


Abstract
DNA replication fidelity in Streptomyces bacteria, prolific producers of many medically important secondary metabolites, is understudied, while in Escherichia coli it is controlled by DnaQ, the ϵ subunit of DNA polymerase III (DNA PolIII). Manipulation of dnaQ paralogues in Streptomyces lividans TK24, did not lead to increased spontaneous mutagenesis in this bacterium suggesting that S. lividans DNA PolIII uses an alternative exonuclease activity for proofreading. In Mycobacterium tuberculosis, such activity is attributed to the DnaE protein representing α subunit of DNA PolIII. Eight DnaE mutants designed based on the literature data were overexpressed in S. lividans, and recombinant strains overexpressing two of these mutants displayed markedly increased frequency of spontaneous mutagenesis (up to 1000-fold higher compared to the control). One of these ‘mutators’ was combined in S. lividans with a biosensor specific for antibiotic coelimycin, which biosynthetic gene cluster is present but not expressed in this strain. Colonies giving a positive biosensor signal appeared at a frequency of ca 10–5, and all of them were found to produce coelimycin congeners. This result confirmed that our approach can be applied for chemical- and radiation-free mutagenesis in Streptomyces leading to activation of orphan biosynthetic gene clusters and discovery of novel bioactive secondary metabolites.
INTRODUCTION
Maintaining DNA replication fidelity is of outmost importance for genome integrity of every living organism. At the same time, eventual DNA replication errors can lead to low-frequency random mutagenesis, which in bacteria plays an important evolutionary role (1). In Escherichia coli, DnaQ, representing the ϵ subunit of the DNA polymerase III complex, has been shown to have proofreading function that corrects replication errors by means of its 3′ to 5′ exonuclease activity (2). Certain mutations in DnaQ cause a ‘mutator’ phenotype in E. coli and other bacteria, leading to a much higher frequency of spontaneous mutagenesis that can yield antibiotic resistance phenotypes or activate expression of certain enzymes, for example nitrous oxide reductase in Bradyrhizobium japonicum (3,4). Apparently, specific mutations in DnaQ that affect exonuclease activity do not influence its ability to form complex with other subunits of DNA polymerase III. If overexpressed, mutant DnaQ can outcompete the wild-type protein in binding to its partner subunits, α and τ, thus causing a dominant ‘mutator’ phenotype (3). Some mutations in DnaQ that do not affect its exonuclease activity per se can induce SOS response, suggesting that this protein may have functions other that proofreading, e.g., in complex disassembly and/or resolution of stalled replication forks (5).
In some bacteria, e.g., Mycobacterium tuberculosis, DnaQ does not perform a proofreading function, which is apparently taken over by a histidinol phosphatase (PHP) domain of DnaE representing the α subunit of DNA polymerase III that has 3′ to 5′ exonuclease activity (6). Certain mutations in E. coli DnaE have also been reported to yield a ‘mutator’ phenotype, while these alterations were not localized to the PHP domain (7). Since DnaE in E. coli is known to interact with DnaQ, it has been suggested that such mutations in the α subunit may affect this interaction, thus preventing proper editing of errors during the DNA replication.
Streptomyces are filamentous Gram-positive bacteria of significant industrial importance. These bacteria produce over 30% of all antibiotics currently used to treat bacterial and fungal infections, but the interest in Streptomyces as sources for new drugs has stagnated due to the frequent re-discovery of already known compounds. Recent advances in Streptomyces genomics and ‘genome mining’ uncovered as yet unrealized potential of these bacteria to biosynthesize potentially novel and valuable compounds. The genome mining approach is focused on targeted activation of silent or poorly expressed biosynthetic gene clusters (BGCs) for secondary metabolites using various techniques (8). However, due to the complexity of regulatory mechanisms that control expression of BGCs, and the crosstalk between different biosynthetic pathways, genome mining strategies often fail to deliver expected results. On the other hand, untargeted mutagenesis using physical and chemical mutagens has been used for many decades to improve productivity of industrial antibiotic-producing strains. Such mutagenesis has an advantage in generating a combination of many mutations that may lead to a desired phenotype if such phenotype can be selected for. Purposeful interference with DNA replication fidelity in Streptomyces without using exogenous mutagens would provide an environmentally friendly alternative for strain development, and possibly also activation of silent BGCs.
