPP+ for 2, 4, 8, 16, or 24 h. The protein level of the UPRmt-associated protease ClpP significantly increased and peaked 4 h after MPP+ treatment and then gradually decreased (Fig. 1A, B). The protein level of ClpX significantly decreased after 16-h treatment with MPP+ in SH-SY5Y cells (Fig. 1A, D). The protein level of LONP, an essential mitochondrial matrix protease that is not involved in the canonical UPRmt pathway, remained unchanged by MPP+ treatment (Fig. 1A, E), suggesting selective responses of UPRmt components under mitochondrial stress that was induced by MPP+. The expression of UPRmt-related chaperones and proteases is mediated by co-transcription factors, such as UBL5. Similar to changes in ClpP, the protein level of UBL5 significantly increased but then decreased with MPP+ treatment in SH-SY5Y cells (Fig. 1A, C). These findings indicate activation of the UPRmt that was induced by MPP+ at the early stage, which was then diminished in SH-SY5Y cells.
Fig. 1. MPP+ treatment affected the expression of UPRmt-related components.
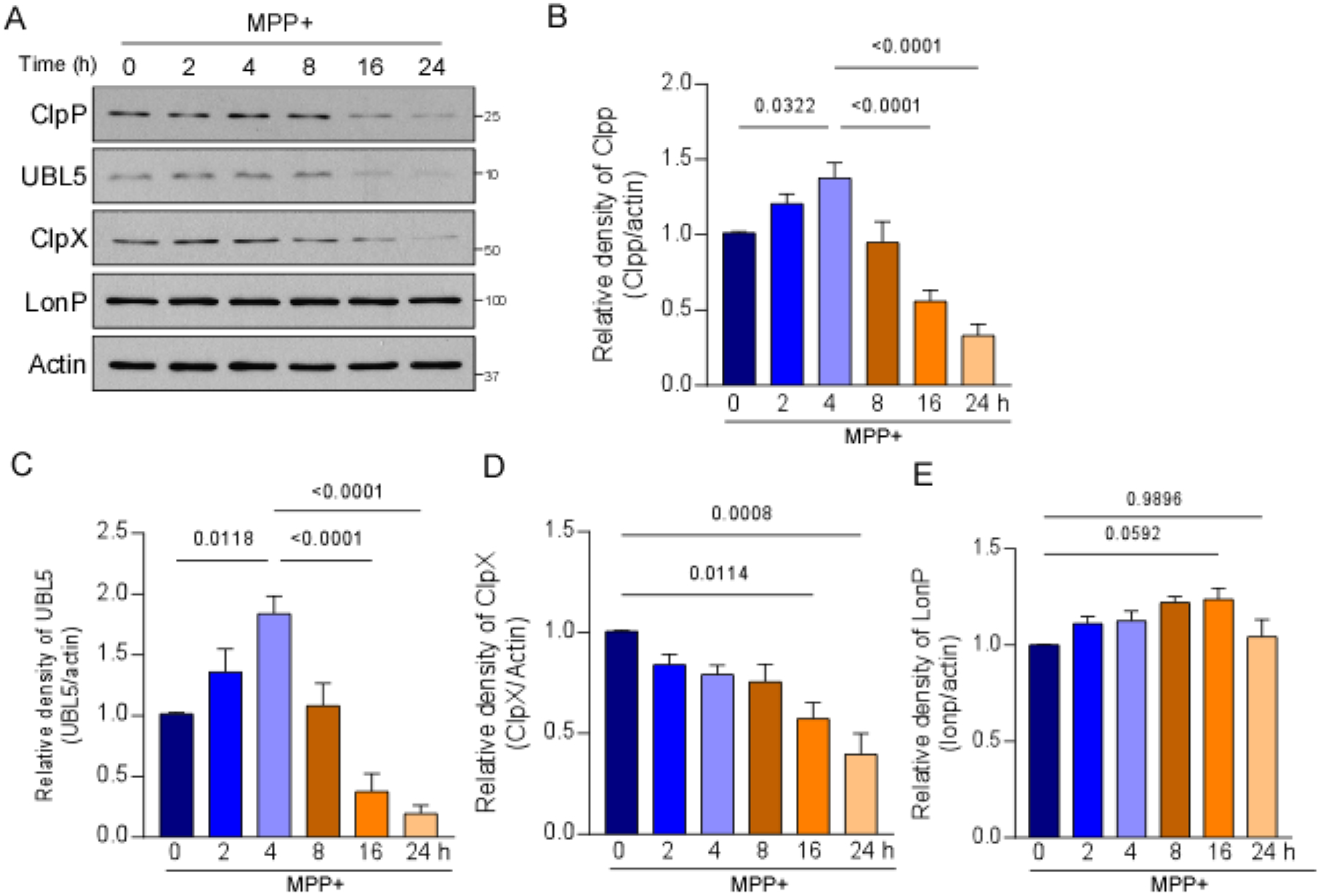
SH-SY5Y cells were treated with 2 mM MPP+ for 2, 4, 8, 16, and 24 h. (A) Total cell lysates were harvested after MPP+ treatment and subjected to Western blot analyses with the indicated antibodies. Histograms show the relative density of (B) ClpP, (C) UBL5, (D) ClpX, and (E) LONP protein levels relative to total protein levels. The data are expressed as the mean ± SEM of three independent experiments. Data are compared with one-way ANOVA with Tukey’s post hoc test.
