

Abstract
Activation-induced cytidine deaminase (AID) initiates antibody diversification by mutating immunoglobulin loci in B lymphocytes. AID and related APOBEC3 (A3) enzymes also induce genome-wide mutations and lesions implicated in tumorigenesis and tumor progression. The most prevalent mutation signatures across diverse tumor genomes are attributable to the mistargeted mutagenic activities of AID/A3s. Thus, inhibiting AID/A3s has been suggested to be of therapeutic benefit. We previously used a computational-biochemical approach to gain insight into the structure of AID’s catalytic pocket, which resulted in the discovery of a novel type of regulatory catalytic pocket closure that regulates AID/A3s that we termed the “Schrodinger’s CATalytic pocket”. Our findings were subsequently confirmed by direct structural studies. Here, we describe our search for small molecules that target the catalytic pocket of AID. We identified small molecules that inhibit purified AID, AID in cell extracts, and endogenous AID of lymphoma cells. Analogue expansion yielded derivatives with improved potencies. These were found to also inhibit A3A and A3B, the two most tumorigenic siblings of AID. Two compounds exhibit low micromolar IC50 inhibition of AID and A3A, exhibiting the strongest potency for A3A. Docking suggests key interactions between their warheads and residues lining the catalytic pockets of AID, A3A, and A3B and between the tails and DNA-interacting residues on the surface proximal to the catalytic pocket opening. Accordingly, mutants of these residues decreased inhibition potency. The chemistry and abundance of key stabilizing interactions between the small molecules and residues within and immediately outside the catalytic pockets are promising for therapeutic development.
Keywords: APOBECs, AID, inhibitor, cancer genome mutations, molecular docking, genome-editing enzymes
The DNA-editing enzyme activation-induced cytidine deaminase (AID) is expressed in activated B lymphocytes. AID mutates deoxycytidine (dC) to deoxyuridine (dU) at immunoglobulin (Ig) genes, triggering somatic hypermutation (SHM) and class switch recombination (CSR) of antibodies.1−7 AID deficiency results in a hyper IgM characterized by a lack of high affinity antibodies of switched isotypes, which is readily treatable by modern pharmaceuticals.2,3,8 AID also mediates significant off-target and genome-wide mutagenesis, some of which result in double stand breaks (DSBs) that mediate chromosomal translocations.9−14 Thus, a wealth of literature over two decades implicates AID in initiation of leukemia/lymphomas including Burkitt’s lymphoma (BL), diffuse large B cell lymphoma (DLCL), follicular lymphoma (FL), multiple myeloma, and chronic lymphocytic leukemia.10,15−19 These tumors arise from centroblasts or postcentroblasts, the narrow stage in a B cell’s life where AID is expressed, and mutations and translocation breakpoints (c-myc/IgH in BL, bcl-2/IgH in FL, bcl-6/IgH in DLCL, IgH-CCND1 in Mantle cell lymphoma) occur at genomic sites that are frequently targeted by AID.18,20−27 The causal role of AID in lymphomagenesis was proven in IL-6 transgenic mice which develop lymphomas that mimic human BL in phenotype/genotype (c-myc/IgH). In this model, DSBs at both the IgH and translocation partner c-myc loci are directly caused by AID.9,15,28−30
Beyond tumor initiation, AID expression can also exacerbate leukemia/lymphomas. Genome-wide AID-mediated mutation signatures are prevalent in leukemia/lymphomas, and numerous studies have shown that AID levels in tumors correlate with poor diagnosis.10,19,31−43 In chronic myeloid leukemia (CML), AID was shown to mutate tumor suppressor and/or DNA repair genes and accelerate imatinib resistance.31 Moreover, recent studies indicate that some therapeutic agents, such as idelalisib and duvelisib, can exacerbate AID-mediated genome mutations in tumors through increased AID expression and chromosomal translocation frequency between the IgH locus and off-target sites.44,45
AID is a member of the apolipoprotein B mRNA editing enzyme catalytic polypeptide-like (APOBEC) family of Zn-dependent, single-stranded polynucleotide-restricted cytidine deaminases.4,5,46 The AID/APOBEC family includes AID, APOBEC1, APOBEC2, APOBEC3A,B,C,D,F,G,H, and APOBEC4.4,5,46 The APOBEC3 (A3) sub-branch carries out antiviral protective functions by targeting viral DNA for mutation in the cytoplasm of infected cells.47−51 This antiviral activity has been the most studied in the context of retroviruses like HIV whose genomes go through a ssDNA replication phase; however, like AID, the A3s (particularly A3A and A3B) are also a major endogenous source of genomic mutations in many human cancers such as lung cancer, gastrointestinal cancer, breast cancer, head and neck cancer, and ovarian cancer2,48,52−67 with prevalent mutational signatures across sequenced tumor genomes. A3 expression associates with poor prognosis, and A3 action is one mechanism for the generation of drug resistance. More recently, the role of A3s as cancer drivers and exacerbators has been confirmed in mouse models.68
Given their prominent roles as mediators of one of the most prevalent tumor genome mutation signatures, inhibiting AID and A3 activity has been suggested as a potentially useful approach to treating AID-expressing malignancies or augmenting other therapies.19,40,69−79 We previously utilized a combined computational–biochemical approach to glean insights into AID’s native and functional structure.80 This approach is based on structure prediction using multiple templates, followed by functional verification of model predictions using a library of AID variants, including point/multiple mutants, orthologues, and chimeric versions with portions of other deaminases exchanged into the AID scaffold, or vice versa. We thus arrived at a map of AID’s functional structure including surface topology, core architecture, and catalytic pocket.80 This map demonstrated that AIDs form a catalytic pocket with the triad of Zn-coordinating residues (H56, C87, and C90 in human AID) and catalytic glutamic acid (E58 in human AID) that can accommodate a dC residue in orientations that support the four-stage deamination chemistry common to cytidine and cytosine deaminase. This was reassuring because the same arrangement of primary catalytic residues directly responsible for cytidine deamination is classically found in cytidine deaminases across evolution. Furthermore, the periphery of AID’s catalytic pocket houses a network of noncatalytic residues, termed the “secondary catalytic residues”. These residues, while not directly involved in deamination catalysis, contact and/or stabilize the dC in deamination-conducive confirmations within the catalytic pocket.80 This network of amino acids consists of G23, R24, R25, E26, T27, L29, N51, K52, N53, G54, C55, V57, T82, W84, S85, P86, D89, Y114, F115, C116, and E122 in human AID.80 These residues form the “walls” and “floors” of the catalytic pocket and interact with substrate dC in several predicted protein conformations through hydrogen bonding, electrostatic interactions, and aromatic base stacking. Since publication of the functional structure of AID using the computational–biochemical method, two partial crystal structures have become available81,82 which confirmed the map of AID’s catalytic pocket, including the aforementioned arrangement of the primary and secondary catalytic residues.
A more detailed observation of the conformational states of AID’s catalytic revealed that it shifts dynamically between open and closed positions and that the majority (∼75%) of conformations at any time are predicted to exhibit a closed pocket.80 This dynamic pocket closure, termed the “Schrodinger’s CATalytic pocket”83 for its duality, was the first demonstration of such an inherent regulatory mechanism in human DNA/RNA-editing enzymes. More recently, this type of catalytic pocket closure was observed by X-ray and NMR on A3A and A3B, two close siblings of AID.84−87
Previous works on small-molecule inhibitors of AID/APOBECs have been largely focused on A3G. Screening of a library of 1280 pharmacologically active compounds against A3G yielded several structurally related small molecules that inhibited A3G at low micromolar concentrations through covalent attachment of a nonconserved cysteine (C321) unique to A3G’s substrate-specificity loop 7.78 As a follow-up, a screening of >300 000 compounds yielded different A3G inhibitors that covalently attached to the same cysteine residue.88 Although useful for studying A3G biology, the electrophilic nature of these inhibitors is too cross-reactive in a cellular context and is thus not suitable for cellular work. As for AID, 5-aza-deoxycytidine incorporated into ssDNA was shown to bind the active site of AID and inhibit expression via proteasomal degradation,75 though 5-aza-deoxycytidine is a transition-state analogue which needs to be in ssDNA, as the free form does not inhibit AID. More recently, 5-fluoro-2′-deoxyzebularine incorporated into ssDNA was also shown to inhibit A3A and an A3B-A3A chimera.89
Thus, to date, there are no reported specific small molecules of AID, A3A, or A3B. As an extension of our previous works on delineating AID’s catalytic pocket, we screened a library of small molecules against multiple highly accessible conformations of its catalytic pocket. We identified first-generation small molecule inhibitors that specifically inhibit the mutagenic activity of purified AID, native AID in whole cell extracts, and endogenous AID of B lymphoma cells. Docking and mutational analyses reveal a network of contacts between the small molecules and AID’s primary and secondary catalytic residues. We found some analogue derivatives of the first-generation small molecules that were even more efficient inhibitors of A3A and A3B compared to AID.
Results
Rationale for Targeting the Catalytic Pocket of AID
AID’s catalytic pocket is an ideal target for small molecule inhibition for several reasons. First, we have previously gleaned detailed insights into its architecture and conformational breathing.80 Second, the majority of structural differences between AID and related cytidine deaminases are concentrated in the catalytic pocket and proximal surface regions at the pocket opening,80,83,90−93 thus allowing maximum specificity. Third, we and others have described the pocket-adjacent main ssDNA binding groove (groove 1) in detail,80,82,83,94,95 thus offering a promising target for future derivatization.
We reasoned that the functional and breathing structure of AID’s catalytic pocket as first described through our computational-biochemical approach80 is an advantageous template for inhibitor search for several reasons. First, the architecture of its catalytic pocket was extensively functionally verified by testing of mutants and chimeras.83 Second, using multiple catalytic pocket conformations is advantageous since AID/APOBEC enzymes contain highly flexible loops that compose their catalytic pocket leading to a range of different catalytic pocket conformations for each enzyme.80,83,90,91,96 Third, purely homology modeled structures, using the same homology modeling methodology used to arrive at AID’s functional structure, were shown to be as reliable for generating inhibitor “hits”, as crystal structures.97 Considering that the functional AID structure, especially of its inhibitor-target catalytic pocket, was backed by extensive biochemical validation of homology modeling predictions using AID mutants and chimeras80,83 and was also confirmed by crystal structures (Figure S1),81,82 we reasoned that it ought to represent an even more high confidence template for inhibitor design.
Structure-Based Virtual Screening of Small Molecules against the Catalytic Pocket of AID
To carry out a structure-based docking search for small molecules that bind in its catalytic pocket, we included five different conformations of AID which are representative of the full range of conformations with accessible catalytic pockets (Figure 1A–C).80,83 This strategy would allow for the selection of compounds that bind all active AID conformations. For small molecules, we utilized the ZINC database which contains >100 million structures of commercially available compounds.98 We restricted our search to the “clean lead” subset of 4.6 million compounds because this set contains compounds that are lead-like defined by pharmacological properties such as Lipinski’s rule of 5 and properties generally amenable for oral intake. This set is composed of molecules with benign functionality and excludes those with potentially toxic chains such as aldehydes and thiols.99,100
Figure 1.

Virtual high-throughput screening of druglike small molecules against the catalytic pocket of AID. (A) Ensemble of AID conformations covering the range of accessible catalytic pockets dynamics used for virtual high-throughput screening. The structures exhibit conserved overall structure, with conformational changes localized to the secondary catalytic loops that compose the walls and floors of the catalytic pocket. N- to C-termini progression is shown from blue to red. The purple sphere depicts the catalytic pocket-coordinated Zn. (B) Representative surface structure of a catalytically productive AID:ssDNA complex with docked ssDNA (blue) and dC poised for deamination in the catalytic pocket (magenta). (C) dC bound in a catalytically productive configuration in the catalytic pocket highlighting the secondary catalytic residues. Adjacent ssDNA structure was omitted for clarity. This and other energetically similar conformations of the accessible catalytic pocket state of AID served as template for virtual high-throughput screening. (D) The structure-based virtual high-throughput screening scheme. We screened a large library of lead-like compounds from the ZINC library against the catalytic pocket of AID. Using several independent and complementary docking protocols to the catalytic pocket alone or the entire surface of AID, we identified 10 low-energy compounds predicted to bind in the catalytic pocket. (E) Structures of the 10 first-generation inhibitor candidates (C1–C10).
Using DOCK Blaster,101 we screened for candidates that bind AID in a search space restricted to the catalytic pocket and proximal surface region (∼10 Å; Figure 1C and D). For each AID conformation, we identified the 500 lowest-energy compounds. We then prioritized compounds that bound several AID conformations over those that bound only to a single conformation and selected the 40 lowest binding energy compounds (Table S1). To confirm specificity for the catalytic pocket, unrestricted docking was repeated using the entire surface of AID, rather than just the catalytic pocket region. In addition, we employed a second independent docking algorithm, AutoDock Vina,102 to substantiate the 40 candidates identified by Dock Blaster. Even with access to AID’s whole surface, 27 of 40 compounds bound preferentially in the pocket, thus validating our screening methodology while further refining the list of hits. The 10 inhibitor candidates with the most favorable docking energies (C1–C10) were selected for functional testing (Figure 1E).
First Generation Hits That Inhibit Purified and Endogenous AID
Using the standard alkaline cleavage enzyme assay for deamination activity of purified AID/APOBEC enzymes, we measured the catalytic activity of purified GST-AID in the presence of each compound (Figure S2A and B). We found that 2 of 10 compounds (C4 and C8) diminished AID activity (8.3% and 17.4% AID catalytic activity, respectively; Figure 2A). In addition to purified GST-AID, we tested inhibition on whole cell extracts of AID-expressing 293T cells transfected with a CMV-promoter based AID-His expression vector (Figure 2B). Akin to our results with purified GST-AID, C4 and C8 inhibited this AID as well (43.9 and 40.3% AID catalytic activity, respectively). To assess potency, we measured the dose–response of C4 and C8 against GST-AID and the AID-expressing 293T cell lysate (Figure 2C and D). C4 and C8 showed a similar potency in GST-AID (IC50 = 290 and 230 μM, respectively) as with AID-His (IC50 = 460 and 390 μM, respectively).
