

Abstract
Botulinum neurotoxins (BoNTs) are extremely toxic and have been deemed a Tier 1 potential bioterrorism agent. The most potent and persistent of the BoNTs is the “A” serotype, with strategies to counter its etiology focused on designing small-molecule inhibitors of its light chain (LC), a zinc-dependent metalloprotease. The successful structure-based drug design of inhibitors has been confounded as the LC is highly flexible with significant morphological changes occurring upon inhibitor binding. To achieve greater success, previous and new cocrystal structures were evaluated from the standpoint of inhibitor enantioselectivity and their effect on active-site morphology. Based upon these structural insights, we designed inhibitors that were predicted to take advantage of π–π stacking interactions present in a cryptic hydrophobic subpocket. Structure–activity relationships were defined, and X-ray crystal structures and docking models were examined to rationalize the observed potency differences between inhibitors.
Keywords: BoNT/A, structure-based drug design, enantioselectivity, hydroxamate, structure−activity relationships, π−π stacking
Botulinum neurotoxins (BoNTs) possess incredible potency and are among the most toxic agents known to humankind. Seven serotypes (A–G) have been identified,1 with serotype A responsible for the majority (52%) of botulism cases within the United States between 1975–2009.2 Its remarkable toxicity (intravenous LD50 = 1–2 ng/kg)3 and persistence within human tissue (t1/2 = months to years),4 coupled with its ease of production and dissemination, has led to categorization by the Centers for Disease Control and Prevention (CDC) as a Tier 1 potential bioterrorism agent.5 BoNTs are produced by a species of diverse family of anaerobic bacteria known as Clostridium botulinum and also pose a risk of foodborne botulism from incorrect handling of food products, particularly canned goods.6,7
The remarkable potency of BoNT/A has been utilized in a multitude of therapeutic and cosmetic applications in which aberrant muscular spasticity is implicated, with several Food and Drug Administration (FDA)-approved formulations available for various and limited indications.8 Debilitating disorders such as cerebral palsy,9 chronic migraines,10 and sialorrhea11 and cosmetic imperfections such as facial wrinkles12 have been successfully treated using BoNT/A formulations. The prolific use of BoNT/A for these therapeutic applications comes with associated risks: misuse, overdose, or poor injection technique of BoNT/A has established side effects such as spread from the injection site, iatrogenic botulism, and respiratory compromise.13
The effects associated with BoNT/A intoxication arise from its ability to prevent exocytotic vesicle fusion at neuronal termini, thus preventing neurotransmitter release and disrupting normal muscular function, which results in the flaccid paralysis that is characteristic of botulism.14 The 150 kDa BoNT/A holotoxin consists of a heavy chain (HC, 100 kDa) and a light chain (LC, 50 kDa) that are connected by a single disulfide bond. Facilitation by the HC binding to cell surface receptor sites, the LC, a zinc-dependent metalloprotease, is translocated to the cytosol.15 Once internalized, the LC selectively cleaves synaptosomal-associated protein 25 (SNAP-25), a crucial component of the soluble N-ethylmaleimide sensitive factor (NSF) attachment protein receptor (SNARE) complex that is responsible for vesicular fusion to the synaptosomal membrane.16 Current FDA-approved postintoxication treatments for BoNT/A are limited to antibody-based antitoxins which have a short therapeutic window (12–24 h postexposure), owing to their inability to enter the neuronal compartment.17 A recent advance in this field has outlined the use of a chimeric toxin-based delivery method which utilizes the intrinsic membrane-penetrating ability of the BoNT/X transmembrane domain to facilitate the internalization of a linked BoNT/A-specific antibody. This strategy was successful in sequestering LC activity in vitro and in vivo.18 Although promising, there remains a large deficit of postintoxication treatments for BoNT/A. An alternative strategy is the development of small-molecule inhibitors.
Currently, several examples of small-molecule inhibitors for the BoNT/A LC have been identified and primarily function through active-site zinc ion chelation.19,20 Some of these inhibitors (Figure 1A) have been cocrystallized within the LC, and distinct morphological changes in the S1′ subsite have been observed (Figure 1B,C). Lipophilic inhibitors 2,4-dichlorocinnamic hydroxamic acid (DCHA) and adamantane-based inhibitor 1 possess almost identical binding poses and occupy the S1′ subsite, a small hydrophobic cleft comprising of residues Phe194, Ile161, Asp370, and Phe369. The phenyl ring of DCHA forms an offset π–π stack with Phe194, with the 370 loop adopting a Phe369-in–Asp370-out conformation with Phe369, forming a partial lid above the inhibitors.21,22 Conversely, hydrophilic inhibitor l-arginine-based hydroxamate 2 causes a conformational inward flip of Asp370, forming H-bond interactions with the guanidinium moiety of 2.23,24 Moreover, Phe194 has also rotated to form a cation−π interaction with 2. Additional examples of small-molecule hydroxamate inhibitors have also outlined a high degree of flexibility within the S1′ region of the BoNT/A LC.25
Figure 1.