MPP+ treatment activated the UPRmt
To confirm activation of the UPRmt by MPP+, we assessed the transcription of ClpP, UBL5, and HSP60 in SH-SY5Y cells. mRNA levels of ClpP, UBL5, and HSP60 significantly and rapidly increased upon 2-h treatment with MPP+ (Fig. 2A), indicating activation of the UPRmt. mRNA levels of ClpP, UBL5, and HSP60 decreased after 8-h treatment with MPP+ (Fig. 2A). We also measured the protein level of ATF5, a transcriptional mediator of mammalian UPRmt signaling. The level of ATF5 increased within 8 h of MPP+ treatment in SH-SY5Y cells and then decreased (Fig. 2B). Overall, these results suggest transcriptional activation of the UPRmt at early time points upon MPP+ treatment.
Fig. 2. MPP+ induced activation of the UPRmt at early time points.
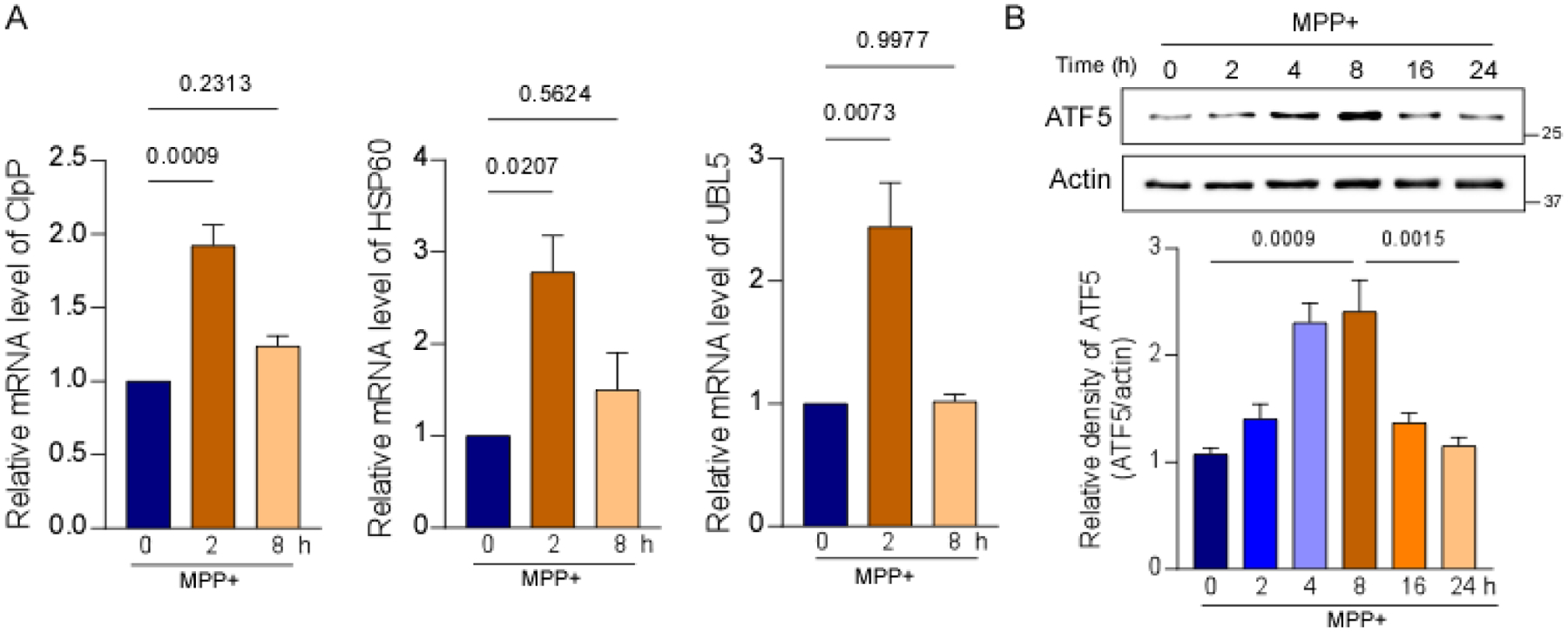
(A) mRNA levels of ClpP, HSP60, and UBL5 were determined by real-time quantitative PCR in SH-SY5Y cells after treatment with 2 mM MPP+ for 2 and 8 h. (B) Total protein lysates were harvested from SH-SY5Y cells after treatment with 2 mM MPP+ for 2, 4, 8, 16, and 24 h and subjected to Western blot analyses with the indicated antibodies. The histogram shows the relative density of ATF5. The data are expressed as the mean ± SEM of three independent experiments. Data are compared with one-way ANOVA with Tukey’s post hoc test.
UPRmt activation induced by Δ-OTC overexpression conferred protection against MPP+-induced toxicity