Figure 2.

First-generation inhibitor candidates inhibit purified AID and AID in whole cell extracts. (A) Catalytic activity of bacterially expressed and purified GST-AID treated with C1–C10 (n = 6 independent experiments conducted with three independently purified preparations of GST-AID). (B) Catalytic activity of eukaryotic-expressed AID in whole 293T cell lysate treated with C1–C10 (n = 3 independently prepared AID-expressing whole cell extracts). (C) Catalytic activity of GST-AID on C4 and C8 as a function of log inhibitor concentration. (D) Catalytic activity of AID-His 293T lysate as a function of log inhibitor concentration. (E) Catalytic activity of eukaryotic-expressed and purified GST-A3A, GST-A3B, GST-A3F, and GST-A3G treated with 700 μM C8. (F) Catalytic activity of GST-A3A, GST-A3B, and GST-A3G in comparison to bacterially expressed and purified GST-AID across a concentration range of C8. All experiments contained a negative control vehicle-only (140 mM DMSO) reaction which was designated as 100% AID activity. All AID reactions were performed at 37 °C for 2–4 h at pH 7.2 using 2 nM of the standard bubble oligonucleotide substrate TGCbub7 which has previously been demonstrated to be AID’s most favored substrate in the alkaline cleavage assay. GST-A3A, GST-A3B, GST-A3F, and GST-A3G reactions were incubated at 37 °C for 2 h in pH 6.0 using 2 nM of standard single-stranded oligonucleotide substrates containing a single target TTCA motif for A3A, A3B, and A3F and a single target CCC motif for A3G.
To evaluate toxicity, we incubated multiple cell lines originating from different tissues (A549, MCF-7, 293T, and Raji) as well as primary peripheral blood monocytes (PBMC) with C4 or C8 and measured viability using the standard MTT assay (Figure S2C). C4 caused minor toxicity toward all anchorage-dependent cells (A549, MCF-7, and 293T) but not Raji or PBMCs, while C8 was found not to be toxic. We thus focused on C8 for further development.
To confirm that C8 was a bona fide AID inhibitor we measured off-target inhibition of UDG, an enzyme used downstream of AID in the alkaline cleavage deamination assay and found C8 did not inhibit UDG (Figure S2E). To test specificity, we examined whether C8 could inhibit the catalytic activity of homologous A3A, A3B, A3F, and A3G. We found GST-A3A, GST-A3B, and GST-A3G were inhibited (5%, 5%, and 37% activity, respectively), while GST-A3F was unaffected (Figure 2E). Compared to bacterially expressed GST-AID, 293T-expressed GST-A3A and GST-A3B were more potently inhibited, and GST-A3G was inhibited to a lesser degree (IC50 = 220, 60, 120, and 280 μM, respectively; Figure 2F). Thus, C8 acts as an AID/A3 inhibitor capable of blocking catalytic activity of specific family members.
C8 inhibition of multiple forms of AID (purified GST-AID and AID-His in whole cell lysates of 293T cells) was reassuring. We then examined the ability of C8 to inhibit native (no fusion tag) full-length AID expressed in 293T cells and found that surprisingly it was even more effective at inhibiting untagged AID in whole cell lysates (IC50 = 11 μM) than purified AID was (Figure 3A). This result prompted us to examine the ability of C8 to inhibit the endogenous AID of AID-expressing lymphoma cell lines (Raji, Daudi, and Ramos; Figure 3B). Given that endogenous AID levels are significantly lower than that of 293T cells transfected with CMV promoter-driven AID expression vectors, we were unable to detect AID activity from cell lysates in the alkaline cleavage assay. Instead, we employed a more sensitive semiquantitative deamination-specific PCR assay (Deam-PCR) which we have previously established for measuring AID activity (Figure S3).7,94,103−105 In this assay, a substrate plasmid is incubated with AID and subjected to PCR using deamination-specific primers that amplify DNA deaminated by AID (Figure S4A). Using this assay on C8-treated extracts of all three lymphoma cells, we also observed inhibition of endogenous AID in the whole cell extracts of three different lymphoma cell lines (Figure 3B). The assay also further confirmed the inhibition of purified AID (Figure 3B, bottom gel). In addition, we observed inhibition of exogenous AID expressed in 293T cells, in the whole cell extract (Figure 3C).
Figure 3.

C8 inhibition of native untagged AID and endogenous AID from B lymphoma cells. (A) Representative alkaline cleavage experiment demonstrating C8 inhibition of untagged native AID in 293T whole cell lysate (left panel). Catalytic activity of eukaryotic-expressed native untagged AID as a function of log C8 concentration, using 140 mM DMSO as control for 100% AID activity (right panel). (B) Deam-specific PCR was used to detect activity of endogenous AID in extracts of AID-expressing B lymphoma cell lines, with or without addition of C8. The deamination substrate plasmid was incubated with cell extracts containing endogenously expressed AID from Raji (top panel), Ramos (middle panel), and Daudi (bottom panel) with added vehicle (140 mM DMSO) or C8. Incubation with C8, but not DMSO, abrogated any detectable PCR product. The bottom gel is a representative experiment demonstrating inhibition of AID-His using Deam-PCR. (C) Deam-PCR was used to demonstrate inhibition of AID expressed in 293T cells, in whole cell extract incubated with the target plasmid.
Inhibition Is Sensitive to Mutation of Catalytic Pocket Residues
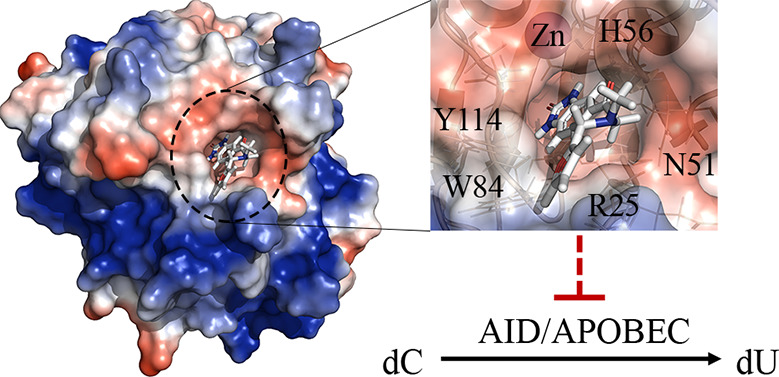
Docking revealed that C8 binds into the catalytic pocket of AID (Figure 4A) and that akin to AID’s native substrate dC (Figure 1C) C8 is stabilized through multiple interactions with secondary catalytic residues in the catalytic pocket.80 These secondary catalytic residues are housed on four secondary catalytic loops: Loops 1 (L1), 3 (L3), 5 (L5), and 7 (L7) (L1, L3, L5, and L7 correspond to the β1-α1 loop, β2-α2 loop, β3-α3 loop, and β4-α4 loop, respectively; Figure 4A, left panel).80Table 1 describes the C8-stabilizing residues across multiple AID conformations, showing π–π stacking and hydrogen-bonding residues. In all AID-C8 complexes, the C8 benzimidazol-2-one is anchored deep in the catalytic pocket where it can π–π stack with H56 and/or Y114 and hydrogen bond with T27, E58 backbone, and S85 backbone. Notably, these are the same residues that interact with AID’s native substrate dC for stabilization and deamination in the catalytic pocket.80,82 The C8-tail bound the ssDNA binding grooves with some flexibility; AID-C8 complexes bound the C8-tail either in the L1-L7 interface (between R25, W84 and Y114; Figure 4A, middle panel) or in the L1-L3 interface (between R25, E26, N51, and K52; Figure 4A, right panel), with a slight preference for the former. The tail-carbonyl hydrogen bonded with N51, H56, or Y114, while the furan group π–π stacked with W84 or Y114 and hydrogen bonded with R25 or Y114 (Table 1). Thus, the benzimidazol-2-one was firmly bound in the catalytic pocket, while the flexible tail was predicted to adopt several configurations interacting with the ssDNA binding grooves.
Figure 4.

Predicted binding modes of AID-C8 and inhibition-resistant mutations (A) Representative AID-C8 docked complex illustrating a low-energy binding mode (left), with the AID surface colored according to surface charges (blue = positive, red = negative, purple = Zn-coordinating residues). The C8 benzimidazol-2-one scaffold was bound deep in the catalytic pocket with the tail region bound outside, stabilized by several ssDNA binding residues in either the L1-L7 (middle) or L1-L3 (right) interface. For each inhibitor, the carbon backbone is colored green, with nitrogen, oxygen, and hydrogen colored blue, red, and white, respectively. (B) C8 inhibition of AID mutants targeting residues predicted to interact with C8 by docking analysis, demonstrating that mutation of ssDNA binding groove residues that directly stabilize C8 results in resistance to C8 inhibition. Dr-AID, representing an evolutionary-distant AID orthologue was unaffected by C8.
Table 1. Inhibitor Binding Residues across All Conformations of AID, A3A, A3B, and A3G.
| C8 | benzimidazol-2-one (π–π stacking) | benzimidazol-2-one (H-bonding) | linker carbonyl (H-bonding) | tail furan (π–π stacking) | tail (H-bonding) |
|---|---|---|---|---|---|
| AID | H56, Y114 | T27, H56, E58,a S85,a C87a | N51, H56, Y114 | W84, Y114 | R25, Y114 |
| A3A | H70, Y130 | T31, N57, S99a | H29, N57 | H29, W98, Y130, Y132 | R28, Y130 |
| A3B | Y250, H253, Y313, Y315, intramolecular | R212,a T214, N240, R252,a S282,a P283,a C284,a D316 | H253, Y313, Y315 | Y250, H253, F285, Y313, Y315, intramolecular | R211, Y250, H253, C284, Y313, Y315. |
| A3G | H257, W285, Y315, intramolecular | H257, E259, S286,a T218 | none | H216, W285, Y315, intramolecular | R213 |
| C8.5 | benzimidazol-2-one (π–π stacking) | benzimidazol-2-one (H-bonding) | linker carbonyl (H-bonding) | tail phenylpropane (π–π stacking) | tail (H-bonding) |
|---|---|---|---|---|---|
| AID | H56, Y114 | T27, H56, C87,a W84,a S85a | N51, H56, Y114 | W84, Y114, F115 | Y114 |
| A3A | H70, W98, Y130 | T31, A71,a E72,a S99,a D131 | R28, T31, N57 | H29, W98, F102, Y132 | Y130 |
| A3B | Y250, H253, Y313 | T214, N240, S282a, R212a | Y315 | F285, W287, Y315 | Y250 |
| A3G | H257, W285, Y315 | T218 and S286a | none | W285, Y315 | none |
| C8.12 | benzimidazol-2-one (π–π stacking) | benzimidazol-2-one (H-bonding) | linker carbonyl (H-bonding) | tail 2,3,4,5-tetrahydro-1,4-benzoxazepine (π–π stacking) | tail (H-bonding) |
|---|---|---|---|---|---|
| AID | H56, Y114 | T27, E58,a W84,a S85a | N51, Y114 | W84, Y114 | G23a |
| A3A | H70, Y130 | T31, A71,a E72,a S99,a C101a | N57 | H29, W98, Y130, Y132 | R28, Y132 |
| A3B | H253, Y313 | R212,a T214, S282,a S282, P283,a D316 | R211, Y315 | F285, W287, Y313, Y315 | R212a |
| A3G | H257, W285, Y315 | T218, N244, H257,a E259, S286a | Y315 | H257, W285, Y315 | none |
Indicates the peptide backbone of that residue.
To verify these docking results, we measured C8 inhibition on a panel of AID mutated at residues predicted to interact with C8 (Figure 4B). We were limited in testing AID mutations, as many of the predicted C8-stabilizing residues are key catalytic residues, wherein even conservative mutations (e.g., N51Q and Y114F) result in catalytically dead mutants;80 however, we tested the ability of C8 to inhibit AID mutated at each of the other contact residues located at the opening of the catalytic pocket (K22A, E26R, R25H, and R25N) and found that all were resistant to C8 inhibition when compared to wild type (IC50 = 500, > 1000, 890, and 770 μM, respectively). Zebrafish AID (Dr-AID) which naturally has a R25H equivalent, was also resistant to C8 inhibition (IC50 > 1000 μM). These data provide functional validation for the predicted AID-C8 interactions and highlight the specificity of C8 for the catalytic pocket and adjacent regions of human AID.
Structural analogues of C8 also exhibit AID inhibition
Using the ZINC database, we identified 948 structural analogues of C8 using a 60% structural similarity cutoff. We docked each analogue with several conformations of catalytically accessible AID, using the same approach described in Figure 1. Analogous to our initial screening approach for identifying C1–C10, we ranked C8 analogues based upon docking energy and chemical diversity and selected 15 analogues (C8.1–C8.15; Figure S5A). C8 analogues were tested for inhibition of purified AID (Figure S5B and C), and several were found to inhibit AID. Compared to C8, C8.5 and C8.12 inhibited purified GST-AID (230, 140, and 160 μM, respectively) and 293T-expressed AID-His (390, 340, and 370 μM, respectively) by a relatively similar degree (Figure 5A and B). However, C8 potently inhibited AID (untagged) to a far greater extent in comparison to C8.5 and C8.12 (IC50 = 11, 90, and 130 μM, respectively; Figure 5C). We performed a similar screen for C4 analogues (C4.1–C4.5), but none were capable of inhibiting AID (Figure S6A and B). Thus, C8, C8.5, and C8.12 inhibit AID activity to a similar degree, except for untagged AID in whole cell extracts which was most potently inhibited by C8. We then carried out a modified intracellular version of the Deam-PCR assay described above, wherein the plasmid substrate was transfected into the lymphoma cells that were either treated or untreated with C8 and C8.5, followed by cell lysis and Deam-PCR to measure mutations mediated by endogenous AID (Figure S4A). Here we also observed that C8 and C8.5 treatment of transfected Raji cells inhibited AID-mediated mutations (Figure S4B)
Figure 5.