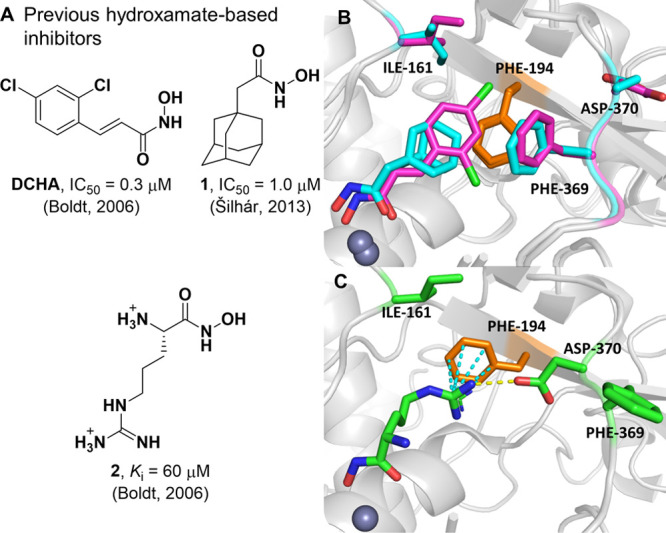
(A) Previous hydroxamate-based inhibitors of the BoNT/A LC. (B) Overlay of X-ray crystal structures of DCHA (purple, PDB 2IMA)24 and 1 (cyan, PDB 4HEV)22 bound within the active site of the LC. The 370 loop adopts an Phe369-in–Asp370-out conformation. (C) X-ray crystal structure of 2 (green, PDB 2IMB)24 bound within the active site of the LC. The 370 loop adopts an Phe369-out–Asp370-in conformation. Hydrogen bonds and cation−π interactions are outlined in yellow and cyan dashed lines, respectively.
The advancement of structural information for the BoNT/A LC has enabled structure-guided design of enantioselective chiral inhibitors, as outlined by Stowe et al. (Figure 2A).26 Analysis of the active-site binding pose of DCHA identified two proximal water molecules involved in an H-bonding network with amino acid residues His223, Glu351, and Arg363 (Figure 2B). Compounds 3 and 4 were designed with two purposes in mind: (1) extend additional chemical composition into the polar space of the active site, and (2) investigate the effect of chiral inhibitors upon BoNT/A LC inhibition.26 Specifically, the hydroxyethyl appendage was hypothesized to displace one of the ordered water molecules within the enzyme active site. Moreover, previous BoNT/A LC inhibition data revealed a ∼4- and 21-fold preference for the R over the S enantiomer of 3 and 4, respectively.26 To investigate this selectivity, we have determined the cocrystal structure of racemic 3 bound within the BoNT/A LC (Figure 2C).
Figure 2.
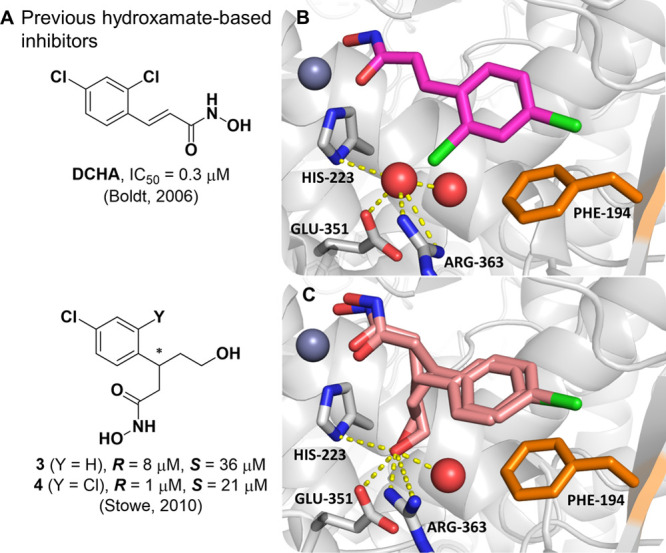
(A) DCHA and previous branched derivatives, values for 3 and 4 are IC50. (B) Alternative view of DCHA bound within the active site of the LC. Structural water molecules are outlined in red spheres, with nearby polar contacts indicated by yellow dashed lines. (C) Cocrystal structure of 3 bound within the active site of the LC (PDB 7N18) determined at a resolution of 2.1 Å. Both R and S enantiomers are shown. The hydroxyethyl group has displaced one of the water molecules observed in the structure of the complex with DCHA. Nearby polar contacts are indicated by yellow dashed lines.
Although the inhibitor could be unambiguously placed within the active site, electron density at the stereogenic center of 3 was poorly defined (Figure S1). As we crystallized with the racemic mixture, and that both enantiomers had comparable potencies, we concluded that each enantiomer had similar occupancies within the LC crystal lattice and an average of the two structures was observed. Despite this, electron density for the phenyl ring, the hydroxamate, and the hydroxyethyl appendage was well-defined, with the hydroxyethyl OH group displacing one water molecule (Figure 2C) to form a H-bond network with residues His223, Glu351, and Arg363. Notably, the inclusion of the 2-chloro substituent in 4 greatly improved enantiomeric selectivity;26 evaluation of the binding poses of both DCHA and 3 show the phenyl ring making a π–π stacking interaction with Phe194. Based on these insights, we posit that the 2-chloro substituent of 4 acts as a “molecular brake” and reduces the free rotation of the phenyl ring, causing a greater chiral effect at the stereocenter.
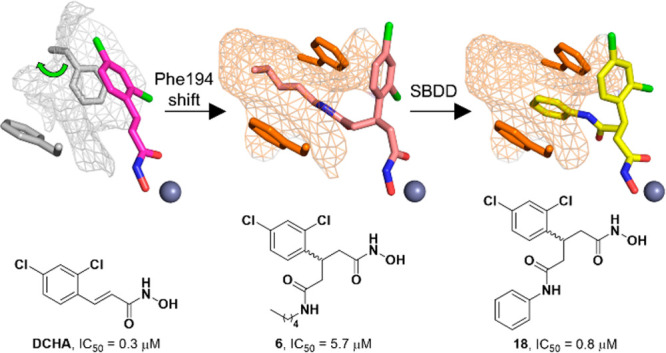
Recently, a central focus of the development of BoNT/A LC inhibitors has been the irreversible inhibition of the enzyme. Success in this area has been found through a bifunctional strategy, wherein coordination to the active-site zinc ion allows proximal positioning of a pendent electrophilic warhead for the simultaneous irreversible modification of a noncatalytic cysteine.27,28 Previously synthesized 5 possessed an electrophilic methanethiosulfonate warhead and the same branched pharmacophore as 3 and 4 and exhibited modest irreversible inhibition against the BoNT/A LC with a kinact/Ki value of 19.4 M–1·s–1 (Figure 3A).27 As proof-of-concept for this covalent approach, previously synthesized 6 was included as a noncovalent control and was crystallized in complex with the BoNT/A LC (Figure 3B).27 This structure revealed a new cryptic hydrophobic subpocket adjacent to the main S1′ binding pocket that was absent in the crystal structure of DCHA bound to the BoNT/A LC (Figure 3C). The π–π stacking interaction observed between the 2,4-dichlorophenyl ring in DCHA and the side chain of Phe194 is not present upon binding of 6. The induced fit caused by the alkyl moiety results in a large shift and rotation of the side chain of Phe194, and together with Phe163, form the edges of the new hydrophobic subpocket effectively “sandwiching” the alkyl chain.