Previous studies suggested that activation of the UPRmt mitigates mitochondrial dysfunction and promotes dopaminergic neuronal survival in nematode models of PD [8]. We hypothesized that UPRmt activation is beneficial during MPP+ treatment. To activate the UPRmt, ΔOTC was expressed in SH-SY5Y cells before MPP+ treatment [23] (Fig. 3A). MPP+ treatment significantly reduced the expression of ClpP and ClpX in cells that expressed the control vector. The overexpression of Myc-tagged Δ-OTC significantly increased protein levels of ClpP and ClpX after treatment with MPP+ (Fig. 3A–C). Notably, the protein level of LONP did not change in either empty vector- or Myc-tagged-Δ-OTC-expressing cells upon MPP+ treatment (Fig. 3A, C). To determine whether UPRmt activation can protect mitochondria against MPP+ toxicity, we measured the production of mitochondrial ROS in SH-SY5Y cells. The overexpression of Δ-OTC significantly reduced the MPP+-induced production of mitochondrial ROS (Fig. 3E). UPRmt activation also significantly improved the viability of SH-SY5Y cells that were treated with MPP+ (Fig. 3F). Notably, Δ-OTC expression did not affect the production of mitochondrial ROS or cell viability (Fig. 3E, F). These findings indicate a protective effect of UPRmt activation against MPP+-induced mitochondrial and cellular stress.
Fig. 3. UPRmt activation conferred protection against MPP+-induced toxicity.