Up to now, relatively little is known about the DNA replication and repair machinery in Streptomyces. Flett et al. have reported functional characterization of two mutants of Streptomyces coelicolor A3(2), a model streptomycete, which growth was affected at elevated temperature. They were able to localize both ts mutations to DnaE protein, which sequence was more similar to that of E. coli than to those of the Gram-positive Bacillus (9). Another study from the Chen group on the same organism revealed that this bacterium harbors two DNA polymerase III gene sets encoding DnaE and DnaQ proteins, and demonstrated that DnaE1 is essential (10). So far, a ‘mutator’ phenotype has not been reported for Streptomyces bacteria, while it may be beneficial for strain selection programs due to bypassing the need for mutagenesis using dangerous chemicals (e.g. nitrosoguanidine) or radiation. In this study we aimed to investigate possible involvement of ϵ (DnaQ) and α (DnaE) subunits of DNA polymerase III in maintaining DNA replication fidelity in Streptomyces lividans TK24, a model streptomycete closely related to S. coelicolor A3(2). Using overexpression of mutated genes and gene deletions we demonstrate importance of DnaQ1and DnaQ2 for SOS-response and generate a ‘mutator’ phenotype by combining different mutations in DnaE1. Coupling the ‘mutator’ to the biosensor for antibiotic coelimycin not normally produced by S. lividans allowed easy selection of coelimycin-producing mutants. This principle can be used to activate silent BGCs in Streptomyces and revitalize drug discovery pipeline based on these bacteria.
MATERIALS AND METHODS
Plasmids, bacterial strains, and growth conditions
Plasmids, bacterial strains and oligonucleotide primers used or constructed during this study are described/provided in Supplementary Tables S1–S4 (Supplementary Data). Streptomyces lividans TK24 was used as the wild-type strain. E. coli strains were grown in Luria-Bertani (LB) broth or on LB agar. XL1-blue was used for general cloning. ET12567 (pUZ8002) was used for intergeneric transfer of plasmids to Streptomyces with the standard procedure of conjugation (11). Integration of transformed plasmid to specific attachment sites of Streptomyces genome were confirmed by PCR. The gene bpsA encoding a peptide synthetase for the biosynthesis of a blue pigment indigoidine in Streptomyces was used as indicator for double crossover (the color of colonies turned white from blue) after conjugation (12). Streptomyces spore preparation was obtained by growing on CP6 or soy flour mannitol medium (SFM) plates at 30°C for 4−7 days. Antibiotics were supplemented to growth medium at the following concentrations when applicable: ampicillin (Amp) 100 μg/ml; apramycin (Am) 50 or 100 μg/ml; chloramphenicol (Cm) 20 μg/ml; hygromycin (Hyg) 100 μg/ml; kanamycin (Kan) 25 μg/ml; mitomycin C (Mtc) 5 μg/ml, nalidixic acid (Nal) 30 μg/ml, rifampicin (Rif) 100 μg/ml.
DNA manipulation
DNA digestion with restriction enzymes, general DNA cloning and plasmid transformation into E. coli were performed as described in Sambrook et al. (13). Streptomyces genomic DNA was isolated using Wizard® Genomic DNA Purification Kit (Promega, USA). PCR fragments were amplified using MasterAmp™ Extra-Long DNA Polymerase Mix and buffers (Epicentre, USA), GC-rich PCR System (Roche) and Q5 High Fidelity 5X Master Mix (New England Biolab). Primers for PCR or sequencing were designed using Clone Manager Professional Version 9 (Sci-Ed Software) and are described in Supplementary Table S3 (Supplemental Data). To construct new vectors, PCR and vector fragments were assembled either by T4 DNA ligase (New England BioLabs) or Gibson ligation (14). Recombinant constructs were verified with DNA sequencing by GATC Biotech.