Figure 3.
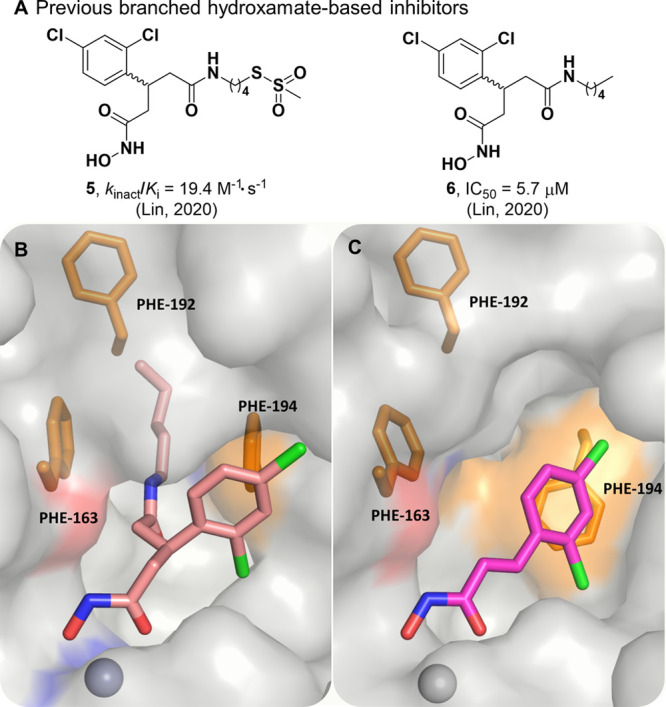
(A) Previous branched hydroxamate-based inhibitors of the BoNT/A LC. (B) X-ray crystal structure of 6 bound in the active site of the LC (PDB 6XCF).27 Accommodation of the alkyl moiety in the hydrophobic subpocket results in disruption of the face-to-face π–π stack between the 2,4-dichlorocinnamic phenyl ring and Phe194. (C) Alternative view of DCHA (purple) bound with the LC. The 2,4-dichlorocinnamic phenyl ring and Phe194 form an offset face-to-face π–π stack.
Formation of this cryptic hydrophobic subpocket has not previously been observed; to quantify the size of this additional binding pocket and the main S1′ pocket, we used Dpocket, a module within the Fpocket suite, to calculate pocket volumes (Table S1) for DCHA, 1–3, and 6.29,30 Dpocket defines the pocket using atoms within 4 Å of the ligand, thus it is expected that larger ligands give rise to larger binding pockets. With the exception of 2, the results clearly reflected this expectation, with 6 filling additional subpockets within the active site compared to other ligands that only occupied the S1′ pocket. Compound 2 is the smallest ligand, but had a comparable pocket size to that of 6. Upon visual inspection of the binding poses of DCHA and 2, Pro69 exhibited a large shift toward solvent, resulting in a larger binding pocket for 2 (Figure S2).
We envisioned that we could design inhibitors based upon the branched pharmacophore of 6 to take advantage of this unexplored, cryptic, hydrophobic subpocket. Specifically, we postulated that we could create a tripartite π–π stacking interaction with Phe163/194 and a phenyl ring by replacement of the alkyl chain in 6. To bolster this hypothesis, we conducted docking studies of the benzylamine-based derivative 7 using Glide (Figure 4A).
Figure 4.
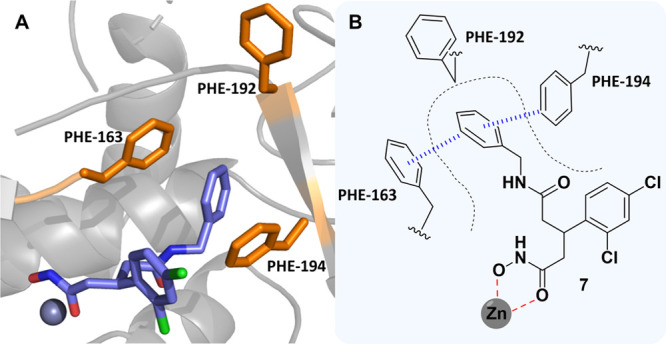
(A) Docking pose of 7 bound to the BoNT/A LC. (B) 2D representation of the predicted binding mode of 7 in the BoNT/A LC active site. Coordination of the hydroxamate and π–π stacking interactions are outlined in red and blue, respectively.
As posited, the benzylamine ring adopted a pose which occupied the hydrophobic subpocket with both Phe163 and 194 forming offset π–π stacking interactions. We further strengthened our docking strategy by showing that the predicted docking pose of 6 had excellent overlap with the crystallized binding mode of 6, with the alkyl chain occupying the cryptic hydrophobic subpocket (Figure S3). To investigate this hypothesis, we designed a series of aniline- and benzylamine-based derivatives (Figure 5). The design included potential hydrophobic/hydrogen-bonding contacts derived through either methyl/ethyl groups as well as OH and NH2 moieties that were strategically placed on the aromatic ring.
Figure 5.