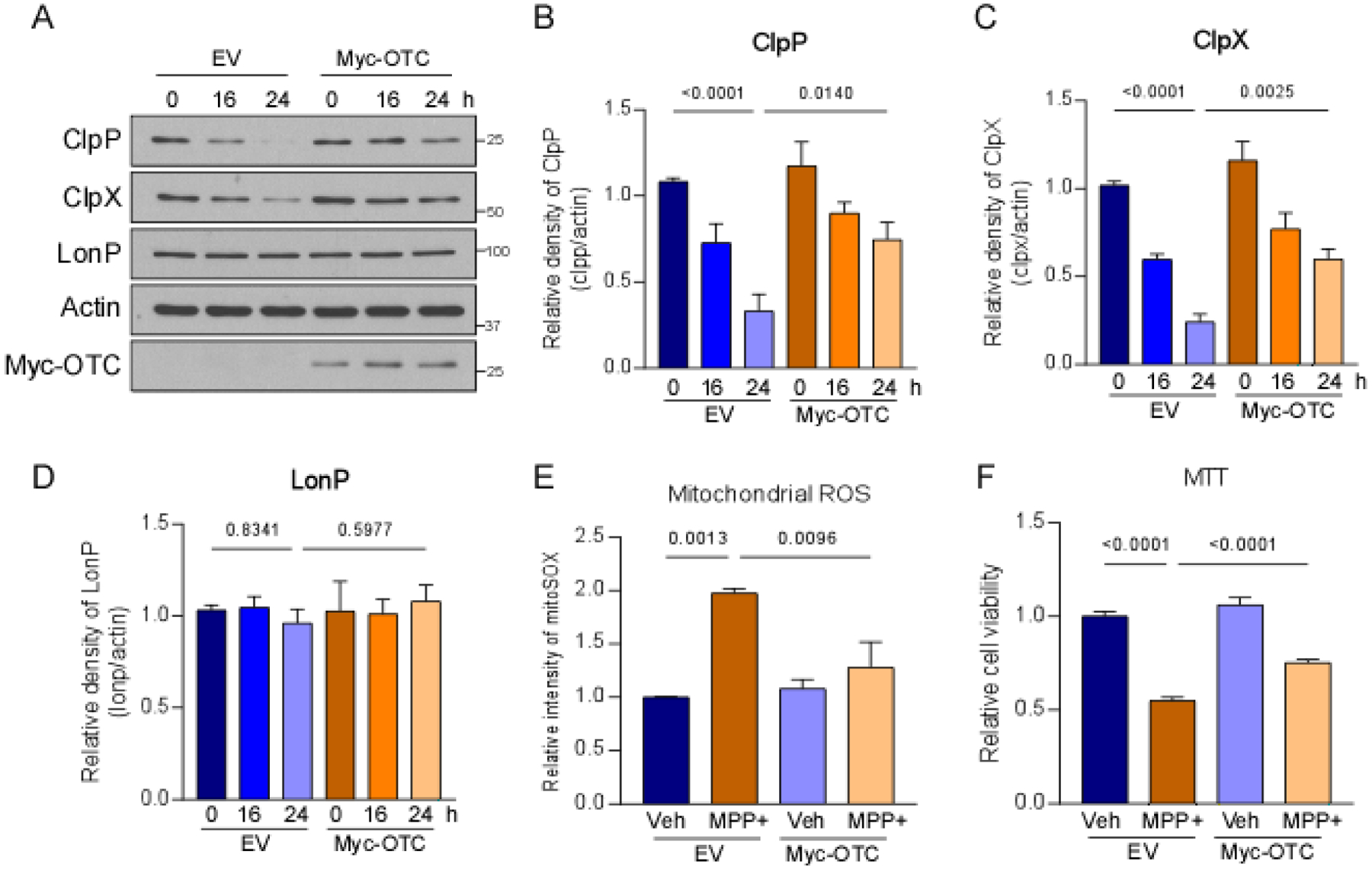
SH-SY5Y cells were treated with 2 mM MPP+ after the overexpression of Δ-OTC. (A) Total protein lysates were harvested after MPP+ treatment and subjected to Western blot analyses with the indicated antibodies. The Histograms show the density of (B) ClpP, (C) ClpX, and (D) LONP protein levels relative to total protein levels. (E) Cells were stained with MitoSOX™ dye to assess mitochondrial superoxide production after 24 h treatment with MPP+. The histogram shows the relative fluorescence density of MitoSOX. (F) Cell viability was examined by the MTT assay after 24 h treatment with MPP+. The histogram shows relative cell survival. The data are expressed as the mean ± SEM of three independent experiments. Data are compared with one-way ANOVA with Tukey’s post hoc test.
Upregulation of ATF5 ameliorated mitochondrial dysfunction and cell toxicity
MPP+ treatment induced the expression of ATF5 in SH-SY5Y cells (Fig. 2B). ATF5 is a critical transcriptional mediator of mammalian UPRmt signaling [17]. We sought to determine whether the upregulation of ATF5 protects against MPP+-induced neuronal toxicity. The overexpression of ATF5 significantly increased protein levels of ClpP and ClpX in SH-SY5Y cells after treatment with MPP+ (Fig. 4A–C). The level of LONP did not change in either control vector- or ATF5-expressing cells that were treated with MPP+ (Fig. 4A, D). The overexpression of ATF5 significantly reduced the production of mitochondrial ROS in cells that were treated with MPP+ (Fig. 4E). We also found ATF5 overexpression improved cell viability in SH-SY5Y cells after MPP+ treatment (Fig. 4F). ATF5 expression did not affect the production of mitochondrial ROS or cell viability in normal cells (Fig. 4E, F). These results indicate that ATF5 upregulation conferred protection in MPP+-treated cells.