Structural analogues of C8 exhibit variable potency against AID, A3A, A3B, and A3G. Each panel measured catalytic activity in the presence of C8, C8.5, or C8.12. (A) Bacterially expressed and purifiedGST-AID. (B) Eukaryotic-expressed AID in whole 293T cell lysate. (C) Eukaryotic-expressed native untagged AID in whole 293T cell lysate. (D) Eukaryotic-expressed and purified GST-A3A. (E) Eukaryotic-expressed and purified GST-A3B. (F) Eukaryotic-expressed and purified GST-A3G. (G) Eukaryotic-expressed native untagged A3A in 293T cell lysate. (H) List of C8, C8.5, and C8.12 IC50 values across each enzyme. All experiments used 140 mM DMSO as a negative control, designating 100% enzyme activity. All AID reactions were performed at 37 °C for 2–4 h at pH 7.2 using 2 nM of the standard bubble oligonucleotide substrate TGCbub7. GST-A3A, GST-A3B, GST-A3F, and GST-A3G reactions were incubated at 37 °C for 2 h in pH 5.5 for A3A/B and pH 6.0 for A3G/F using 2 nM of standard single-stranded oligonucleotide substrates containing a single target TTCA motif for A3A, A3B, and A3F and a single target CCC motif for A3G.
C8.5 Inhibits the Catalytic Activities of A3A, A3B, and A3G
Given inhibition by C8 on A3A/B/G (Figure 2E and F), we next sought to examine inhibition with C8.5 and C8.12 (Figure 5D–G). We found that C8.5 inhibits GST-A3A, GST-A3B, and GST-A3G and is remarkably more potent on inhibition of GST-A3A than any other inhibition value measured thus far in our assays (IC50 = 3, 40, and 70 μM, respectively). In contrast, C8.12 moderately inhibited GST-A3A but was a poor inhibitor of GST-A3B and GST-A3G (IC50 = 290, 700, and 740 μM, respectively). We also performed a similar screen of C8, C8.5, and C8.12 on GST-A3F but did not detect any inhibition (Figure S6C). Like the AID (untagged) expression system, we also attempted to express untagged versions of A3A and A3B but were only successful with A3A. We found that untagged A3A was rendered completely resistant against C8 (IC50 > 1000 μM), while C8.5 most potently inhibited its activity (IC50 = 9 μM), and C8.12 moderately inhibited its activity (IC50 = 170 μM; Figure 5G). Thus, of all the inhibition values measured in this study, the most potent inhibition was in the case of C8.5 and A3A, in single digit micromolar values for two independent forms of the enzyme.
Predicted AID/APOBEC3-Inhibitor Complexes Vary by Structural Differences between Secondary Catalytic Loops
Given the varying degrees of inhibition by C8, C8.5, and C8.12 on AID, A3A, A3B, and A3G, we sought a structural rationale for the observed differences. C8, C8.5, and C8.12 conserve the warhead benzimidazol-2-one group, the tail carbonyl, and a flexible, largely hydrophobic tail containing a planar ring available for π–π stacking (Figure 6, top panel). For each enzyme, we examined inhibitor binding variability across two to three catalytically accessible conformations and tabulated the most frequently contacted residues by the inhibitors (Table 1).80,83
Figure 6.

Favored binding modes of C8, C8.5, and C8.12 with AID, A3A, A3B, and A3G. Top panel: Conserved benzimidazol-2-one scaffold (blue) among C8, C8.5, and C8.12. Each inhibitor contains a tail-carbonyl and a flexible tail containing a largely hydrophobic aromatic ring. Each enzyme–inhibitor pair depicts the benzimidazol-2-one group bound deep in the catalytic pocket, with the flexible tail stabilized by different combinations of the secondary catalytic loops (L1, L3, L5, and L7). Stabilizing residues are transparent, colored, and labeled. The Zn-coordinating residues are colored purple. For each inhibitor, the carbon backbone is colored green, with nitrogen, oxygen, and hydrogen colored blue, red, and white, respectively.
Figure 6 illustrates the dominant binding mode (shown in stick), while stabilizing residues are shown in colored transparent surfaces. In AID, the C8-tail frequently bound the L1-L7 interface with some conformations in the L1-L3 interface, while the C8.5 tail bound the L5-L7-α4 region and the C8.12-tail bound the L1-L3 interface (Figure 6, AID panel). In A3A, we found conformations of inhibitors where the benzimidazol-2-one group bound facing the Zn-coordinating residues and some that were slightly tilted toward L3 (shown for A3A-C8.5). For A3A, the C8-tail and C8.12-tail both bound between L1-L3-L7, with most interactions in the L1-L7 interface. The C8.5-tail preferentially bound the L5-L7-α4 region, with some conformations in the L1-L7 interface (Figure 6, A3A panel). Like A3A, we also noted different configurations of the benzimidazol-2-one group bound in the catalytic pocket of A3B, either slightly tilting toward L3 or L7. Furthermore, among all enzyme–inhibitor pairs, we noted the most inhibitor-tail flexibility when bound with A3B. For A3B, the C8-tail most frequently bound the L1-L3 interface, with other configurations bound with L1-L7, L1-L5-α4, and L3-L5-L7. Akin to AID and A3A, in A3B, the C8.5-tail frequently bound the L5-L7-α4 interface, with some binding also observed on the L1-L7 interface. The C8.12-tail primarily bound the L3-L5-L7-α3 interface, with some bound in the L1-L7 interface (Figure 6, A3B panel). Unlike A3A or A3B, we found A3G bound C8, C8.5, and C8.12 with limited variability, whereby each tail only bound in the L1-L7 interface, except for the C8.12-tail, which had some conformations bound in the L1-L3 interface (Figure 6, A3G panel). Across AID, A3A, A3B, and A3G, we noted the benzimidazol-2-one was consistently stabilized by the Zn-coordinating histidine (H56, H70, H253, and H257, respectively) as well as several secondary catalytic residues, including the L1 threonine (T27, T31, T214, and T218, respectively), the L5 serine backbone (S85, S99, S282, and S286, respectively), and the L7 tyrosine (Y114, Y130, Y313, and Y315, respectively; Table 1). Interestingly, the C8.5-tail preferentially bound the L5-L7-α4 interface of AID, A3A, and A3B, whereby the tail phenylpropane π–π stacked with one or more aromatic residues.
Discussion
Since AID expression drives and exacerbates tumorigenesis, an AID inhibitor has been suggested to be of benefit; however, development of such an agent has not been possible since structural insights into AID were not gleaned until recently. The functional and native structure of AID described in 2015 revealed two structural features that explained the unusually low catalytic rate and high ssDNA binding affinity of AID.80 These include frequent catalytic pocket closure and sporadic ssDNA binding by a highly positively charged surface, in positions that are not deamination-viable. This structural analysis, which has since been confirmed by two partial AID crystal structures,81,82 provided an understanding of AID’s catalytic pocket conformational dynamics and ssDNA stabilization interactions proximal to the pocket (Figure 1C), thus offering an opportunity for structure-based inhibitor design.
Using these AID–DNA interactions, we scanned the ZINC database of lead-like compounds and identified 10 compounds for testing against AID (Figure 1D and E). We identified two compounds, C4 and C8, that inhibit the enzymatic activity of AID (Figure 2). MTT assay showed that C8 was noncytotoxic across several cell lines and primary healthy donor cells tested (Figure S2C). This suggested that C8 or derivatizations could be used successfully in future in vivo studies, and hence, we focused on C8. Although purified GST-AID was inhibited by C8 with modest IC50 values (230 μM), C8 could inhibit multiple forms of purified AID (both bacterially expressed GST-AID and 293T-expressed AID-His) as well as AID in whole cell lysates of 293T cells (Figure 2). It was particularly encouraging that native untagged AID in whole cell 293T extract, which is the best representative of endogenous cellular AID, was inhibited by C8 with the most effective IC50 of 11 μM, ∼21-fold more effectively than purified AID (Figure 3A). This increased susceptibility of C8 inhibition is likely due to the higher specific activity of native untagged AID, thus requiring a lower amount of enzyme to achieve similar catalytic rates when compared with purified GST- or His-tagged AID.
Since the topological features of the AID catalytic pocket and surrounding region are unique, we rationalized that these residues ought to act as specific anchors for small molecule placement. Furthermore, inhibitor binding in this region would impart a level of structural specificity to our strategy. Akin to dC stabilization (Figure 1C), we noted several stabilizing interactions with secondary catalytic residues and DNA binding residues in and proximal to the catalytic pocket, respectively (Figures 4A and 6). The benzimdazol-2-one group was anchored in the catalytic pocket, while the tail group adopted several conformations in the DNA binding groove. To bolster our understanding of the AID-C8 complexes, we probed several nonlethal surface mutants predicted to destabilize C8 binding. DNA binding groove mutants (K22A, R25H, R25N, and E26R) as well as evolutionary-distant Dr-AID were all resistant to C8 inhibition with increased IC50 values (Figure 4B).
Using C8 as a parent compound, we examined 948 structural analogues, obtaining 15 (C8.1–C8.15) for analysis. Analogues lacking the protruding carbonyl characteristic of the benzimidazol-2-one group (C8.2, C8.3, C8.4, C8.13, and C8.14) resulted in a major loss of AID inhibition (Figure S5), likely due to a loss of stabilizing hydrogen bonds. C8.5 and C8.12 inhibited AID with similar potencies relative to C8 (Figure 5A and B), except in the case of native AID (untagged), whereby C8 was superior with an IC50 of 11 μM (Figure 5C). Akin to C8, C8.5 and 8.12 conserve the benzimidazol-2-one group, a tail carbonyl, and a flexible, mostly hydrophobic tail with a planar ring available for π–π stacking interactions (Figure 6, top panel).
To assess the specificity of C8 within the AID/A3 family, we measured the inhibition of A3A, A3B, A3F, and A3G. Surprisingly, A3A was potently inhibited by C8, A3B and A3G were moderately inhibited, and A3F was unaffected (Figure 2E and F). Even more surprising is the fact that C8.5, a C8 analogue screened against the catalytic pocket of AID, potently inhibited A3A, A3B, and A3G while it moderately inhibited AID (Figure 5). However, in retrospect, we believe this result was achieved for two reasons: First, the catalytic pockets of AID, A3A, A3B, and A3G each evolved to be conducive to a polynucleotide containing dC, and, as such, many of the secondary catalytic residues that stabilize dC also stabilize C8.5 (e.g., hydrogen bonding with the conserved L1 threonine residue T27, T31, T214, and T218, respectively; Table 1). Second, our screening of AID exclusively included accessible catalytic pockets, which are predicted to represent the minority of conformations at any given time.80,83 A3A and A3B-CTD have high sequence similarity, with the largest differences observed in the L1 region, with identical sequences for L5, L7, and α4 (Figure S6D). Given the dominant binding modes of the C8-tail (L1-L7 vs L1-L3 interface for A3A and A3B, respectively) and the C8.12-tail (L1-L7 vs L3-L5-L7-α3 interface for A3A and A3B, respectively; Figure 6), we were not surprised to find differences in inhibition potencies. In support of the importance of these interactions, we note that A3F, which is unaffected by C8, C8.5, or C8.12 (Figure S6C), has major sequence differences of L1/L3/L5/L7 when compared to A3A/B/G as well as notable differences in its catalytic pocket chemistry that may explain a lack of inhibition (Figure S6D). Such differences include alternative Zn-coordination106 and differences among key inhibitor-stabilizing residues (e.g., L1 threonine stabilizing residue is instead a serine, S216; Figure S6D). We noticed that A3A-inhibitor and A3B-inhibitor complexes with the benzimidazol-2-one group bound directly facing toward either Zn or Zn-coordinating residues, but also a tilted angle toward L3 or L5 residues (an example is shown in Figure 6 for A3A-C8.5), a feature not observed for AID or A3G. We postulate that these tilted binding modes observed for A3A and A3B may bestow increased conformational freedom of inhibitors, thus allowing for overall enhanced stabilization of the benzimidazol-2-one group in the catalytic pocket.
For both AID and A3s, we ensured that multiple enzyme versions are tested for inhibition, including GST-tagged purified and untagged in whole cell extracts and, in some cases, several other forms expressed in different cell hosts. In our view, this approach provides critical confidence that the observed inhibition is not a property of one version of the enzyme, as fusion tags and expression hosts are well-known to impact protein conformations and, in the case of enzymes, specific activity levels.107 However reassuring the results were that in each case multiple forms of AID and A3s were indeed inhibited, the differences in IC50 values between the various versions are a testament to such conformational differences due to expression hosts and fusion tags.
In the future, to maximize binding affinity and specificity among AID/A3 family members, several strategies could be undertaken to generate more efficacious inhibitors. Derivatization of the C8/C8.5/C8.12 benzimidazol-2-one scaffold could introduce additional hydrogen bonding pairs, conducive to the secondary catalytic residues of the AID/A3 catalytic pocket to improve affinity. Additionally, these derivatives could be modified to take advantage of the multiple regions bound by the tail of each inhibitor. Instead of a single, flexible tail connected to the benzimidazol-2-one scaffold, a 2- or 3-tail inhibitor could be constructed, such that the L1-L3, L1-L7, and L5-L7-α4 regions are bound simultaneously. Because most structural differences reside within these secondary catalytic loops, in particular the L1-L7 interface, such multitailed derivatives could achieve improved affinity and specificity to the desired AID/A3 target. An alternative approach for AID/A3 inhibitor design might include screening for inhibitors that bind accessible and/or partially occluded conformations of the catalytic pocket. This would help improve the specificity of inhibitors toward AID/A3 family members, based upon their catalytic pocket dynamics. For example, inhibitors restricted to the partially occluded conformations of A3B would be less likely to bind A3A, due to A3A’s superior catalytic pocket accessibility.83−86,96 Such inhibitors would prove useful as probes to study AID/A3 biology and as initial scaffolds for future drug design. This effort could achieve either specific inhibition of A3A or A3B, or pan inhibition of AID, A3A and A3B, three highly tumorigenic AID/APOBEC family member enzymes. Unlike C8, the C8.5 and C8.12 tail-structures were much more hydrophobic (Figure 6) and required sonication to coax dissolution (see Methods). Despite our best efforts, our working aliquots were only partially dissolved in solution and thus, the true IC50 of C8.5 and C8.12 may likely be even lower than the experimentally observed IC50. We suggest that future modifications of tail structures that include hydrogen bond donor/acceptor groups should dramatically improve solubility.