Aniline- and benzylamine-based target library.
Synthesis of target compounds 7–28 (Scheme 1) began with ring opening of previously synthesized cyclic anhydride 29(27) with O-tetrahydropyran-protected hydroxylamine to afford the carboxylic acid intermediate 30 in a moderate yield. Acid 30 was subjected to amide coupling conditions with various benzylamines to afford protected hydroxamates (31) that were then deprotected using pyridinium p-toluenesulfonate (PPTS) to yield the final target compounds. Notably, amide coupling conditions for 30 using less-reactive/ortho-substituted aniline reagents resulted in no formation of the desired product. Fortunately, a change in order of steps (route 2) reacting aniline reagents with 29 directly to give carboxylic acids (32) was successful. Furthermore, optimization of workup procedures allowed both the amide coupling and deprotection step to be combined, allowing rapid access to final compounds 7–28. Further details on the synthetic routes employed can be found in SI-4.0.
Scheme 1. General Route to Benzylamine- and Aniline-Based Final Compounds.
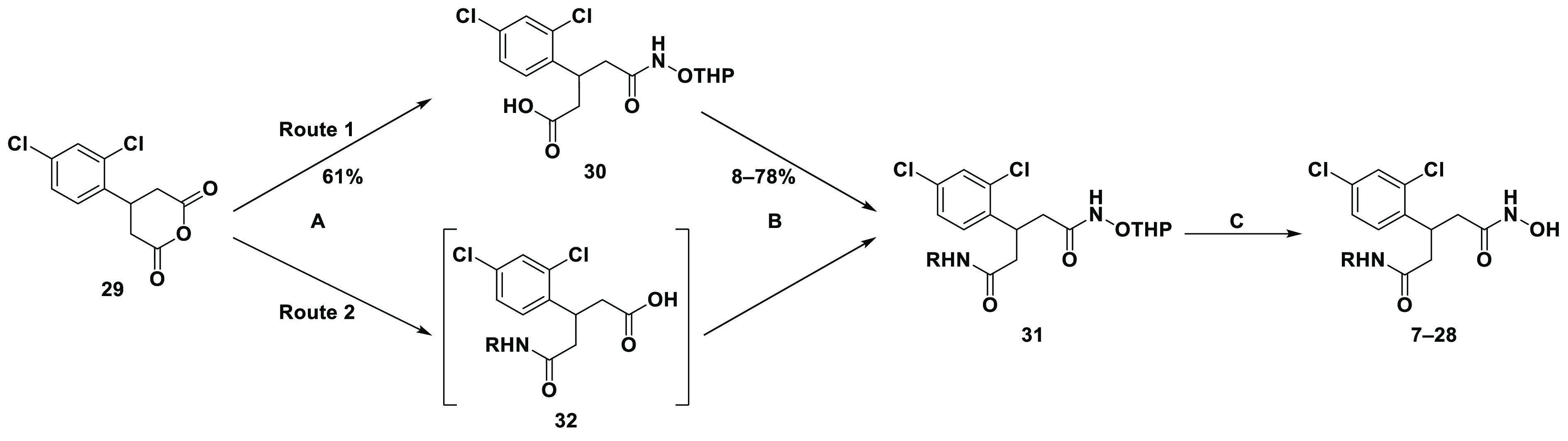
31 and 32 represent multiple intermediates. Reagents and conditions: (A) RNH2, CHCl3, or CH2Cl2, rt; (B) RNH2, HATU or EDC, DIPEA, DMF, rt; (C) PPTS, EtOH, 65 °C or 10% TFA–CH2Cl2, rt.
Having established solid synthetic routes, hydroxamates 7–28 were evaluated for BoNT/A LC inhibition (Table 1) using a previously described27 fluorescence resonance energy transfer (FRET)-based assay that uses a truncated form of the BoNT/A LC (amino acids 1–425) and a SNAPtide substrate.31,32 Compounds were preincubated for 30 min at various concentrations with 10 nM BoNT/A LC, followed by addition of 4 μM substrate, and reaction progress fluorescently monitored at λex/em = 490/523 nm.
Table 1. IC50 Values for 7–28 When Evaluated against the FRET-Based SNAPtide Assay; See SI-6.1 for Dose Response Curves.