Fig. 4. Overexpression of ATF5 reduced MPP+-induced cellular stress.
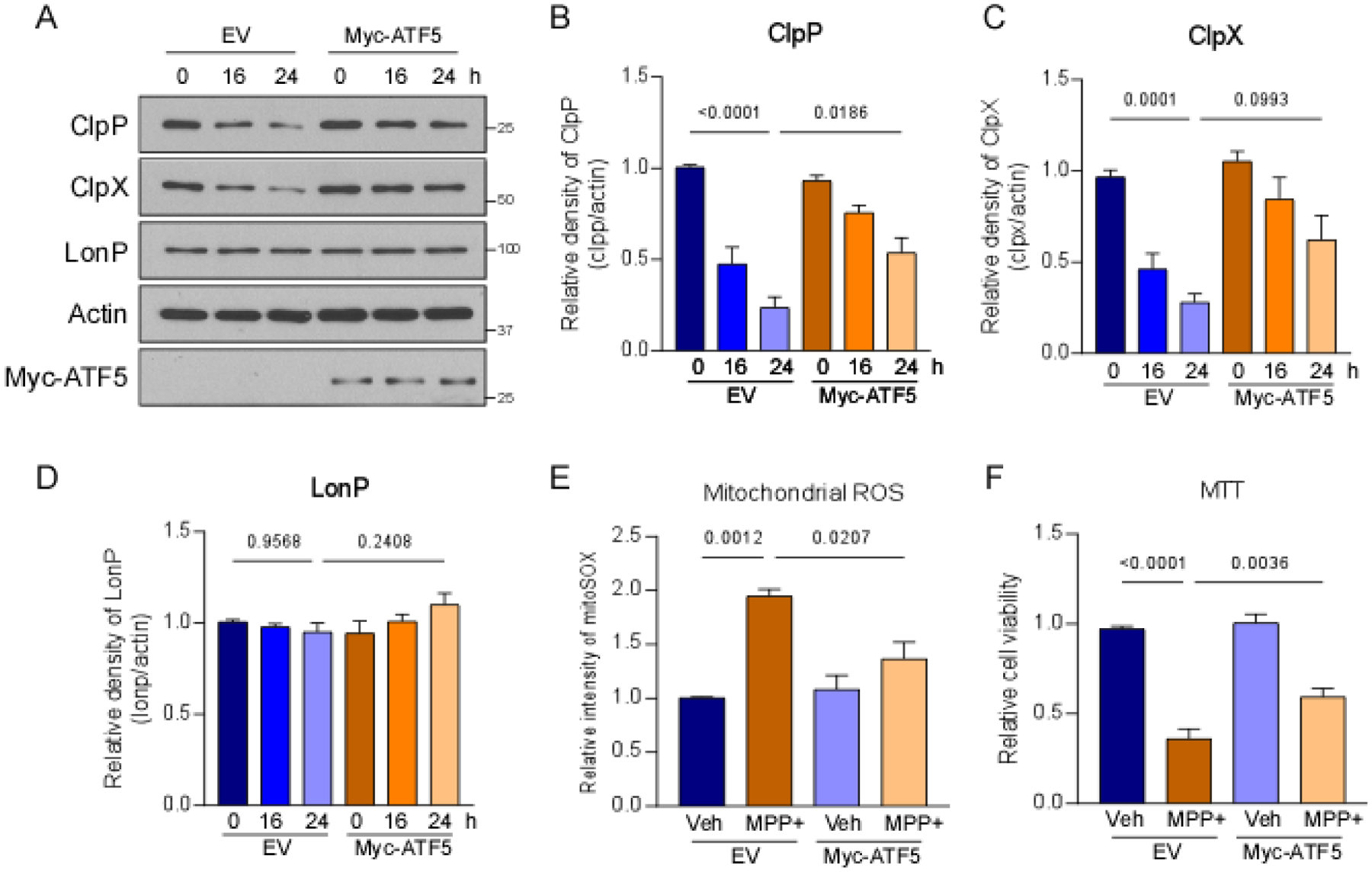
SH-SY5Y cells were treated with 2 mM MPP+ after the overexpression of ATF5. (A) Total protein lysates were harvested after MPP+ treatment and subjected to Western blot analyses with the indicated antibodies. Histograms show the relative density of (B) ClpP, (C) ClpX, and (D) LONP protein levels. (E) Cells were stained with MitoSOX™ dye to assess mitochondrial superoxide production after 24 h treatment with MPP+. The histogram shows the relative fluorescence density of MitoSOX. (F) Cell viability was examined by the MTT assay after 24 h treatment with MPP+. The histogram shows relative cell survival. The data are expressed as the mean ± SEM of three independent experiments. Data are compared with one-way ANOVA with Tukey’s post hoc test.