Recently, several studies of SARS-CoV-2 have suggested that APOBECs may contribute to viral mutagenesis.108−112 Similar to their role in promoting immune and drug escape in HIV,47,113−115 APOBEC-induced mutations could generate SARS-CoV-2 variants with enhanced immune evasion and drug resistance. It is tempting to consider APOBEC inhibitors in this context, in addition to therapeutics that thwart cancer genome mutations. Beyond their potential for therapeutic development, the demonstration that C8 and analogues, which were identified based on docking into the Schrodinger’s CATalytic pocket, could indeed functionally inhibit AID and A3s provides further verification of the pocket structure itself. In the future, these and other derivatized inhibitors could also be useful as biochemical probes for studying the interaction of AID and A3s with DNA/RNA and as novel tools for studying their biology.
Methods
Virtual High-Throughput Screening of Small Molecules against the Catalytic Pocket of AID
The AID structure used for high throughput in silico identification of first generation hits is based on the functional and native AID structure described previously though a combined computational–biochemical method.80 This structure has been verified by a partial AID crystal structure.81,82 In addition, since the catalytic pocket of AID was designated as the inhibitor target for this study, this structure is advantageous because it includes several dynamic conformations of AID’s catalytic pocket, verified by biochemical analysis of AID variants.80 Briefly, this structure was generated by modeling full-length AID based on eight X-ray or APOBEC structures as templates for homology modeling: A2 NMR (PDB: 2RPZ), A241–224 chain A and B X-ray (PDB: 2NYT:A and 2NYT:B, respectively), A3A NMR (PDB: 2M65), A3C X-ray (PDB: 3VOW), A3F-CTD X-ray (PDB: 4IOU), A3G-CTD NMR (PDB: 3E1U), and A3G-CTD NMR (PDB: 2KBO).84,116−121 All AID/APOBEC X-ray/NMR structures were obtained from the protein databank (http://www.rcsb.org) and visualized using PyMOL v1.7.6 (http://www.pymol.org). Using the default parameters of I-TASSER (http://zhanglab.ccmb.med.umich.edu/I-TASSER/),122,123 full-length human AID (Hs-AID) and variants were modeled from APOBEC templates. The catalytic pocket is defined as the indented space containing Zn and the catalytic residues (H56, E58, C87, and C90 in Hs-AID).
Using DOCK Blaster v1.6.0 (http://blaster.docking.org/),101 we virtually screened 4.6 × 106 “clean-lead” small molecules from the ZINC database (http://zinc.docking.org/) against the catalytic pocket of AID. We used several AID-DNA complexes containing dC in the catalytic pocket as a template for screening.80 In total, we screened five low energy conformations of AID, representative of the range of catalytically accessible catalytic pocket conformations. As a result, small molecules were screened for their ability to bind to the catalytic pocket and surrounding DNA binding grooves across the ensemble of catalytically active AID structures. Compounds were docked and ranked based on binding energy. The 500 lowest energy compounds bound to each AID conformation were cross-referenced, and compounds predicted to bind only one catalytically active AID conformation were excluded. The 40 lowest-energy compounds (Table S1) bound across several catalytically active AID conformations were selected for additional docking using Autodock VINA (http://vina.scripps.edu)102 via PyRx (https://pyrx.sourceforge.io)124 to confirm specificity to the catalytic pocket and ranked energies. The top 10 compounds (C1–C10) were then selected for testing based on binding energy ranking as well as the chemical diversity of structure side chains. Compounds were purchased from Molport, and purities were > 90%. C4, C8, C8.5 and C8.12. Molport IDs are, respectively, 008-366-081, 009-139-310, 005-764-107, 020-119-012. For analogue expansion, we used the same approach against the ZINC database to identify 948 structural analogues of C4 and C8 considering a 60% similarity cutoff. Using Autodock VINA via PyRx, we screened the five AID conformations and identified the top ranking structural analogues of C4 and C8. Additionally, we constructed a full-length AID model based the crystal structure of near-native AID (PDB: 5W1C)82 for binding with inhibitors to confirm AID-inhibitor binding modes. Two A3A conformations were selected from PDB 2M65 (apo) and PDB 5KEG (ssDNA-bound), so the catalytic pockets represent both unbound and bound states (with respective rotations of Y130/Y132, etc). For A3B-CTD, we chose two NMR conformations (PDB: 2NBQ) that had rotations of the equivalent tyrosine residues (Y313 and Y315), albeit different from the A3A structures. For A3G-CTD, we used two structures to represent different conformations (PDB: 3E1U and 3IR2). When examining AID/A3–inhibitor binding mode interactions, we examined three or more low-energy clusters per conformation for a total of six or more docked complexes per enzyme–inhibitor pair.
Expression and Purification of AID/APOBECs
Expression and purification of GST-AID and AID-His in bacteria and HEK 293T cells have previously been described.80,125,126 Briefly, for bacterially expressed GST-AID, the expression and purification of human (Hs-AID), Hs-AID mutants, and zebrafish (Dr-AID) GST-AID were carried out as previously described, using the pGEX5.3 expression system.92,126 Point mutants were generated by site-directed mutagenesis using appropriate GST-AID constructs as templates, as previously described.80 Briefly, GST-AID was expressed in E. coli (DE3-Bl21) and purified using GST-column chromatography as per the manufacturer’s guidelines. In total, six independent preparations of GST-AID and two independent preparations of each mutant/chimeric/orthologous AID were made and tested in parallel. The expression and catalytic activities of AID-His and untagged native AID in HEK 293T cells were carried out as previously described, using the pcDNA3.1 expression system.94,107 Briefly, 50 × 10 cm plates of 293T cells were transfected with 5 μg of plasmid per plate using Polyjet transfection reagent (Froggabio), incubated for 48 h, resuspended in 50 mM phosphate buffer (pH 8.2 for AID-His and pH 7 for untagged AID) + 500 mM NaCl, 0.2 mM PMSF, 50 μg/mL RNase A, and lysed using a French pressure cell press. Whole cell extracts expressing either AID-His or native untagged AID were flash frozen in liquid nitrogen and stored for activity analysis. GST-tagged versions of A3A/B/F/G expressed in HEK-293T were purified using GST-beads per the manufacturer’s guidelines as previously described.107,127 The GST-tagged A3 enzymes were stored in 20 mM Tris-HCl pH 7.5, 100 mM NaCl, 1 mM DTT, 5% glycerol, and 50 μg/mL BSA. Whole cell extracts expressing native untagged A3A obtained as described above were flash frozen in liquid nitrogen. Expression of AID/APOBECs was verified using Western blotting probed with anti-GST (SantaCruz) antibodies, followed by the secondary detection by goat anti-rabbit IgG (SantaCruz). Western blots to verify the expression of untagged native AID/APOBECs were probed with rabbit polyclonal anti-AID antibody (Abcam) or anti-A3A antibody (Abcam), respectively, followed by the aforementioned secondary IgG. The relative yield and purity of each purified AID/APOBEC preparation were evaluated using standard SDS Coomassie staining. The concentration of 293T-purified AID/APOBECs ranged from 10 to 50 ng/μL. The concentration of native untagged AID ranged from 0.7 to 7 ng/μL, and for untagged native A3A it was 4 ng/μL.
Inhibition of AID/APOBEC Using the Alkaline Cleavage Assay
The standard alkaline cleavage assay for AID/APOBEC-mediated deamination was used to screen compounds for inhibition of AID, A3A, A3B, A3F, and A3G. For AID reactions, we used the standard seven-nucleotide bubble substrate containing the WRC motif TGC (5′-TTTGCTT-3′) as a substrate, because it has previously been optimized for the highest levels of AID activity in this enzyme assay.80,125,128 For A3A, A3B, and A3F, we used a single-stranded oligo containing the preferred 5′-TC-3′ dinucleotide;106,129,130 and for A3G, we used a single-stranded 5′-CCC-3′ oligo (Figure S7). Substrates were labeled and purified as described previously.80,92,125 For AID reactions, purified substrate (1.7 nM) was incubated with AID enzyme (∼0.9 μg of bacterially expressed GST-AID, 1–10 ng of 293T-expressed AID-His or untagged AID) in phosphate buffer (100 mM, pH 7.2) with H2O, 140 mM DMSO as a vehicle control or compound (see below). All AID reactions were incubated at 37 °C, except for Dr-AID, which was incubated at 25 °C, which was previously shown to be its optimal temperature,92,126 in a total volume of 10 μL. For A3A, A3B, A3F, and A3G, alkaline cleavage reactions were conducted in the same manner (12–30 ng of GST-tagged A3s and 12 ng of native untagged A3A used per reaction), except they were incubated in buffers of more acidic pH since the A3 family enzymes are optimally active at a more acidic pH as compared to AID.131,132 The A3G/A3F activity buffer was 100 mM phosphate buffer (pH 6.0), 1 mM DTT, and 50 μg/mL RNase A. The A3A/A3B activity buffer was 25 mM HEPES (pH 5.5), 100 mM NaCl, 1 mM DTT, 0.1% Triton X-100, and 100 ng/mL RNase A. Concentrated stocks of compound were sonicated at 37 °C for up to 4 h in 1–10% DMSO to promote dissolution. Due to solubility differences between compounds, the highest achieved concentration in 140 mM DMSO was used for initial screening. For initial C1–C10, C4.1–C4.5, and C8.1–C8.15 screening, final alkaline cleavage concentrations (in 140 mM DMSO) ranged from 500 to 840 μM, except for C9, C8.4, C8.5, and C8.9 which had lower concentrations (310, 400, 170, and 270 μM, respectively). The A3 enzymes tolerated a higher [DMSO] as compared to AID which began to show reduced activity at 10% DMSO; therefore, for the A3 inhibition assays, we were able to use a 1:10 dilution of C8, C8.5, and C8.12 which were dissolved in 100% DMSO, thus bringing the final reaction to 10% (1.4 M) [DMSO].
Inhibition of Endogenous AID in Lymphoma Cells Measured by the Deamination-Specific PCR Assay
Daudi, Raji, and Ramos cells (ATCC = CCL-213, CCL-86, and CRL-1596, respectively) were suspension cultured for 48 h in RPMI 1640 growth media containing 10% FBS. To lyse the cells, cultures were centrifuged and cells (2.5–3 × 106 cells) were washed twice with PBS. Cell were lysed using glass beads (425–600 μm) (Sigma). The volume of glass beads used was twice the volume of the cell pellet. The mixture was then vortexed for 20–30 s and then incubated on ice for 30 s. This was repeated two to three more times in PBS with 0.2 mM PMSF, 50 μg/mL RNase A. To detect AID expression in these cells, we employed quantitative real-time PCR. Total RNA was extracted from 106 cultured cells using TRIzol solution (Invitrogen) according to the manufacturer’s instructions. The quality and quantity of the extracted RNA were estimated by spectrometry. Total RNA (1 μg) was used for cDNA synthesis with the ProtoScript First Strand cDNA Synthesis Kit (NEB, UK). Quantitative real-time PCR was performed in triplicate using SYBR Green I. The amount of GAPDH housekeeping gene transcripts was used as a reference for the level of AID gene expression. Amplification was carried out in a total volume of 20 μL containing 0.5 μg of cDNA prepared as described above, 0.3 μM each of GAPDH- and AID-specific primers, and 1× reaction mixture consisting of RNase-free water and 2× QuantiTect SYBR Green PCR Master Mix (Qiagen). Thermal cycling for both genes was initiated with a denaturation step at 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 15 s, annealing at 50 °C for 30 s, and elongation at 72 °C for 1 min. Melting curve analysis of the amplification products was performed at the end of each PCR by cooling the samples to 60 °C and then increasing the temperature to 95 °C at 0.2 °C/s. The experiment was repeated three times for statistical analysis.