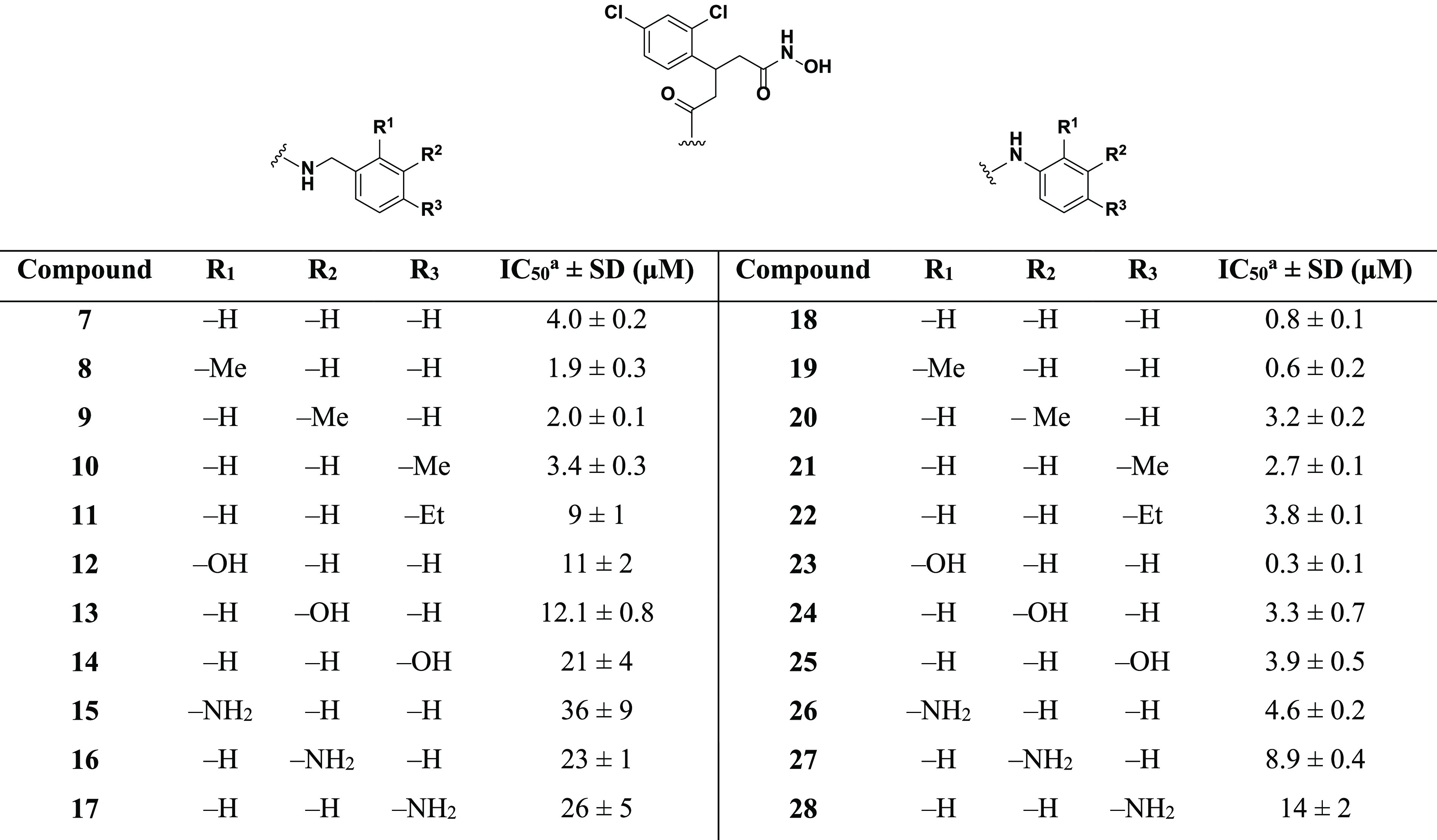
Average of at least three experimental replicates.
The initial target hydroxamate 7 had an IC50 value of 4.0 μM, thus showing no real improvement when compared to the alkyl-containing counterpart 6 (IC50 = 5.7 μM). This suggested that the hypothesized π–π stacking interaction with residues Phe163 and 194 did not provide significant binding stabilization or was unlikely to be occurring. Addition of a methyl group in the ortho (8) and meta (9) position produced a ∼2-fold increase in potency, whereas the para-substituted compound 10 exhibited minimal effect on the potency. Of note, addition of an ethyl group (11) in the para position resulted in a ∼2-fold loss in potency when compared to unsubstituted benzylamine 7. With the docked structure of 7 (Figure 4A) in mind, this suggested that 11 may be too large, potentially clashing with Phe192 deep within the hydrophobic subpocket.
Hydroxamates 12–17 were designed to explore hydrogen-bonding potential within the hydrophobic subpocket. Substitution at either the ortho, meta, or para position with polar OH and NH2 groups for benzylamine-based inhibitors resulted in a large decrease in potency. This was readily interpreted as repulsive interactions between the hydrophobic residues that line the subpocket and the appended polar groups, and that H-bonding contacts may be untenable. Unsubstituted aniline 18 exhibited a submicromolar IC50 value of 0.8 μM, a ∼7- and 5-fold increase over 6 and 7, respectively. This indicated optimum positioning and alignment of the aniline ring for the predicted π–π stacking interaction with Phe163/194. To further explore these differences, we conducted docking of 18 and compared the docked pose to that of 7 (Figure 6).
Figure 6.
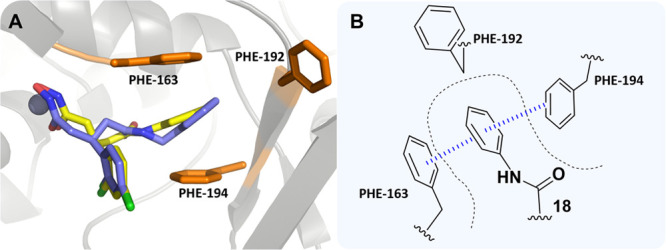
(A) Docking poses of 7 (blue) and 18 (yellow) bound within the BoNT/A LC. (B) 2D representation of the predicted π-sandwich interaction between Phe163 and 194 and the aniline ring of 18. The π–π stacking interactions are outlined in blue.
The aniline phenyl ring in 18 was predicted to form a double offset π–π interaction with Phe163 and 194 (Figure S4) and exhibited a more planar orientation with respect to the phenylalanine ring systems than what was observed for 7. The additional methylene group present in 7 was foreseen to disrupt the planarity of the π–π stacking interactions and could account for the lower potency observed for the benzylamine-based compounds. Addition of a methyl group in the ortho position (19) improved the potency, whereas the meta-substituted (20) and para-substituted (21) derivatives exhibited a loss of potency when compared to unsubstituted aniline 18. As might be posited from the docking models (Figure 6), 7 was expected to extend further into the hydrophobic subpocket than 18; hence, the polar groups would be better tolerated when placed toward the entrance of the hydrophobic subpocket. This premise was bolstered by the superior potency of ortho-substituted compounds 19, 23, and 26 when compared to their meta-substituted (20, 24, and 27) and para-substituted (21, 25, and 28) analogues. Ethyl-containing aniline 22 also exhibited a loss in potency but was still superior to its benzylamine counterpart 11. The reduced extendibility of the aniline compounds may allow for larger groups in the para position to be better accommodated deep within the hydrophobic subpocket. Compound 23 exhibited an IC50 of 0.3 μM and was the most potent compound evaluated. The improved potency of this compound when compared to that of 24 and 25 indicated that there may be a H-bond forming between the phenol and residues near the entrance of the hydrophobic subpocket. However, this potency increase was not observed for dianiline 26, which also possessed the capacity to H-bond. Therefore, it was unclear what the precise reasoning was for the superior potency of 23. In summary, aniline-based molecules 18–28 exhibited superior inhibitory potency when compared to that of benzylamine-based structures 7–17, which we propose is facilitated through π–π stacking interactions with the aniline phenyl ring and Phe163/194.