Measurement of spontaneous mutation frequency
Spontaneous mutation frequency was measured by counting rifampicin resistant colonies. Spore suspension collected from 6 days CP6 plates were spread on SFM plates supplemented with rifampicin 100 μg/ml. For each strain 100 μl spore suspension was spread on each of three plates as one biological replicate, and 2–12 biological replicates were carried out for each strain. Colony forming unit (CFU/ml) was calculated by spreading serial diluted spore suspension on CP6 plate. The rifampicin resistant colonies were counted after 3 days, and spontaneous mutation frequency was measured as number of rifampicin resistant colonies on three plates divided by total number of plated Streptomyces. The comparisons of means of lg(mutation frequency) were analyzed by one-way ANOVA with Bonferroni post hoc correction. P < 0.05 is regarded as statistically significant.
Induction of SOS response in Streptomyces lividans
In order to investigate the roles of DnaQ in SOS response in S. lividans, spores growing on CP6 plates supplemented with mitomycin C (5 μg/ml, sub-inhibitory) for 6 days were collected and tested subsequently for spontaneous mutation frequency on SFM plates supplemented with rifampicin 100 μg/ml.
Identification of rifampicin resistant colonies
It has been shown that rifampicin resistance in Streptomyces lividans results from mutations in the rpoB gene, which encodes the beta subunit of RNA polymerase (15). All mutations in the rpoB gene associated with RifR phenotype are located in three conserved regions. We therefore PCR amplified and sequenced these three conserved regions of the rpoB gene from eight mutant isolates and compared them to the wild-type strain.
RNAseq-based transcriptomics
The data used was taken from (16), the raw sequence data sets are available at the NCBI SRA under study ID SRP144344, SRA accessions SRR7093720-SRR7093723, SRR7093726, SRR7093727. In short, samples of bacterial culture grown in minimal medium were taken during the mid-log and late-log growth phase as well as after entry into the stationary phase. TruSeq Stranded mRNA Library Prep Kit from Illumina were prepared and sequenced in paired-end mode on an Illumina HiSeq 1500 system with 28 respectively 70 bases read length. After mapping, reads per kilo base per million mapped reads (RPKM) were calculated using ReadXplorer v.2.2 (17).
Genome sequencing and analyses
Genomic DNA was isolated using the NucleoSpin Microbial DNA kit (Macherey&Nagel) according to the manufacturer's instructions. Sequencing and assembly of the genomes of the reference strain and four derivatives were performed in a hybrid. For each strain, two sequencing libraries were prepared, one for sequencing on the MiSeq platform (Illumina Inc., NL) and one for sequencing on the MinION platform (Oxford Nanopore Technologies, UK). The former consisted of a TruSeq DNA PCR-free library which was run in a 2 × 150 nt run using a 300 cycle MiSeq reagent kit v3 (Illumina Inc., NL). For ONT sequencing, the ligation-based sequencing kit SQK-LSK109 was used to prepare the library, which was in turn run on a R9.4.1 flow cell. Base-calling of the raw data was performed with ont-guppy-for-gridion v4.0.11. The Canu assembler v2.1 (18) was used to assemble the ONT data into contigs, which were subsequently polished using the Illumina data and the Pilon polisher v1.22 (19) for a total of 10 rounds. For the first 5, Bwa Mem (20) was used as a mapper; for the final 5 Bowtie2 (21) was applied. In addition, the Illumina data were assembled using Newbler v2.8 (22). For each strain, both assemblies were combined and manually curated using Consed (23). The software snippy (Seemann, T. (2017) Snippy: Fast Bacterial Variant Calling from NGS Reads.) was used for fast bacterial variant calling from NGS raw read data as well as from the assembled genomes. Larger rearrangements were search for via sequence alignment in SnapGene (from Insightful Science; available at snapgene.com). Detected SNPs along with other mutations are listed in Supplementary Table S6 provided as an Excel file in Supplementary Data.
All DNA sequencing data is available via NCBI/ANE/DDBJ linked under BioProject PRJNA699418, the complete genome sequence of S. lividans TK24-YQS040 is accessible via BioProject PRJNA713129.
Detection of indigoidine and coelimycin congeners
100 μl of streptomycetes spore suspensions were inoculated into 10 ml YEME media and incubated at 28oC overnight in shaking incubator, 200 rpm, to produce seed cultures. Next day 2.5 ml of seed cultures were used to start 25 ml fermentation in YEME media, at the same incubation conditions. The blue color indicating the expression of bspA gene appeared after 3–4 days of incubation. Spectrophotometric indigoidine detection was done as described previously (24) using culture supernatant. Fermentation continued for 7 days, and then the whole fermentation cultures were quickly frozen in a cold ethanol bath and lyophilized.