To detect AID activity in cell extracts, we utilized a deamination-specific PCR (Deam-PCR) as previously described for measuring the mutational patterns of purified AID.7,94,103−105 In this assay, a plasmid DNA substrate is incubated with AID and mutations are detected by amplification using deamination-specific primers. Here, we applied this assay in three iterations: first, to measure inhibition of purified AID incubated with the substrate plasmid; second, to measure inhibition of AID in whole cell extracts of lymphoma cells incubated with the substrate plasmid; and third, we extended the application of this assay to detecting intracellular AID-mediated mutations on transfected plasmid substrate. Briefly, the plasmid used as a substrate for the deamination specific PCR assay was pcDNA3.1 containing a random WRC-rich target sequence as previously described. An amount of 50 ng of supercoiled plasmid was denatured at 98 °C in 100 mM phosphate buffer pH 7.2 for 10 min followed by snap-cooling in an ice bath to generate ssDNA targets for AID to mutate. A volume of 4 μL of either purified AID or AID-expressing cell lysates (from Daudi, Raji, or Ramos which are naturally AID+ B lymphomas, or from 293T cell transfected with an AID expression vector as described above) was mixed either with C8 or vehicle, added to the target plasmid, and incubated for 4 h at 32 °C. To detect AID-mediated mutations, 1 μL of each reaction was amplified by deamination-specific nested PCR using mutation-specific primers, as previously described.7,94,103,104 The inner primers used in the second nested PCR reaction generate a 451 nt-product. PCR amplicons were subsequently TA-cloned, and 10 from each reaction were sequenced to confirm AID-mutated mutations C to T or G to A on the sense and nonsense strands. Negative control deamination-specific reactions were conducted on the substrate alone with no added extract or AID. Positive control Deam-specific PCRs were conducted on reactions containing substrate plasmid and extracts of AID-expressing 293T cells, as described above. An additional negative control PCR reaction was carried out by adding C8 directly to the PCR reaction of the aforementioned positive control, in order to make sure that our observation of inhibition of endogenous lymphoma cell AID by C8 is not due to interference of C8 with the deamination-specific PCR step itself. For the intracellular endogenous AID inhibition assay, Raji cells were suspended for 48 h in RPMI 1640 growth media containing 10% FBS. Then 1–2 × 106 cells were transfected with 5 μg of the same substrate plasmid DNA as above. Cells were treated with either C8 or C8.5 at a final concentration of 700 μM or treated with vehicle immediately prior to transfection with the AID substrate plasmid. Untreated transfected Raji cells and untreated untransfected Raji cells were used as negative controls. To lyse the cells 24 h post transfection, cultures were centrifuged and cells were washed twice with PBS. Cell were lysed using glass beads (425–600 μm) (Sigma). The volume of glass beads used was twice the volume of the cell pellet, followed by vortexing for 20–30 s and incubation on ice for 30 s. This procedure was repeated three times. Then 1 μL of the cell lysate or 1 μL of each condition diluted 100 times was subject to Deam-PCR as described above. To confirm that compounds do not interfere with Taq polymerase, either inhibitor-treated or untreated/transfected Raji cell lysate was added to the positive PCR reaction.
MTT Assay
A breast cancer cell line (MCF-7; ATCC = HTB22), lung cancer cell line (A549; ATCC = CCL185), embryonic kidney cell line, (293T; ATCC = CRL3216), B cell lymphoma cell line (Raji; ATCC = CCl-86), and primary peripheral blood mononuclear cells (PBMCs) from healthy donors were used to test the toxicity of C4 and C8. MCF-7, A549, and 293T cells were gifted from Dr. Kao (Memorial University of Newfoundland), while Raji cell lines were gifted from Dr. Hirasawa (Memorial University of Newfoundland). The B cell lymphoma cell line and PBMCs were grown in RPMI-1640 medium (Invitrogen, USA) supplemented with 10% fetal calf serum (Invitrogen, USA), 200 IU/mL penicillin/streptomycin (Invitrogen, USA), 1% 1 M HEPES (Invitrogen, USA), 1% l-glutamine (Invitrogen, USA), and 2.0 × 10–5 M 2-mercaptoethanol (Sigma-Aldrich, USA), whereas MCF-7, A549, and 293T cell lines were maintained in DMEM containing 100 U/mL penicillin and 100 μg/mL streptomycin (Invitrogen, USA) and supplemented with 10% fetal calf serum (Invitrogen, USA). PBMCs from an anonymous healthy donor was obtained with approval from the Health Research Ethics Authority of Newfoundland and Labrador, Canada, carried out in accordance with the recommendations of the Canadian Tri-Council Policy Statement: Ethical Conduct for Research Involving Humans. All cells were grown at 37 °C in a humidified condition containing 5% CO2. Cells were transferred into a 96-well plate (104 cells per well) and treated in eight replicates with C4 and C8 (100, 250, and 450 μM) separately or with vehicle alone (140 mM DMSO). Untreated cells were also considered as the negative control. After 24 and 48 h incubation at 37 °C, 10 μL of 12 mM MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) solution (Molecular Probes) was added to each well and incubated for 4 h. The reaction was terminated by the addition of 100 μL of detergent reagent and incubation for 4 h in the dark. Colorimetric evaluation was performed using a spectrophotometer at 490 nm. The percentage of viable cells was calculated from the absorbance values of untreated and treated cells as %Viable Cells = (OD490 treated/OD490 untreated) × 100.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.1c00091.
Additional data on compound structures, inhibition assays of purified and intracellular versions of the enzymes, toxicity tests, and additional details of experimental conditions and controls (PDF)
Author Contributions
§ J.J.K. and F.B.: These authors made equal contributions.
Author Contributions
J.J.K. and M.L. designed and conceptualized the work. J.J.K. conducted the computational search for the inhibitors based on previous insights generated by J.J.K. into the catalytic pocket structure of AID. J.J.K. carried out the initial enzyme assays, with technical assistance from J.I. and C.P.D. who contributed to the enzyme assays of purified AID, A3A , and A3B. F.B. conducted enzyme assays on purified and native untagged AID, A3A, and A3B and the inhibition assays of endogenous AID in AID-expressing B cells. M.A. conducted the MTT assays. A.G., H.F., and L.B. contributed to the preparation of reagents and purified enzymes.
This work was supported by grants from the Canadian Institutes of Health Research (CIHR) (MOP111132), The Leukemia and Lymphoma Society of Canada (LLSC), and the International Development Research Center (IDRC) (108405-001) to M.L. J.J.K. received support from a CIHR Frederick Banting and Charles Best Canada Doctoral Scholarship and a Beatrice Hunter Cancer Research Institute Cancer Research Training Program (CRTP) fellowship. F.B. was supported by a doctoral graduate scholarship from the Beatrice Hunter Cancer Research Institute Cancer Research Training Program (CRTP).
The authors declare no competing financial interest.
Supplementary Material
References
- Muramatsu M.; Sankaranand V. S.; Anant S.; Sugai M.; Kinoshita K.; Davidson N. O.; Honjo T. (1999) Specific Expression of Activation-Induced Cytidine Deaminase (AID), a Novel Member of the RNA-Editing Deaminase Family in Germinal Center B Cells. J. Biol. Chem. 274 (26), 18470–18476. 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- Muramatsu M.; Kinoshita K.; Fagarasan S.; Yamada S.; Shinkai Y.; Honjo T. (2000) Class Switch Recombination and Hypermutation Require Activation-Induced Cytidine Deaminase (AID), a Potential RNA Editing Enzyme. Cell 102 (5), 553–63. 10.1016/S0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- Revy P.; Muto T.; Levy Y.; Geissmann F.; Plebani A.; Sanal O.; Catalan N.; Forveille M.; Dufourcq-Labelouse R.; Gennery A.; Tezcan I.; Ersoy F.; Kayserili H.; Ugazio A. G.; Brousse N.; Muramatsu M.; Notarangelo L. D.; Kinoshita K.; Honjo T.; Fischer A.; Durandy A. (2000) Activation-Induced Cytidine Deaminase (AID) Deficiency Causes the Autosomal Recessive Form of the Hyper-IgM Syndrome (HIGM2). Cell 102 (5), 565–575. 10.1016/S0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- Larijani M.; Martin A. (2012) The Biochemistry of Activation-Induced Deaminase and Its Physiological Functions. Semin. Immunol. 24 (4), 255–263. 10.1016/j.smim.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Conticello S. G. (2008) The AID/APOBEC Family of Nucleic Acid Mutators. Genome Biol. 9 (6), 229. 10.1186/gb-2008-9-6-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohail A.; Klapacz J.; Samaranayake M.; Ullah A.; Bhagwat A. S. (2003) Human Activation-Induced Cytidine Deaminase Causes Transcription-Dependent, Strand-Biased C to U Deaminations. Nucleic Acids Res. 31 (12), 2990–2994. 10.1093/nar/gkg464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larijani M.; Frieder D.; Basit W.; Martin A. (2005) The Mutation Spectrum of Purified AID Is Similar to the Mutability Index in Ramos Cells and in Ung(−/−)Msh2(−/−) Mice. Immunogenetics 56 (11), 840–845. 10.1007/s00251-004-0748-0. [DOI] [PubMed] [Google Scholar]
- Durandy A.; Peron S.; Fischer A. (2006) Hyper-IgM Syndromes. Curr. Opin. Rheumatol. 18 (4), 369–376. 10.1097/01.bor.0000231905.12172.b5. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L.; Bhagat G.; Jankovic M.; Compagno M.; Smith P.; Muramatsu M.; Honjo T.; Morse H. C.; Nussenzweig M. C.; Dalla-Favera R. (2008) AID Is Required for Germinal Center-Derived Lymphomagenesis. Nat. Genet. 40 (1), 108–112. 10.1038/ng.2007.35. [DOI] [PubMed] [Google Scholar]
- McCarthy H.; Wierda W. G.; Barron L. L.; Cromwell C. C.; Wang J.; Coombes K. R.; Rangel R.; Elenitoba-Johnson K. S.; Keating M. J.; Abruzzo L. V. (2003) High Expression of Activation-Induced Cytidine Deaminase (AID) and Splice Variants Is a Distinctive Feature of Poor-Prognosis Chronic Lymphocytic Leukemia. Blood 101 (12), 4903–4908. 10.1182/blood-2002-09-2906. [DOI] [PubMed] [Google Scholar]
- Okazaki I. M.; Hiai H.; Kakazu N.; Yamada S.; Muramatsu M.; Kinoshita K.; Honjo T. (2003) Constitutive Expression of AID Leads to Tumorigenesis. J. Exp. Med. 197 (9), 1173–1181. 10.1084/jem.20030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strout M. P.; Schatz D. G. (2009) Imatinib Resistance and Progression of CML to Blast Crisis: Somatic Hypermutation AIDing the Way. Cancer Cell 16 (3), 174–176. 10.1016/j.ccr.2009.08.012. [DOI] [PubMed] [Google Scholar]
- Liu M.; Duke J. L.; Richter D. J.; Vinuesa C. G.; Goodnow C. C.; Kleinstein S. H.; Schatz D. G. (2008) Two Levels of Protection for the B Cell Genome during Somatic Hypermutation. Nature 451 (7180), 841–845. 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- Klasen M.; Spillmann F. J.; Marra G.; Cejka P.; Wabl M. (2005) Somatic Hypermutation and Mismatch Repair in Non-B Cells. Eur. J. Immunol. 35 (7), 2222–2229. 10.1002/eji.200526221. [DOI] [PubMed] [Google Scholar]
- Robbiani D. F.; Bothmer A.; Callen E.; Reina-San-Martin B.; Dorsett Y.; Difilippantonio S.; Bolland D. J.; Chen H. T.; Corcoran A. E.; Nussenzweig A.; Nussenzweig M. C. (2008) AID Is Required for the Chromosomal Breaks in C-Myc That Lead to c-Myc/IgH Translocations. Cell 135 (6), 1028–1038. 10.1016/j.cell.2008.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greeve J.; Philipsen A.; Krause K.; Klapper W.; Heidorn K.; Castle B. E.; Janda J.; Marcu K. B.; Parwaresch R. (2003) Expression of Activation-Induced Cytidine Deaminase in Human B-Cell Non-Hodgkin Lymphomas. Blood 101 (9), 3574–3580. 10.1182/blood-2002-08-2424. [DOI] [PubMed] [Google Scholar]