To further investigate the effect of chirality upon inhibitor binding, we employed chiral supercritical fluid chromatography (SFC) to separate enantiomers of the three best inhibitors 18, 19, and 23. This approach allowed us rapid access to enantiopure material without undertaking the cumbersome asymmetric synthetic approach outlined previously;26 details pertaining to SFC are outlined in SI-4.4. Relative stereochemistry of enantiomers was assigned through their specific rotation of plane-polarized light and their BoNT/A LC inhibition was assessed using the FRET-based assay (Table 2).
Table 2. IC50 Values for Enantiopure Compounds 18, 19, and 23.
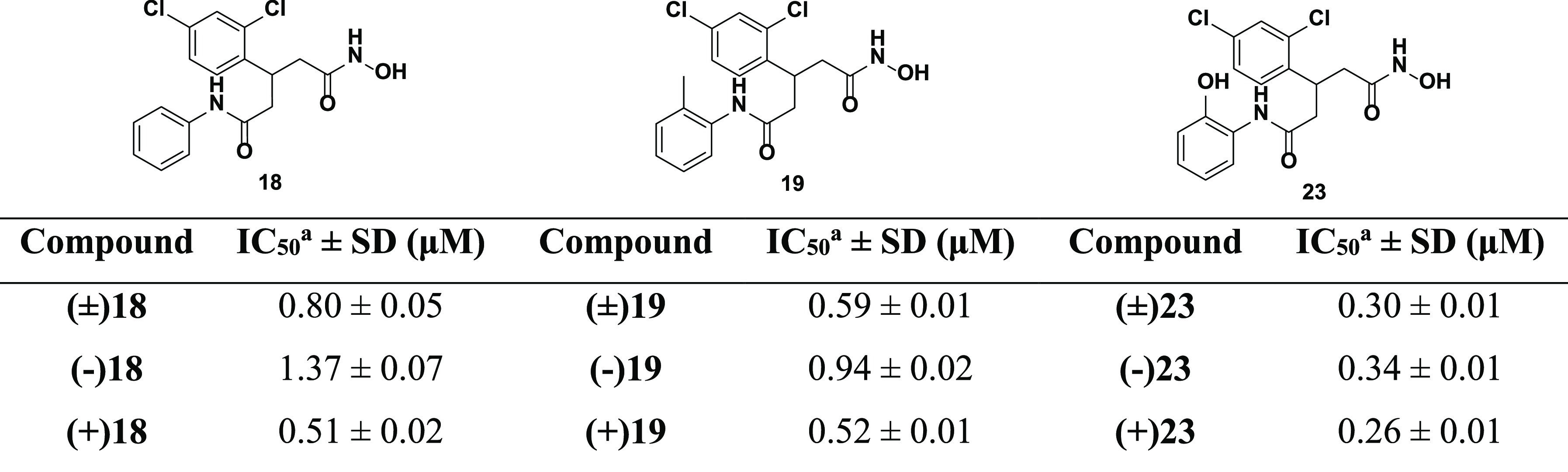
Average of at least three independent experiments.
Differences in inhibitory potency was observed, with the (+) configuration being slightly preferred. These hydroxamates did not exhibit the same magnitude of enantiomer selectivity that was observed for 3 and 4.26 The lack of the hydroxyethyl moiety and subsequent loss of the H-bond network with amino acids His223, Glu351, and Arg363 (Figure 2C) for inhibitors 18, 19, and 23 suggested that this interaction was crucial for the development of exquisitely enantioselective inhibitors of BoNT/A LC using this pharmacophore. The fluid morphology of the BoNT/A LC active site, coupled with the fact that both groups flanking the stereocenter are aromatic, suggests that both ring systems may have the capacity to occupy both the main S1′ pocket and the hydrophobic subpocket.
In summary, we have designed, synthesized, and biochemically evaluated a comprehensive series of chiral benzylamine- and aniline-based inhibitors of the BoNT/A LC. With the aid of cocrystal structures and docking, molecules were designed to take advantage of a π–π stacking interaction to improve inhibitor potency. Aniline-based hydroxamates possessed superior potency compared to that of the benzylamine-based hydroxamates, which was attributed to a more planar orientation of the π–π stacking interaction, as predicted by docking. Moreover, the cocrystal structure of 3 has revealed that displacement of an active-site water molecule by the hydroxyethyl moiety is crucial to enantiomer selectivity. We envision that both occupation of the hydrophobic subpocket by aniline functionalities and the crucial H-bond network interaction obtained from the hydroxyethyl moiety can be combined into one entity that will possess not only improved potency but also greater enantioselectivity; work on such compounds is currently being conducted.
Acknowledgments
The authors thank James Conley for his assistance regarding crystallography, Dr. Joseph Barbieri for providing truncated BoNT/A LC, and Emily Sturgell and Brittany Sanchez for their contribution to SFC. This work was supported by the National Institutes of Health Grant No. R01 AI153298, the Fulbright Scholar Program (A.L.N.), the Natural Sciences and Engineering Research Council of Canada PGSD3-502274 (L.L.), and the Skaggs Institute for Chemical Biology (L.L. and K.D.J.). This is Scripps Research manuscript #30105.