Discussion
In the present study, we found that the UPRmt was activated by MPP+ treatment in dopaminergic SH-SY5Y cells, reflected by upregulation of the expression of UPRmt-related proteases, chaperones, and transcription factors. We also observed the downregulation of these UPRmt-related components at the late stage of MPP+ treatment. The overexpression of Myc-tagged Δ-OTC, which triggers UPRmt activation, significantly reduced the production of mitochondrial ROS and enhanced cell survival in SH-SY5Y cells that were treated with MPP+. By expressing the UPRmt transcriptional factor ATF5, we also found a decrease in mitochondrial ROS production and an increase in SH-SY5Y cell viability. Altogether, these findings suggest a protective role for UPRmt activation in maintaining mitochondrial function and cellular function in a cell culture model of MPP+-induced PD.
The UPRmt is an adaptive stress response that maintains mitochondrial proteostasis and regulates other mitochondrial functions, including OXPHOS, ROS detoxification, mitochondrial biogenesis, and innate immune responses [15, 16, 21]. The pathological relevance of the UPRmt in PD has been recently demonstrated. In PRKN and PINK1 mutant nematodes, the UPRmt was activated, and the deletion of atfs-1 decreased lifespan and resulted in the loss of dopaminergic neurons [8]. Our findings are consistent with these results, suggesting that the UPRmt is required to mitigate the detrimental effect of mitochondrial stressors that can lead to neuronal death. We also observed the downregulation of selective UPRmt-related components, including ClpP, ClpX, UBL5, and ATF5, after 8-h treatment with MPP+. This downregulation did not result from the loss of mitochondrial mass or mitophagy because LONP protein levels remained steady during MPP+ treatment. A previous study reported that ATF5 is localized to mitochondria, degraded by matrix protease, and transported into the nucleus to trigger the transcription of UPRmt-related genes under mitochondrial stress [17]. Considering the alteration of ATF5 expression that we observed in the present study, we propose that MPP+ might indirectly affect retrograde communication between mitochondria and the nucleus, in addition to impairing OXPHOS complex I.
The accumulation of unfolded/misfolded proteins activates the UPRmt to trigger the expression of mitochondrial proteases and chaperones [18, 24]. Thereafter, perturbations of the UPRmt lead to compromised resistance to excessive protein load, thereby sensitizing mitochondria to the pathological environment. Our previous study demonstrated the protective role of the matrix protease ClpP, an essential UPRmt component, in A53T-α-syncline (αSyn)-associated PD models [7]. The overexpression of ClpP significantly reduced the load of mitochondrial unfolded protein and abolished the expression of pathogenic phosphorylated S129 αSyn species in A53T-αSyn transgenic mice [7]. Similarly, in the present study, we found that ATF5 overexpression protected both mitochondrial function and cell viability against MPP+-induced toxicity. These results further support our previous findings and suggest that upregulation of the UPRmt can protect against the progression of PD. Notably, A53T-αSyn can be translocated into mitochondria and interact with and impair the activity of OXPHOS complex I, which is also attacked by MPP+. OXPHOS complex I is composed of subunit proteins that are encoded by both nuclear and mitochondrial genomes [25]. Impairments in OXPHOS complex I cause an imbalance of mitochondrial and nuclear protein load, resulting in UPRmt activation. Future studies that explore connections between OXPHOS complex I impairments and UPRmt activation will provide a better understanding of the molecular pathways that underlie the pathogenesis of PD. Moreover, the administration of MPTP or rotenone in vivo can selectively kill dopaminergic neurons and induce Parkinsonism [11, 26]. Understanding the ways in which the UPRmt responds to complex I damage will likely provide insights into the selective vulnerability of dopaminergic neurons in PD.