- Tsukamoto T.; Nakano M.; Sato R.; Adachi H.; Kiyota M.; Kawata E.; Uoshima N.; Yasukawa S.; Chinen Y.; Mizutani S.; Shimura Y.; Kobayashi T.; Horiike S.; Yanagisawa A.; Taniwaki M.; Tashiro K.; Kuroda J. (2017) High-Risk Follicular Lymphomas Harbour More Somatic Mutations Including Those in the AID-Motif. Sci. Rep. 7 (1), 14039. 10.1038/s41598-017-14150-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesi M.; Robbiani D. F.; Sebag M.; Chng W. J.; Affer M.; Tiedemann R.; Valdez R.; Palmer S. E.; Haas S. S.; Stewart A. K.; Fonseca R.; Kremer R.; Cattoretti G.; Bergsagel P. L. (2008) AID-Dependent Activation of a MYC Transgene Induces Multiple Myeloma in a Conditional Mouse Model of Post-Germinal Center Malignancies. Cancer Cell 13 (2), 167–180. 10.1016/j.ccr.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patten P. E.; Chu C. C.; Albesiano E.; Damle R. N.; Yan X. J.; Kim D.; Zhang L.; Magli A. R.; Barrientos J.; Kolitz J. E.; Allen S. L.; Rai K. R.; Roa S.; Mongini P. K.; MacCarthy T.; Scharff M. D.; Chiorazzi N. (2012) IGHV-Unmutated and IGHV-Mutated Chronic Lymphocytic Leukemia Cells Produce Activation-Induced Deaminase Protein with a Full Range of Biologic Functions. Blood 120 (24), 4802–4811. 10.1182/blood-2012-08-449744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai A. G.; Lu H.; Raghavan S. C.; Muschen M.; Hsieh C. L.; Lieber M. R. (2008) Human Chromosomal Translocations at CpG Sites and a Theoretical Basis for Their Lineage and Stage Specificity. Cell 135 (6), 1130–1142. 10.1016/j.cell.2008.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greisman H. A.; Lu Z.; Tsai A. G.; Greiner T. C.; Yi H. S.; Lieber M. R. (2012) IgH Partner Breakpoint Sequences Provide Evidence That AID Initiates t(11;14) and t(8;14) Chromosomal Breaks in Mantle Cell and Burkitt Lymphomas. Blood 120 (14), 2864–2867. 10.1182/blood-2012-02-412791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon M. S.; Kanegai C. M.; Doerr J. R.; Wall R. (2003) Somatic Hypermutation of the B Cell Receptor Genes B29 (Igbeta, CD79b) and Mb1 (Igalpha, CD79a). Proc. Natl. Acad. Sci. U. S. A. 100 (7), 4126–31. 10.1073/pnas.0735266100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L.; Kitaura Y.; Gu H.; Dalla-Favera R. (2006) PKA-Mediated Phosphorylation Regulates the Function of Activation-Induced Deaminase (AID) in B Cells. Proc. Natl. Acad. Sci. U. S. A. 103 (2), 395–400. 10.1073/pnas.0509969103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H. M.; Peters A.; Baron B.; Zhu X.; Storb U. (1998) Mutation of BCL-6 Gene in Normal B Cells by the Process of Somatic Hypermutation of Ig Genes. Science 280 (5370), 1750–1752. 10.1126/science.280.5370.1750. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L.; Guglielmino R.; Houldsworth J.; Mohr J.; Aoufouchi S.; Polakiewicz R.; Chaganti R. S.; Dalla-Favera R. (2004) Expression of the AID Protein in Normal and Neoplastic B Cells. Blood 104 (10), 3318–3325. 10.1182/blood-2004-04-1558. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L.; Bereschenko O.; Niu H.; Klein U.; Basso K.; Guglielmino R.; Cattoretti G.; Dalla-Favera R. (2003) Molecular Pathogenesis of Non-Hodgkin’s Lymphoma: The Role of Bcl-6. Leuk. Lymphoma 44 (Suppl 3), S5–S12. 10.1080/10428190310001621588. [DOI] [PubMed] [Google Scholar]
- Muschen M.; Re D.; Jungnickel B.; Diehl V.; Rajewsky K.; Kuppers R. (2000) Somatic Mutation of the CD95 Gene in Human B Cells as a Side-Effect of the Germinal Center Reaction. J. Exp. Med. 192 (12), 1833–40. 10.1084/jem.192.12.1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramiro A. R.; Jankovic M.; Eisenreich T.; Difilippantonio S.; Chen-Kiang S.; Muramatsu M.; Honjo T.; Nussenzweig A.; Nussenzweig M. C. (2004) AID Is Required for C-Myc/IgH Chromosome Translocations In Vivo. Cell 118 (4), 431–438. 10.1016/j.cell.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Dorsett Y.; Robbiani D. F.; Jankovic M.; Reina-San-Martin B.; Eisenreich T. R.; Nussenzweig M. C. (2007) A Role for AID in Chromosome Translocations between C-Myc and the IgH Variable Region. J. Exp. Med. 204 (9), 2225–2232. 10.1084/jem.20070884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbiani D. F.; Bunting S.; Feldhahn N.; Bothmer A.; Camps J.; Deroubaix S.; McBride K. M.; Klein I. A.; Stone G.; Eisenreich T. R.; Ried T.; Nussenzweig A.; Nussenzweig M. C. (2009) AID Produces DNA Double-Strand Breaks in Non-Ig Genes and Mature B Cell Lymphomas with Reciprocal Chromosome Translocations. Mol. Cell 36 (4), 631–641. 10.1016/j.molcel.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm L.; Duy C.; Iacobucci I.; Kuchen S.; von Levetzow G.; Feldhahn N.; Henke N.; Li Z.; Hoffmann T. K.; Kim Y.; Hofmann W.-K.; Jumaa H.; Groffen J.; Heisterkamp N.; Martinelli G.; Lieber M. R.; Casellas R.; Müschen M. (2009) The B Cell Mutator AID Promotes B Lymphoid Blast Crisis and Drug Resistance in Chronic Myeloid Leukemia. Cancer Cell 16 (3), 232–245. 10.1016/j.ccr.2009.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelmez M. Y.; Coskunpinar E.; Saracoglu B.; Deniz G.; Aktan M. (2017) Investigation of AID, Dicer, and Drosha Expressions in Patients with Chronic Lymphocytic Leukemia. Immunol. Invest. 46 (5), 433–446. 10.1080/08820139.2017.1288241. [DOI] [PubMed] [Google Scholar]
- Kawamura K.; Wada A.; Wang J. Y.; Li Q.; Ishii A.; Tsujimura H.; Takagi T.; Itami M.; Tada Y.; Tatsumi K.; Shimada H.; Hiroshima K.; Tagawa M. (2016) Expression of Activation-Induced Cytidine Deaminase Is Associated with a Poor Prognosis of Diffuse Large B Cell Lymphoma Patients Treated with CHOP-Based Chemotherapy. J. Cancer Res. Clin. Oncol. 142 (1), 27–36. 10.1007/s00432-015-2001-7. [DOI] [PubMed] [Google Scholar]
- Kuppers R.; Dalla-Favera R. (2001) Mechanisms of Chromosomal Translocations in B Cell Lymphomas. Oncogene 20 (40), 5580–94. 10.1038/sj.onc.1204640. [DOI] [PubMed] [Google Scholar]
- Aguilera N. S.; Auerbach A.; Barekman C. L.; Lichy J.; Abbondanzo S. L. (2010) Activation-Induced Cytidine Deaminase Expression in Diffuse Large B-Cell Lymphoma with a Paracortical Growth Pattern: A Lymphoma of Possible Interfollicular Large B-Cell Origin. Arch. Pathol. Lab. Med. 134 (3), 449–456. 10.5858/134.3.449. [DOI] [PubMed] [Google Scholar]
- Palacios F.; Moreno P.; Morande P.; Abreu C.; Correa A.; Porro V.; Landoni A. I.; Gabus R.; Giordano M.; Dighiero G.; Pritsch O.; Oppezzo P. (2010) High Expression of AID and Active Class Switch Recombination Might Account for a More Aggressive Disease in Unmutated CLL Patients: Link with an Activated Microenvironment in CLL Disease. Blood 115 (22), 4488–4496. 10.1182/blood-2009-12-257758. [DOI] [PubMed] [Google Scholar]
- Hancer V. S.; Kose M.; Diz-Kucukkaya R.; Yavuz A. S.; Aktan M. (2011) Activation-Induced Cytidine Deaminase MRNA Levels in Chronic Lymphocytic Leukemia. Leuk. Lymphoma 52 (1), 79–84. 10.3109/10428194.2010.531410. [DOI] [PubMed] [Google Scholar]
- Kano C.; Wang J. Y. (2013) High Levels of AID Cause Strand Bias of Mutations at A versus T in Burkitt’s Lymphoma Cells. Mol. Immunol. 54 (3–4), 397–402. 10.1016/j.molimm.2013.01.005. [DOI] [PubMed] [Google Scholar]
- Huemer M.; Rebhandl S.; Zaborsky N.; Gassner F. J.; Hainzl S.; Weiss L.; Hebenstreit D.; Greil R.; Geisberger R. (2014) AID Induces Intraclonal Diversity and Genomic Damage in CD86(+) Chronic Lymphocytic Leukemia Cells. Eur. J. Immunol. 44 (12), 3747–3757. 10.1002/eji.201344421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arima H.; Fujimoto M.; Nishikori M.; Kitano T.; Kishimoto W.; Hishizawa M.; Kondo T.; Yamashita K.; Hirata M.; Haga H.; Takaori-Kondo A. (2018) Prognostic Impact of Activation-Induced Cytidine Deaminase Expression for Patients with Diffuse Large B-Cell Lymphoma. Leuk. Lymphoma 59, 2085–2095. 10.1080/10428194.2017.1410884. [DOI] [PubMed] [Google Scholar]
- Lossos I. S.; Levy R.; Alizadeh A. A. (2004) AID Is Expressed in Germinal Center B-Cell-like and Activated B-Cell-like Diffuse Large-Cell Lymphomas and Is Not Correlated with Intraclonal Heterogeneity. Leukemia 18 (11), 1775–1779. 10.1038/sj.leu.2403488. [DOI] [PubMed] [Google Scholar]
- Willis T. G.; Dyer M. J. (2000) The Role of Immunoglobulin Translocations in the Pathogenesis of B-Cell Malignancies. Blood 96 (3), 808–822. 10.1182/blood.V96.3.808. [DOI] [PubMed] [Google Scholar]
- Marantidou F.; Dagklis A.; Stalika E.; Korkolopoulou P.; Saetta A.; Anagnostopoulos A.; Laoutaris N.; Stamatopoulos K.; Belessi C.; Scouras Z.; Patsouris E. (2010) Activation-Induced Cytidine Deaminase Splicing Patterns in Chronic Lymphocytic Leukemia. Blood Cells, Mol., Dis. 44 (4), 262–267. 10.1016/j.bcmd.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Compagno M.; Wang Q.; Pighi C.; Cheong T. C.; Meng F. L.; Poggio T.; Yeap L. S.; Karaca E.; Blasco R. B.; Langellotto F.; Ambrogio C.; Voena C.; Wiestner A.; Kasar S. N.; Brown J. R.; Sun J.; Wu C. J.; Gostissa M.; Alt F. W.; Chiarle R. (2017) Phosphatidylinositol 3-Kinase Delta Blockade Increases Genomic Instability in B Cells. Nature 542 (7642), 489–493. 10.1038/nature21406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman D. A.; O’Brien S. (2017) Cancer: A Targeted Treatment with off-Target Risks. Nature 542 (7642), 424–425. 10.1038/nature21504. [DOI] [PubMed] [Google Scholar]
- Conticello S. G.; Langlois M. A.; Yang Z.; Neuberger M. S. (2007) DNA Deamination in Immunity: AID in the Context of Its APOBEC Relatives. Adv. Immunol. 94, 37–73. 10.1016/S0065-2776(06)94002-4. [DOI] [PubMed] [Google Scholar]
- Monajemi M.; Woodworth C. F.; Benkaroun J.; Grant M.; Larijani M. (2012) Emerging Complexities of APOBEC3G Action on Immunity and Viral Fitness during HIV Infection and Treatment. Retrovirology 9, 35. 10.1186/1742-4690-9-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borzooee F.; Asgharpour M.; Quinlan E.; Grant M. D.; Larijani M. (2018) Viral Subversion of APOBEC3s: Lessons for Anti-Tumor Immunity and Tumor Immunotherapy. Int. Rev. Immunol. 37 (3), 151–164. 10.1080/08830185.2017.1403596. [DOI] [PubMed] [Google Scholar]
- Smith H. C. (2011) APOBEC3G: A Double Agent in Defense. Trends Biochem. Sci. 36 (5), 239–244. 10.1016/j.tibs.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albin J. S.; Harris R. S. (2010) Interactions of Host APOBEC3 Restriction Factors with HIV-1 in Vivo: Implications for Therapeutics. Expert Rev. Mol. Med. 12, e4 10.1017/S1462399409001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissing S.; Galloway N. L. K.; Greene W. C. (2010) HIV-1 Vif versus the APOBEC3 Cytidine Deaminases: An Intracellular Duel between Pathogen and Host Restriction Factors. Mol. Aspects Med. 31 (5), 383–397. 10.1016/j.mam.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu Y. L.; Greene W. C. (2006) APOBEC3 Cytidine Deaminases: Distinct Antiviral Actions along the Retroviral Life Cycle. J. Biol. Chem. 281 (13), 8309–8312. 10.1074/jbc.R500021200. [DOI] [PubMed] [Google Scholar]