Glossary
Abbreviations
- BoNT/A
botulinum neurotoxin A
- CDC
Center for Disease Control and Prevention
- DCHA
2,4-dichlorocinnamic hydroxamic acid
- DIPEA
diisopropylethylamine
- DMF
dimethylformamide
- EDC
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- FDA
Food and Drug Administration
- FRET
fluorescence resonance energy transfer
- HATU
1-[bis(dimethlyamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- HC
heavy chain
- LC
light chain
- NSF
N-ethylmaleimide sensitive factor
- PDB
protein data bank
- PPTS
pyridinium p-toluenesulfonate
- SFC
supercritical fluid chromatography
- SI
Supporting Information
- SNARE
NSF attachment protein factor
- SNAP-25
synaptosomal-associated protein-25
- TFA
trifluoroacetic acid
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00325.
Supporting figures, tables, biological materials and methods, chemistry experimental details, IC50 curves, compound HPLC chromatograms, and NMR spectra (PDF)
Accession Codes
X-ray diffraction data, coordinates, and structure factors for the X-ray crystal structure of 3 bound within the BoNT/A LC is deposited on the PDB under accession code 7N18.
Author Present Address
△ Department of Chemistry and Biochemistry, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, United States
Author Contributions
L.D.T. conceptualized the study; L.D.T., A.L.N., and A.J.C. carried out chemical synthesis and characterization; L.D.T., L.L., and A.J.C. carried out biochemical assays and data analysis; N.R.S. carried out crystallization studies and refinement; A.L.N. and A.E.W. performed computational modeling and calculations; J.S.C. carried out and overlooked SFC; L.D.T. wrote the original draft of the manuscript, and all authors revised the final version; K.N.A. and K.D.J. acquired funding and supervised the study.
The authors declare no competing financial interest.
Supplementary Material
References
- Collins M. D.; East A. K. Phylogeny and taxonomy of the food-borne pathogen Clostridium botulinum and its neurotoxins. J. Appl. Microbiol. 1998, 84, 5–17. 10.1046/j.1365-2672.1997.00313.x. [DOI] [PubMed] [Google Scholar]
- Jackson K. A.; Mahon B. E.; Copeland J.; Fagan R. P. Botulism mortality in the USA, 1975–2009. Botulinum J. 2015, 3, 6–17. 10.1504/TBJ.2015.078132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnon S. S.; Schechter R.; Inglesby T. V.; Henderson D. A.; Bartlett J. G.; Ascher M. S.; Eitzen E.; Fine A. D.; Hauer J.; Layton M.; Lillibridge S.; Osterholm M. T.; O'Toole T.; Parker G.; Perl T. M.; Russell P. K.; Swerdlow D. L.; Tonat K.; for the Working Group on Civilian Biodefense Botulinum toxin as a biological weapon: Medical and public health management. JAMA 2001, 285, 1059–1070. 10.1001/jama.285.8.1059. [DOI] [PubMed] [Google Scholar]
- Foran P. G.; Mohammed N.; Lisk G. O.; Nagwaney S.; Lawrence G. W.; Johnson E.; Smith L.; Aoki K. R.; Dolly J. O. Evaluation of the therapeutic usefulness of botulinum neurotoxin B, C1, E, and F compared with the long lasting type A - Basis for distinct durations of inhibition of exocytosis in central neurons. J. Biol. Chem. 2003, 278, 1363–1371. 10.1074/jbc.M209821200. [DOI] [PubMed] [Google Scholar]
- Oliveira M.; Mason-Buck G.; Ballard D.; Branicki W.; Amorim A. Biowarfare, bioterrorism and biocrime: A historical overview on microbial harmful applications. Forensic Sci. Int. 2020, 314, 110366. 10.1016/j.forsciint.2020.110366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S. K.; Whiting R. C. Methods for detection of Clostridium botulinum toxin in foods. J. Food Prot. 2005, 68, 1256–1263. 10.4315/0362-028X-68.6.1256. [DOI] [PubMed] [Google Scholar]
- CDC . Home canning and botulism; https://www.cdc.gov/foodsafety/communication/home-canning-and-botulism.html (accessed 03-18-2021).