In addition to PD, the UPRmt is also engaged in a spectrum of neurodegenerative diseases. Amyloid β accumulation activated the UPRmt in amyloid precursor protein (APP)-expressing SH-SY5Y cells and APP/presenilin 1 mouse models of AD [27]. The expression of UPRmt-related genes increased in post mortem samples from Alzheimer’s disease patients [28], suggesting UPRmt activation. Evidence from multiple independent studies demonstrated activation of the UPRmt in mutant superoxide dismutase 1 (SOD1)-, TAR DNA-binding protein-43 (TDP43)-, and coiled-coil-helix-coiled-coil-helix domain-containing protein 10 (CHCHD10)-associated models of amyotrophic lateral sclerosis [29–31]. Moreover, mutant huntingtin protein can inhibit the UPRmt by impairing the mRNA stability of mitochondrial adenosine triphosphate-binding cassette subfamily B member 10 (ABCB10) in Huntington’s disease [32]. These findings emphasize the critical role of the UPRmt in maintaining mitochondrial function under pathological conditions. Nevertheless, although an extensive body of literature suggests a role for the UPRmt in protecting mitochondrial function and promoting cellular homeostasis under stress conditions, some evidence indicates the toxic potential of UPRmt signaling that can promote the accumulation of damaged mitochondria and shorten lifespan [33]. Notably, a recent study in nematodes found that UPRmt activation accelerated dopaminergic neurodegeneration [34]. Thus, it is necessary to investigate the duality of the UPRmt in cellular health, especially in dopaminergic neurons, and links with PD pathogenesis.
In summary, MPP+ treatment increased the expression of UPRmt-related proteins in SH-SY5Y cells. Treatment with MPP+ also induced the transcription of UPRmt-related genes and stimulated the expression of ATF5, which is a transcriptional mediator that is required for UPRmt activation in mammalian cells. Although ATF5 overexpression conferred protection against MPP+-induced toxicity, further studies are needed to explain the ways in which MPP+ treatment modulates the expression of ATF5, which might reveal potential therapeutic targets for PD.
Supplementary Material
Highlights.
MPP+ treatment induces activation of UPRmt
Activation of UPRmt provides resistance to MPP+-induced toxicity
ATF5 overexpression confers protection against MPP+ toxicity
Funding
This study was supported by grants from the United States National Institutes of Health (R01AG065240, R01NS115903, and R21NS107897 to X.Q.), a Dr. Ralph and Marian Falk Medical Research Trust-Transformative Award, and Harrington Rare Disease Scholar Award (to X.Q.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Availability
The datasets that were generated during and/or analyzed during the present study are available from the corresponding author upon reasonable request.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Marras C, et al. , Prevalence of Parkinson’s disease across North America. NPJ Parkinsons Dis, 2018. 4: p. 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ascherio A and Schwarzschild MA, The epidemiology of Parkinson’s disease: risk factors and prevention. Lancet Neurol, 2016. 15(12): p. 1257–1272. [DOI] [PubMed] [Google Scholar]
- 3.de Lau LM and Breteler MM, Epidemiology of Parkinson’s disease. Lancet Neurol, 2006. 5(6): p. 525–35. [DOI] [PubMed] [Google Scholar]
- 4.Schapira AH, et al. , Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem, 1990. 54(3): p. 823–7. [DOI] [PubMed] [Google Scholar]
- 5.Dias V, Junn E, and Mouradian MM, The role of oxidative stress in Parkinson’s disease. J Parkinsons Dis, 2013. 3(4): p. 461–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ryan BJ, et al. , Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci, 2015. 40(4): p. 200–10. [DOI] [PubMed] [Google Scholar]
- 7.Hu D, et al. , Alpha-synuclein suppresses mitochondrial protease ClpP to trigger mitochondrial oxidative damage and neurotoxicity. Acta Neuropathol, 2019. 137(6): p. 939–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper JF, et al. , Activation of the mitochondrial unfolded protein response promotes longevity and dopamine neuron survival in Parkinson’s disease models. Sci Rep, 2017. 7(1): p. 16441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kopin IJ and Markey SP, MPTP toxicity: implications for research in Parkinson’s disease. Annu Rev Neurosci, 1988. 11: p. 81–96. [DOI] [PubMed] [Google Scholar]
- 10.Jackson-Lewis V and Przedborski S, Protocol for the MPTP mouse model of Parkinson’s disease. Nat Protoc, 2007. 2(1): p. 141–51. [DOI] [PubMed] [Google Scholar]