- Alexandrov L. B.; Nik-Zainal S.; Wedge D. C.; Aparicio S. A. J. R.; Behjati S.; Biankin A. V.; Bignell G. R.; Bolli N.; Borg A.; Børresen-Dale A.-L.; Boyault S.; Burkhardt B.; Butler A. P.; Caldas C.; Davies H. R.; Desmedt C.; Eils R.; Eyfjörd J. E.; Foekens J. A.; Greaves M.; Hosoda F.; Hutter B.; Ilicic T.; Imbeaud S.; Imielinski M.; Imielinsk M.; Jäger N.; Jones D. T. W.; Jones D.; Knappskog S.; Kool M.; Lakhani S. R.; López-Otín C.; Martin S.; Munshi N. C.; Nakamura H.; Northcott P. A.; Pajic M.; Papaemmanuil E.; Paradiso A.; Pearson J. V.; Puente X. S.; Raine K.; Ramakrishna M.; Richardson A. L.; Richter J.; Rosenstiel P.; Schlesner M.; Schumacher T. N.; Span P. N.; Teague J. W.; Totoki Y.; Tutt A. N. J.; Valdés-Mas R.; van Buuren M. M.; van ’t Veer L.; Vincent-Salomon A.; Waddell N.; Yates L. R.; Zucman-Rossi J.; Futreal P. A.; McDermott U.; Lichter P.; Meyerson M.; Grimmond S. M.; Siebert R.; Campo E.; Shibata T.; Pfister S. M.; Campbell P. J.; Stratton M. R. (2013) Signatures of Mutational Processes in Human Cancer. Nature 500 (7463), 415–421. 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts S. A.; Lawrence M. S.; Klimczak L. J.; Grimm S. A.; Fargo D.; Stojanov P.; Kiezun A.; Kryukov G. V.; Carter S. L.; Saksena G.; Harris S.; Shah R. R.; Resnick M. A.; Getz G.; Gordenin D. A. (2013) An APOBEC Cytidine Deaminase Mutagenesis Pattern Is Widespread in Human Cancers. Nat. Genet. 45 (9), 970–976. 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts S. A.; Gordenin D. A. (2014) Hypermutation in Human Cancer Genomes: Footprints and Mechanisms. Nat. Rev. Cancer 14 (12), 786–800. 10.1038/nrc3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S.; Davies H.; Staaf J.; Ramakrishna M.; Glodzik D.; Zou X.; Martincorena I.; Alexandrov L. B.; Martin S.; Wedge D. C.; Van Loo P.; Ju Y. S.; Smid M.; Brinkman A. B.; Morganella S.; Aure M. R.; Lingjærde O. C.; Langerød A.; Ringnér M.; Ahn S.-M.; Boyault S.; Brock J. E.; Broeks A.; Butler A.; Desmedt C.; Dirix L.; Dronov S.; Fatima A.; Foekens J. A.; Gerstung M.; Hooijer G. K. J.; Jang S. J.; Jones D. R.; Kim H.-Y.; King T. A.; Krishnamurthy S.; Lee H. J.; Lee J.-Y.; Li Y.; McLaren S.; Menzies A.; Mustonen V.; O’Meara S.; Pauporté I.; Pivot X.; Purdie C. A.; Raine K.; Ramakrishnan K.; Rodríguez-González F. G.; Romieu G.; Sieuwerts A. M.; Simpson P. T.; Shepherd R.; Stebbings L.; Stefansson O. A.; Teague J.; Tommasi S.; Treilleux I.; Van den Eynden G. G.; Vermeulen P.; Vincent-Salomon A.; Yates L.; Caldas C.; van’t Veer L.; Tutt A.; Knappskog S.; Tan B. K. T.; Jonkers J.; Borg Å.; Ueno N. T.; Sotiriou C.; Viari A.; Futreal P. A.; Campbell P. J.; Span P. N.; Van Laere S.; Lakhani S. R.; Eyfjord J. E.; Thompson A. M.; Birney E.; Stunnenberg H. G.; van de Vijver M. J.; Martens J. W. M.; Børresen-Dale A.-L.; Richardson A. L.; Kong G.; Thomas G.; Stratton M. R. (2016) Landscape of Somatic Mutations in 560 Breast Cancer Whole-Genome Sequences. Nature 534 (7605), 47–54. 10.1038/nature17676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K.; Roberts S. A.; Klimczak L. J.; Sterling J. F.; Saini N.; Malc E. P.; Kim J.; Kwiatkowski D. J.; Fargo D. C.; Mieczkowski P. A.; Getz G.; Gordenin D. A. (2015) An APOBEC3A Hypermutation Signature Is Distinguishable from the Signature of Background Mutagenesis by APOBEC3B in Human Cancers. Nat. Genet. 47 (9), 1067–1072. 10.1038/ng.3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knisbacher B. A.; Gerber D.; Levanon E. Y. (2016) DNA Editing by APOBECs: A Genomic Preserver and Transformer. Trends Genet. 32 (1), 16–28. 10.1016/j.tig.2015.10.005. [DOI] [PubMed] [Google Scholar]
- Venkatesan S.; Rosenthal R.; Kanu N.; McGranahan N.; Bartek J.; Quezada S. A.; Hare J.; Harris R. S.; Swanton C. (2018) Perspective: APOBEC Mutagenesis in Drug Resistance and Immune Escape in HIV and Cancer Evolution. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 29 (3), 563–572. 10.1093/annonc/mdy003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petljak M.; Maciejowski J. (2020) Molecular Origins of APOBEC-Associated Mutations in Cancer. DNA Repair 94, 102905. 10.1016/j.dnarep.2020.102905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper N.; Gao S.; Maity T. K.; Banday A. R.; Zhang X.; Venugopalan A.; Cultraro C. M.; Patidar R.; Sindiri S.; Brown A.-L.; Goncearenco A.; Panchenko A. R.; Biswas R.; Thomas A.; Rajan A.; Carter C. A.; Kleiner D. E.; Hewitt S. M.; Khan J.; Prokunina-Olsson L.; Guha U. (2019) APOBEC Mutagenesis and Copy-Number Alterations Are Drivers of Proteogenomic Tumor Evolution and Heterogeneity in Metastatic Thoracic Tumors. Cell Rep. 26 (10), 2651–2666. 10.1016/j.celrep.2019.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson S.; Chakravarthy A.; Su X.; Boshoff C.; Fenton T. R. (2014) APOBEC-Mediated Cytosine Deamination Links PIK3CA Helical Domain Mutations to Human Papillomavirus-Driven Tumor Development. Cell Rep. 7 (6), 1833–1841. 10.1016/j.celrep.2014.05.012. [DOI] [PubMed] [Google Scholar]
- Du Y.; Tao X.; Wu J.; Yu H.; Yu Y.; Zhao H. (2018) APOBEC3B Up-Regulation Independently Predicts Ovarian Cancer Prognosis: A Cohort Study. Cancer Cell Int. 18, 78. 10.1186/s12935-018-0572-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker B. A.; Wardell C. P.; Murison A.; Boyle E. M.; Begum D. B.; Dahir N. M.; Proszek P. Z.; Melchor L.; Pawlyn C.; Kaiser M. F.; Johnson D. C.; Qiang Y.-W.; Jones J. R.; Cairns D. A.; Gregory W. M.; Owen R. G.; Cook G.; Drayson M. T.; Jackson G. H.; Davies F. E.; Morgan G. J. (2015) APOBEC Family Mutational Signatures Are Associated with Poor Prognosis Translocations in Multiple Myeloma. Nat. Commun. 6, 6997. 10.1038/ncomms7997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law E. K.; Sieuwerts A. M.; LaPara K.; Leonard B.; Starrett G. J.; Molan A. M.; Temiz N. A.; Vogel R. I.; Meijer-Van Gelder M. E.; Sweep F. C. G. J.; Span P. N.; Foekens J. A.; Martens J. W. M.; Yee D.; Harris R. S. (2016) The DNA Cytosine Deaminase APOBEC3B Promotes Tamoxifen Resistance in ER-Positive Breast Cancer. Sci. Adv. 2 (10), e1601737 10.1126/sciadv.1601737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petljak M.; Alexandrov L. B.; Brammeld J. S.; Price S.; Wedge D. C.; Grossmann S.; Dawson K. J.; Ju Y. S.; Iorio F.; Tubio J. M. C.; Koh C. C.; Georgakopoulos-Soares I.; Rodríguez-Martín B.; Otlu B.; O’Meara S.; Butler A. P.; Menzies A.; Bhosle S. G.; Raine K.; Jones D. R.; Teague J. W.; Beal K.; Latimer C.; O’Neill L.; Zamora J.; Anderson E.; Patel N.; Maddison M.; Ng B. L.; Graham J.; Garnett M. J.; McDermott U.; Nik-Zainal S.; Campbell P. J.; Stratton M. R. (2019) Characterizing Mutational Signatures in Human Cancer Cell Lines Reveals Episodic APOBEC Mutagenesis. Cell 176 (6), 1282–1294. 10.1016/j.cell.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov L. B.; Kim J.; Haradhvala N. J.; Huang M. N.; Tian Ng A. W.; Wu Y.; Boot A.; Covington K. R.; Gordenin D. A.; Bergstrom E. N.; Islam S. M. A.; Lopez-Bigas N.; Klimczak L. J.; McPherson J. R.; Morganella S.; Sabarinathan R.; Wheeler D. A.; Mustonen V.; Getz G.; Rozen S. G.; Stratton M. R. (2020) PCAWG Consortium. The Repertoire of Mutational Signatures in Human Cancer. Nature 578 (7793), 94–101. 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law E. K.; Levin-Klein R.; Jarvis M. C.; Kim H.; Argyris P. P.; Carpenter M. A.; Starrett G. J.; Temiz N. A.; Larson L. K.; Durfee C.; Burns M. B.; Vogel R. I.; Stavrou S.; Aguilera A. N.; Wagner S.; Largaespada D. A.; Starr T. K.; Ross S. R.; Harris R. S. (2020) APOBEC3A Catalyzes Mutation and Drives Carcinogenesis in Vivo. J. Exp. Med. 217 (12), e20200261. 10.1084/jem.20200261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber T. A.; Chang M. S.; Sposto R.; Muschen M. (2010) Activation-Induced Cytidine Deaminase Accelerates Clonal Evolution in BCR-ABL1-Driven B-Cell Lineage Acute Lymphoblastic Leukemia. Cancer Res. 70 (19), 7411–7420. 10.1158/0008-5472.CAN-10-1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussenzweig A.; Nussenzweig M. C. (2010) Origin of Chromosomal Translocations in Lymphoid Cancer. Cell 141 (1), 27–38. 10.1016/j.cell.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz D. P.; Lee E. L.; Takayama S.; Coppe J. P.; Heo S. J.; Boffelli D.; Di Noia J. M.; Martin D. I. (2013) Activation-Induced Cytidine Deaminase (AID) Is Necessary for the Epithelial-Mesenchymal Transition in Mammary Epithelial Cells. Proc. Natl. Acad. Sci. U. S. A. 110 (32), E2977–86. 10.1073/pnas.1301021110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montamat-Sicotte D.; Litzler L. C.; Abreu C.; Safavi S.; Zahn A.; Orthwein A.; Muschen M.; Oppezzo P.; Munoz D. P.; Di Noia J. M. (2015) HSP90 Inhibitors Decrease AID Levels and Activity in Mice and in Human Cells. Eur. J. Immunol. 45 (8), 2365–2376. 10.1002/eji.201545462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebhandl S.; Geisberger R. (2015) AIDing Cancer Treatment: Reducing AID Activity via HSP90 Inhibition. Eur. J. Immunol. 45 (8), 2208–2211. 10.1002/eji.201545832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methot S. P.; Di Noia J. M. (2015) Pharmacological Manipulation of AID. Oncotarget 6 (29), 26550–26551. 10.18632/oncotarget.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai C. T.; Yang P. M.; Chern T. R.; Chuang S. H.; Lin J. H.; Klemm L.; Muschen M.; Chen C. C. (2014) AID Downregulation Is a Novel Function of the DNMT Inhibitor 5-Aza-Deoxycytidine. Oncotarget 5 (1), 211–223. 10.18632/oncotarget.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald G.; Medina C. O.; Pilichowska M.; Kearney J. F.; Shinkura R.; Selsing E.; Wortis H. H.; Honjo T.; Imanishi-Kari T. (2017) Accelerated Systemic Autoimmunity in the Absence of Somatic Hypermutation in 564Igi: A Mouse Model of Systemic Lupus with Knocked-In Heavy and Light Chain Genes. Front. Immunol. 8, 1094. 10.3389/fimmu.2017.01094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu H. C.; Yang P.; Wu Q.; Wang J. H.; Job G.; Guentert T.; Li J.; Stockard C. R.; Le T. V.; Chaplin D. D.; Grizzle W. E.; Mountz J. D. (2011) Inhibition of the Catalytic Function of Activation-Induced Cytidine Deaminase Promotes Apoptosis of Germinal Center B Cells in BXD2Mice. Arthritis Rheum. 63 (7), 2038–2048. 10.1002/art.30257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Shandilya S. M.; Carpenter M. A.; Rathore A.; Brown W. L.; Perkins A. L.; Harki D. A.; Solberg J.; Hook D. J.; Pandey K. K.; Parniak M. A.; Johnson J. R.; Krogan N. J.; Somasundaran M.; Ali A.; Schiffer C. A.; Harris R. S. (2012) First-in-Class Small Molecule Inhibitors of the Single-Strand DNA Cytosine Deaminase APOBEC3G. ACS Chem. Biol. 7 (3), 506–517. 10.1021/cb200440y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson M. E.; Harris R. S.; Harki D. A. (2018) APOBEC Enzymes as Targets for Virus and Cancer Therapy. Cell Chem. Biol. 25 (1), 36–49. 10.1016/j.chembiol.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King J. J.; Manuel C. A.; Barrett C. V.; Raber S.; Lucas H.; Sutter P.; Larijani M. (2015) Catalytic Pocket Inaccessibility of Activation-Induced Cytidine Deaminase Is a Safeguard against Excessive Mutagenic Activity. Structure 23 (4), 615–627. 10.1016/j.str.2015.01.016. [DOI] [PubMed] [Google Scholar]
- Pham P.; Afif S. A.; Shimoda M.; Maeda K.; Sakaguchi N.; Pedersen L. C.; Goodman M. F. (2016) Structural Analysis of the Activation-Induced Deoxycytidine Deaminase Required in Immunoglobulin Diversification. DNA Repair 43, 48–56. 10.1016/j.dnarep.2016.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Q.; Wang L.; Meng F. L.; Hwang J. K.; Alt F. W.; Wu H. (2017) AID Recognizes Structured DNA for Class Switch Recombination. Mol. Cell 67 (3), 361–373. 10.1016/j.molcel.2017.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King J. J.; Larijani M. (2017) A Novel Regulator of Activation-Induced Cytidine Deaminase/APOBECs in Immunity and Cancer: Schrodinger’s CATalytic Pocket. Front. Immunol. 8, 351. 10.3389/fimmu.2017.00351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byeon I. J.; Ahn J.; Mitra M.; Byeon C. H.; Hercik K.; Hritz J.; Charlton L. M.; Levin J. G.; Gronenborn A. M. (2013) NMR Structure of Human Restriction Factor APOBEC3A Reveals Substrate Binding and Enzyme Specificity. Nat. Commun. 4, 1890. 10.1038/ncomms2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi K.; Carpenter M. A.; Kurahashi K.; Harris R. S.; Aihara H. (2015) Crystal Structure of the DNA Deaminase APOBEC3B Catalytic Domain. J. Biol. Chem. 290 (47), 28120–28130. 10.1074/jbc.M115.679951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byeon I. J.; Byeon C. H.; Wu T.; Mitra M.; Singer D.; Levin J. G.; Gronenborn A. M. (2016) Nuclear Magnetic Resonance Structure of the APOBEC3B Catalytic Domain: Structural Basis for Substrate Binding and DNA Deaminase Activity. Biochemistry 55 (21), 2944–2959. 10.1021/acs.biochem.6b00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi K.; Demir O.; Carpenter M. A.; Wagner J.; Kurahashi K.; Harris R. S.; Amaro R. E.; Aihara H. (2017) Conformational Switch Regulates the DNA Cytosine Deaminase Activity of Human APOBEC3B. Sci. Rep. 7 (1), 17415. 10.1038/s41598-017-17694-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson M. E.; Li M.; Harris R. S.; Harki D. A. (2013) Small-Molecule APOBEC3G DNA Cytosine Deaminase Inhibitors Based on a 4-Amino-1,2,4-Triazole-3-Thiol Scaffold. ChemMedChem 8 (1), 112–117. 10.1002/cmdc.201200411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvach M. V.; Barzak F. M.; Harjes S.; Schares H. A. M.; Kurup H. M.; Jones K. F.; Sutton L.; Donahue J.; D’Aquila R. T.; Jameson G. B.; Harki D. A.; Krause K. L.; Harjes E.; Filichev V. V. (2020) Differential Inhibition of APOBEC3 DNA-Mutator Isozymes by Fluoro- and Non-Fluoro-Substituted 2’-Deoxyzebularine Embedded in Single-Stranded DNA. ChemBioChem 21 (7), 1028–1035. 10.1002/cbic.201900505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli R. M.; Abrams S. R.; Gajula K. S.; Maul R. W.; Gearhart P. J.; Stivers J. T. (2009) A Portable Hot Spot Recognition Loop Transfers Sequence Preferences from APOBEC Family Members to Activation-Induced Cytidine Deaminase. J. Biol. Chem. 284 (34), 22898–22904. 10.1074/jbc.M109.025536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter M. A.; Rajagurubandara E.; Wijesinghe P.; Bhagwat A. S. (2010) Determinants of Sequence-Specificity within Human AID and APOBEC3G. DNA Repair 9 (5), 579–587. 10.1016/j.dnarep.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdouni H.; King J. J.; Suliman M.; Quinlan M.; Fifield H.; Larijani M. (2013) Zebrafish AID Is Capable of Deaminating Methylated Deoxycytidines. Nucleic Acids Res. 41 (10), 5457–5468. 10.1093/nar/gkt212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx A.; Galilee M.; Alian A. (2016) Zinc Enhancement of Cytidine Deaminase Activity Highlights a Potential Allosteric Role of Loop-3 in Regulating APOBEC3 Enzymes. Sci. Rep. 5, 18191. 10.1038/srep18191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdouni H. S.; King J. J.; Ghorbani A.; Fifield H.; Berghuis L.; Larijani M. (2018) DNA/RNA Hybrid Substrates Modulate the Catalytic Activity of Purified AID. Mol. Immunol. 93, 94–106. 10.1016/j.molimm.2017.11.012. [DOI] [PubMed] [Google Scholar]