- Samizadeh S.; De Boulle K. Botulinum neurotoxin formulations: Overcoming the confusion. Clin., Cosmet. Invest. Dermatol. 2018, 11, 273–287. 10.2147/CCID.S156851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farag S. M.; Mohammed M. O.; El-Sobky T. A.; ElKadery N. A.; ElZohiery A. K. Botulinum toxin A injection in treatment of upper limb spasticity in children with cerebral palsy: A systematic review of randomized controlled trials. JBJS Reviews 2020, 8, e0119. 10.2106/JBJS.RVW.19.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton R. B.; Rosen N. L.; Ailani J.; DeGryse R. E.; Gillard P. J.; Varon S. F. OnabotulinumtoxinA improves quality of life and reduces impact of chronic migraine over one year of treatment: Pooled results from the PREEMPT randomized clinical trial program. Cephalalgia 2016, 36, 899–908. 10.1177/0333102416652092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira Filho A. F.; Silva G. A. d. M.; Almeida D. M. X. Application of botulinum toxin to treat sialorrhea in amyotrophic lateral sclerosis patients: A literature review. Einstein (Sao Paulo, Brazil) 2016, 14, 431–434. 10.1590/S1679-45082016RB3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small R. Botulinum toxin injection for facial wrinkles. Am. Fam. Physician 2014, 90, 168–175. [PubMed] [Google Scholar]
- Yiannakopoulou E. Serious and long-term adverse events associated with the therapeutic and cosmetic use of Botulinum toxin. Pharmacology 2015, 95, 65–69. 10.1159/000370245. [DOI] [PubMed] [Google Scholar]
- Pellizzari R.; Rossetto O.; Schiavo G.; Montecucco C. Tetanus and botulinum neurotoxins: mechanism of action and therapeutic uses. Philos. Trans. R. Soc., B 1999, 354, 259–268. 10.1098/rstb.1999.0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rummel A.; Karnath T.; Henke T.; Bigalke H.; Binz T. Synaptotagmins I and II act as nerve cell receptors for botulinum neurotoxin G. J. Biol. Chem. 2004, 279, 30865–30870. 10.1074/jbc.M403945200. [DOI] [PubMed] [Google Scholar]
- Sun S.; Suresh S.; Liu H.; Tepp W. H.; Johnson E. A.; Edwardson J. M.; Chapman E. R. Receptor binding enables botulinum neurotoxin B to sense low pH for translocation channel assembly. Cell Host Microbe 2011, 10, 237–247. 10.1016/j.chom.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulveda J.; Mukherjee J.; Tzipori S.; Simpson L. L.; Shoemaker C. B. Efficient serum clearance of Botulinum neurotoxin achieved using a pool of small antitoxin binding agents. Infect. Immun. 2010, 78, 756–763. 10.1128/IAI.01084-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita S.-I.; Zhang J.; Zhang S.; Shoemaker C. B.; Dong M. Delivery of single-domain antibodies into neurons using a chimeric toxin–based platform is therapeutic in mouse models of botulism. Sci. Transl. Med. 2021, 13, eaaz4197. 10.1126/scitranslmed.aaz4197. [DOI] [PubMed] [Google Scholar]
- Lin L.; Olson M. E.; Eubanks L. M.; Janda K. D. Strategies to counteract Botulinum neurotoxin A: Nature’s deadliest biomolecule. Acc. Chem. Res. 2019, 52, 2322–2331. 10.1021/acs.accounts.9b00261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L.; Turner L. D.; Šilhár P.; Pellett S.; Johnson E. A.; Janda K. D. Identification of 3-hydroxy-1,2-dimethylpyridine-4(1H)-thione as a metal-binding motif for the inhibition of botulinum neurotoxin A. RSC. Med. Chem. 2021, 12, 137–143. 10.1039/D0MD00320D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldt G. E.; Kennedy J. P.; Janda K. D. Identification of a potent Botulinum neurotoxin A protease inhibitor using in situ lead identification chemistry. Org. Lett. 2006, 8, 1729–1732. 10.1021/ol0603211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šilhár P.; Silvaggi N. R.; Pellett S.; Čapková K.; Johnson E. A.; Allen K. N.; Janda K. D. Evaluation of adamantane hydroxamates as botulinum neurotoxin inhibitors: synthesis, crystallography, modeling, kinetic and cellular based studies. Bioorg. Med. Chem. 2013, 21, 1344–1348. 10.1016/j.bmc.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z.; Chen S.; Baldwin M. R.; Boldt G. E.; Crawford A.; Janda K. D.; Barbieri J. T.; Kim J.-J. P. Light chain of Botulinum neurotoxin serotype A: Structural resolution of a catalytic intermediate. Biochemistry 2006, 45, 8903–8911. 10.1021/bi060786z. [DOI] [PubMed] [Google Scholar]
- Silvaggi N. R.; Boldt G. E.; Hixon M. S.; Kennedy J. P.; Tzipori S.; Janda K. D.; Allen K. N. Structures of Clostridium botulinum neurotoxin serotype A light chain complexed with small-molecule inhibitors highlight active-site flexibility. Chem. Biol. 2007, 14, 533–542. 10.1016/j.chembiol.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Thompson A. A.; Jiao G. S.; Kim S.; Thai A.; Cregar-Hernandez L.; Margosiak S. A.; Johnson A. T.; Han G. W.; O’Malley S.; Stevens R. C. Structural characterization of three novel hydroxamate-based zinc chelating inhibitors of the Clostridium botulinum serotype A neurotoxin light chain metalloprotease reveals a compact binding site resulting from 60/70 loop flexibility. Biochemistry 2011, 50, 4019–4028. 10.1021/bi2001483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowe G. N.; Šilhár P.; Hixon M. S.; Silvaggi N. R.; Allen K. N.; Moe S. T.; Jacobson A. R.; Barbieri J. T.; Janda K. D. Chirality holds the key for potent inhibition of the Botulinum neurotoxin serotype A protease. Org. Lett. 2010, 12, 756–759. 10.1021/ol902820z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L.; Olson M. E.; Sugane T.; Turner L. D.; Tararina M. A.; Nielsen A. L.; Kurbanov E. K.; Pellett S.; Johnson E. A.; Cohen S. M.; Allen K. N.; Janda K. D. Catch and anchor approach to combat both toxicity and longevity of Botulinum toxin A. J. Med. Chem. 2020, 63, 11100–11120. 10.1021/acs.jmedchem.0c01006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner L. D.; Nielsen A. L.; Lin L.; Pellett S.; Sugane T.; Olson M. E.; Johnson E. A.; Janda K. D. Irreversible inhibition of BoNT/A protease: Proximity-driven reactivity contingent upon a bifunctional approach. RSC Med. Chem. 2021, 12, 960–969. 10.1039/D1MD00089F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guilloux V.; Schmidtke P.; Tuffery P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinf. 2009, 10, 168. 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtke P.; Le Guilloux V.; Maupetit J.; Tufféry P. Fpocket: Online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res. 2010, 38, W582–W589. 10.1093/nar/gkq383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin M. R.; Bradshaw M.; Johnson E. A.; Barbieri J. T. The C-terminus of botulinum neurotoxin type A light chain contributes to solubility, catalysis, and stability. Protein Expression Purif. 2004, 37, 187–195. 10.1016/j.pep.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Shine N. S. K.New FRET substrate for Botulinum neurotoxin type A. In 7th International Conference on Basic and Therapeutic Aspects of Botulinum and Tetanus Toxins, Santa Fe, NM, USA, 2011.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.