- 11.Sherer TB, et al. , Mechanism of toxicity in rotenone models of Parkinson’s disease. J Neurosci, 2003. 23(34): p. 10756–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pickrell AM and Youle RJ, The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron, 2015. 85(2): p. 257–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lucking CB, et al. , Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med, 2000. 342(21): p. 1560–7. [DOI] [PubMed] [Google Scholar]
- 14.Shpilka T and Haynes CM, The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat Rev Mol Cell Biol, 2018. 19(2): p. 109–120. [DOI] [PubMed] [Google Scholar]
- 15.Lin YF, et al. , Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature, 2016. 533(7603): p. 416–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nargund AM, et al. , Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol Cell, 2015. 58(1): p. 123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fiorese CJ, et al. , The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr Biol, 2016. 26(15): p. 2037–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nargund AM, et al. , Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science, 2012. 337(6094): p. 587–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pellegrino MW, Nargund AM, and Haynes CM, Signaling the mitochondrial unfolded protein response. Biochim Biophys Acta, 2013. 1833(2): p. 410–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Naresh NU and Haynes CM, Signaling and Regulation of the Mitochondrial Unfolded Protein Response. Cold Spring Harb Perspect Biol, 2019. 11(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pellegrino MW, et al. , Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature, 2014. 516(7531): p. 414–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jensen MB and Jasper H, Mitochondrial proteostasis in the control of aging and longevity. Cell Metab, 2014. 20(2): p. 214–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao Q, et al. , A mitochondrial specific stress response in mammalian cells. EMBO J, 2002. 21(17): p. 4411–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haynes CM, et al. , ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell, 2007. 13(4): p. 467–80. [DOI] [PubMed] [Google Scholar]
- 25.Sharma LK, Lu J, and Bai Y, Mitochondrial respiratory complex I: structure, function and implication in human diseases. Curr Med Chem, 2009. 16(10): p. 1266–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watanabe Y, Himeda T, and Araki T, Mechanisms of MPTP toxicity and their implications for therapy of Parkinson’s disease. Medical Science Monitor, 2005. 11(1): p. Ra17–Ra23. [PubMed] [Google Scholar]
- 27.Shen Y, et al. , Activation of Mitochondrial Unfolded Protein Response in SHSY5Y Expressing APP Cells and APP/PS1 Mice. Frontiers in Cellular Neuroscience, 2020. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beck JS, Mufson EJ, and Counts SE, Evidence for Mitochondrial UPR Gene Activation in Familial and Sporadic Alzheimer’s Disease. Curr Alzheimer Res, 2016. 13(6): p. 610–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Riar AK, et al. , Sex specific activation of the ER alpha axis of the mitochondrial UPR (UPRmt) in the G93A-SOD1 mouse model of familial ALS. Human Molecular Genetics, 2017. 26(7): p. 1318–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang P, et al. , TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. Plos Genetics, 2019. 15(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Straub IR, Weraarpachai W, and Shoubridge EA, Multi-OMICS study of a CHCHD10 variant causing ALS demonstrates metabolic rewiring and activation of endoplasmic reticulum and mitochondrial unfolded protein responses. Hum Mol Genet, 2021. 30(8): p. 687–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu Z, et al. , Mutant huntingtin inhibits the mitochondrial unfolded protein response by impairing ABCB10 mRNA stability. Biochim Biophys Acta Mol Basis Dis, 2019. 1865(6): p. 1428–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bennett CF, et al. , Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nature Communications, 2014. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez BA, et al. , Dysregulation of the Mitochondrial Unfolded Protein Response Induces Non-Apoptotic Dopaminergic Neurodegeneration in C. elegans Models of Parkinson’s Disease. J Neurosci, 2017. 37(46): p. 11085–11100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.