- King J. J.; Larijani M. (2021) Structural Plasticity of Substrate Selection by Activation-Induced Cytidine Deaminase as a Regulator of Its Genome-Wide Mutagenic Activity. FEBS Lett. 595 (1), 3–13. 10.1002/1873-3468.13962. [DOI] [PubMed] [Google Scholar]
- Shi K.; Demir O.; Carpenter M. A.; Wagner J.; Kurahashi K.; Harris R. S.; Amaro R. E.; Aihara H. (2017) Conformational Switch Regulates the DNA Cytosine Deaminase Activity of Human APOBEC3B. Sci. Rep. 7 (1), 17415. 10.1038/s41598-017-17694-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H.; Brender J. R.; Zhang J.; Zhang Y. (2015) Protein Structure Prediction Provides Comparable Performance to Crystallographic Structures in Docking-Based Virtual Screening. Methods 71, 77–84. 10.1016/j.ymeth.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J. J.; Sterling T.; Mysinger M. M.; Bolstad E. S.; Coleman R. G. (2012) ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 52 (7), 1757–1768. 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. (2001) Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Delivery Rev. 46 (1–3), 3–26. 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. (2004) Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discovery Today: Technol. 1 (4), 337–341. 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Irwin J. J.; Shoichet B. K.; Mysinger M. M.; Huang N.; Colizzi F.; Wassam P.; Cao Y. (2009) Automated Docking Screens: A Feasibility Study. J. Med. Chem. 52 (18), 5712–5720. 10.1021/jm9006966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. (2010) AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 31 (2), 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larijani M.; Frieder D.; Sonbuchner T. M.; Bransteitter R.; Goodman M. F.; Bouhassira E. E.; Scharff M. D.; Martin A. (2005) Methylation Protects Cytidines from AID-Mediated Deamination. Mol. Immunol. 42 (5), 599–604. 10.1016/j.molimm.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Quinlan E. M.; King J. J.; Amemiya C. T.; Hsu E.; Larijani M. (2017) Biochemical Regulatory Features of Activation-Induced Cytidine Deaminase Remain Conserved from Lampreys to Humans. Mol. Cell. Biol. 37 (20), e00077-17. 10.1128/MCB.00077-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branton S. A.; Ghorbani A.; Bolt B. N.; Fifield H.; Berghuis L. M.; Larijani M. (2020) Activation-Induced Cytidine Deaminase Can Target Multiple Topologies of Double-Stranded DNA in a Transcription-Independent Manner. FASEB J. 34 (7), 9245–9268. 10.1096/fj.201903036RR. [DOI] [PubMed] [Google Scholar]
- Fang Y.; Xiao X.; Li S. X.; Wolfe A.; Chen X. S. (2018) Molecular Interactions of a DNA Modifying Enzyme APOBEC3F Catalytic Domain with a Single-Stranded DNA. J. Mol. Biol. 430 (1), 87–101. 10.1016/j.jmb.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borzooee F.; Larijani M. (2019) Pichia Pastoris as a Host for Production and Isolation of Mutagenic AID/APOBEC Enzymes Involved in Cancer and Immunity. New Biotechnol. 51, 67–79. 10.1016/j.nbt.2019.02.006. [DOI] [PubMed] [Google Scholar]
- Di Giorgio S.; Martignano F.; Torcia M. G.; Mattiuz G.; Conticello S. G. (2020) Evidence for Host-Dependent RNA Editing in the Transcriptome of SARS-CoV-2. Sci. Adv. 6 (25), eabb5813 10.1126/sciadv.abb5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosuge M.; Furusawa-Nishii E.; Ito K.; Saito Y.; Ogasawara K. (2020) Point Mutation Bias in SARS-CoV-2 Variants Results in Increased Ability to Stimulate Inflammatory Responses. Sci. Rep. 10 (1), 17766. 10.1038/s41598-020-74843-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman N.; Jacob-Hirsch J.; Drori Y.; Eran E.; Kol N.; Nayshool O.; Mendelson E.; Rechavi G.; Mandelboim M. (2021) Transcriptomic Profiling and Genomic Mutational Analysis of Human Coronavirus (HCoV)-229E -Infected Human Cells. PLoS One 16 (2), e0247128 10.1371/journal.pone.0247128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siqueira J. D.; Goes L. R.; Alves B. M.; de Carvalho P. S.; Cicala C.; Arthos J.; Viola J. P. B.; de Melo A. C.; Soares M. A. (2021) SARS-CoV-2 Genomic Analyses in Cancer Patients Reveal Elevated Intrahost Genetic Diversity. Virus Evol. 7 (1), veab013 10.1093/ve/veab013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourier T.; Sadykov M.; Carr M. J.; Gonzalez G.; Hall W. W.; Pain A. (2021) Host-Directed Editing of the SARS-CoV-2 Genome. Biochem. Biophys. Res. Commun. 538, 35–39. 10.1016/j.bbrc.2020.10.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant M.; Larijani M. (2017) Evasion of Adaptive Immunity by HIV through the Action of Host APOBEC3G/F Enzymes. AIDS Res. Ther. 14 (1), 44. 10.1186/s12981-017-0173-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squires K. D.; Monajemi M.; Woodworth C. F.; Grant M. D.; Larijani M. (2015) Impact of APOBEC Mutations on CD8+ T Cell Recognition of HIV Epitopes Varies Depending on the Restricting HLA. JAIDS, J. Acquired Immune Defic. Syndr. 70 (2), 172–178. 10.1097/QAI.0000000000000689. [DOI] [PubMed] [Google Scholar]
- Wood N.; Bhattacharya T.; Keele B. F.; Giorgi E.; Liu M.; Gaschen B.; Daniels M.; Ferrari G.; Haynes B. F.; McMichael A.; Shaw G. M.; Hahn B. H.; Korber B.; Seoighe C. (2009) HIV Evolution in Early Infection: Selection Pressures, Patterns of Insertion and Deletion, and the Impact of APOBEC. PLoS Pathog. 5 (5), e1000414 10.1371/journal.ppat.1000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden L. G.; Prochnow C.; Chang Y. P.; Bransteitter R.; Chelico L.; Sen U.; Stevens R. C.; Goodman M. F.; Chen X. S. (2008) Crystal Structure of the Anti-Viral APOBEC3G Catalytic Domain and Functional Implications. Nature 456 (7218), 121–124. 10.1038/nature07357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa A.; Nagata T.; Matsugami A.; Habu Y.; Sugiyama R.; Hayashi F.; Kobayashi N.; Yokoyama S.; Takaku H.; Katahira M. (2009) Structure, Interaction and Real-Time Monitoring of the Enzymatic Reaction of Wild-Type APOBEC3G. EMBO J. 28 (4), 440–451. 10.1038/emboj.2008.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochnow C.; Bransteitter R.; Klein M. G.; Goodman M. F.; Chen X. S. (2007) The APOBEC-2 Crystal Structure and Functional Implications for the Deaminase AID. Nature 445 (7126), 447–451. 10.1038/nature05492. [DOI] [PubMed] [Google Scholar]
- Krzysiak T. C.; Jung J.; Thompson J.; Baker D.; Gronenborn A. M. (2012) APOBEC2 Is a Monomer in Solution: Implications for APOBEC3G Models. Biochemistry 51 (9), 2008–2017. 10.1021/bi300021s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura S.; Ode H.; Nakashima M.; Imahashi M.; Naganawa Y.; Kurosawa T.; Yokomaku Y.; Yamane T.; Watanabe N.; Suzuki A.; Sugiura W.; Iwatani Y. (2012) The APOBEC3C Crystal Structure and the Interface for HIV-1 Vif Binding. Nat. Struct. Mol. Biol. 19 (10), 1005–1010. 10.1038/nsmb.2378. [DOI] [PubMed] [Google Scholar]
- Bohn M. F.; Shandilya S. M.; Albin J. S.; Kouno T.; Anderson B. D.; McDougle R. M.; Carpenter M. A.; Rathore A.; Evans L.; Davis A. N.; Zhang J.; Lu Y.; Somasundaran M.; Matsuo H.; Harris R. S.; Schiffer C. A. (2013) Crystal Structure of the DNA Cytosine Deaminase APOBEC3F: The Catalytically Active and HIV-1 Vif-Binding Domain. Structure 21 (6), 1042–1050. 10.1016/j.str.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. (2008) I-TASSER Server for Protein 3D Structure Prediction. BMC Bioinf. 9, 40. 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A.; Yang J.; Zhang Y. (2012) COFACTOR: An Accurate Comparative Algorithm for Structure-Based Protein Function Annotation. Nucleic Acids Res. 40 (W1), W471–7. 10.1093/nar/gks372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallakyan S.; Olson A. J. (2015) Small-Molecule Library Screening by Docking with PyRx. Methods Mol. Biol. 1263, 243–250. 10.1007/978-1-4939-2269-7_19. [DOI] [PubMed] [Google Scholar]
- Larijani M.; Petrov A. P.; Kolenchenko O.; Berru M.; Krylov S. N.; Martin A. (2007) AID Associates with Single-Stranded DNA with High Affinity and a Long Complex Half-Life in a Sequence-Independent Manner. Mol. Cell. Biol. 27 (1), 20–30. 10.1128/MCB.00824-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancyger A. M.; King J. J.; Quinlan M. J.; Fifield H.; Tucker S.; Saunders H. L.; Berru M.; Magor B. G.; Martin A.; Larijani M. (2012) Differences in the Enzymatic Efficiency of Human and Bony Fish AID Are Mediated by a Single Residue in the C Terminus Modulating Single-Stranded DNA Binding. FASEB J. 26, 1517. 10.1096/fj.11-198135. [DOI] [PubMed] [Google Scholar]
- Diamond C. P.; Im J.; Button E. A.; Huebert D. N. G.; King J. J.; Borzooee F.; Abdouni H. S.; Bacque L.; McCarthy E.; Fifield H.; Berghuis L. M.; Larijani M. (2019) AID, APOBEC3A and APOBEC3B Efficiently Deaminate Deoxycytidines Neighboring DNA Damage Induced by Oxidation or Alkylation. Biochim. Biophys. Acta, Gen. Subj. 1863 (11), 129415. 10.1016/j.bbagen.2019.129415. [DOI] [PubMed] [Google Scholar]
- Larijani M.; Martin A. (2007) Single-Stranded DNA Structure and Positional Context of the Target Cytidine Determine the Enzymatic Efficiency of AID. Mol. Cell. Biol. 27 (23), 8038–8048. 10.1128/MCB.01046-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi K.; Carpenter M. A.; Banerjee S.; Shaban N. M.; Kurahashi K.; Salamango D. J.; McCann J. L.; Starrett G. J.; Duffy J. V.; Demir O.; Amaro R. E.; Harki D. A.; Harris R. S.; Aihara H. (2017) Structural Basis for Targeted DNA Cytosine Deamination and Mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 24 (2), 131–139. 10.1038/nsmb.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouno T.; Silvas T. V.; Hilbert B. J.; Shandilya S. M. D.; Bohn M. F.; Kelch B. A.; Royer W. E.; Somasundaran M.; Kurt Yilmaz N.; Matsuo H.; Schiffer C. A. (2017) Crystal Structure of APOBEC3A Bound to Single-Stranded DNA Reveals Structural Basis for Cytidine Deamination and Specificity. Nat. Commun. 8, 15024. 10.1038/ncomms15024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham P.; Landolph A.; Mendez C.; Li N.; Goodman M. F. (2013) A Biochemical Analysis Linking APOBEC3A to Disparate HIV-1 Restriction and Skin Cancer. J. Biol. Chem. 288 (41), 29294–29304. 10.1074/jbc.M113.504175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harjes S.; Solomon W. C.; Li M.; Chen K. M.; Harjes E.; Harris R. S.; Matsuo H. (2013) Impact of H216 on the DNA Binding and Catalytic Activities of the HIV Restriction Factor APOBEC3G. J. Virol. 87 (12), 7008–7014. 10.1128/JVI.03173-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.