25 ml of methanol was added to the lyophilized material. The suspensions were processed on rotary shaker at room temperature for 60 min, and then centrifuged at 8000 rpm for 10 min. Supernatants were dried completely using rotavapors. The dry material was dissolved in 1 ml of methanol, and 100 μl of extracts were used for MS/MS analysis.
LC–MS analyses were performed on a Vanquish Horizon UHPLC system (Thermo Fisher Scientific) coupled to the ESI source of an LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific). Chromatographic and MS parameters were used as described previously (25), except that MS1 spectra were recorded in a range of m/z 150–1000.
RESULTS
Identification of DNA polymerase III subunits in Streptomyces lividans TK24
Inspection of the S. lividans TK24 genome (26) for the genes encoding main components of the DNA polymerase III revealed those specifying two α subunits (DnaE1 and E2), two β subunits (DnaN1 and N2), and three putative ϵ subunits (DnaQ1, Q2 and Q3). Levels of expression of these and some other DNA PolIII subunits’ genes were examined using RNAseq-based transcriptomics in order to pinpoint those that contribute most to the DNA replication (Table 1). According to the RNAseq data, of the subunits that may be involved in proofreading based on the data from E. coli and M. tuberculosis, only the DnaE1 and DnaQ1 were expressed at high levels in liquid culture. DnaQ1 and DnaQ2 share only 40% identity in their amino acid sequences (Supplementary Figure S1, Supplementary Data). Although DnaQ2 was expressed at much lower level compared to DnaQ1, it was similar to that of DnaX, τ subunit of DNA PolIII (Table 1). DnaQ3 contains an exonuclease domain similar to that of DnaQ1 and DnaQ2, but has an extended C-terminal BRCT domain proposed to be involved in cell cycle progression and DNA damage signaling in eukaryotes (27). Taking these observations together, it appeared likely that DnaQ1 represented the true active ϵ subunit that may be responsible for proofreading during the DNA replication in S. lividans, although we could not completely exclude that DnaQ2 and DnaQ3 may also be important ensuring DNA replication fidelity.
Table 1.
Expression levels of DNA polymerase III major subunits in Streptomyces lividans TK24 based on the RNAseq data
| Subunit | Gene name | Annotation | RNAseq data (Max RPKM) | Product |
|---|---|---|---|---|
| alpha | dnaE1 | SLIV_27385 | 272.2 | DnaE1 |
| alpha | dnaE2 | SLIV_29050 | 3.0 | DnaE2 |
| beta | dnaN1 | SLIV_18870 | 606.6 | DnaN1 |
| beta | dnaN2 | SLIV_31895 | 5.7 | DnaN2 |
| epsilon | dnaQ1 | SLIV_30105 | 2613.5 | DnaQ1 |
| epsilon | dnaQ2 | SLIV_07720 | 72.1 | DnaQ2 |
| epsilon | dnaQ3 | SLIV_28580 | 131.0 | DnaQ3 |
| gamma & tau | dnaX | SLIV_18100 | 91.7 | DnaX |
| delta | holA | SLIV_20580 | 115.5 | HolA |
DnaQ paralogues of S. lividans TK24 are not involved in proofreading of DNA replication
Aiming at generating DnaQ1 mutants that may yield a ‘mutator’ phenotype in S. lividans, we first aligned this protein sequence to that of E. coli DnaQ, and identified amino acid residues corresponding to those shown to be critical for the dominant ‘mutator’ phenotype in E. coli (28). Accordingly, we designed five mutants (M1-M5) for both DnaQ1 and DnaQ2 of S. lividans (29,30), which contained either one or two amino acid changes at different sites. The amino acid substitutions of DnaQ1 mutants were: M1 = Val69Trp; M2 = Ala169Val; M3 = Val69Trp + Ala169Val; M4 = Asp13Ala; M5 = Asp13Ala + Glu15Ala. The amino acid substitutions of DnaQ2 mutants were: M1 = Val67Trp; M2 = Ala168Val; M3 = Val67Trp + Ala168Val; M4 = Asp12Ala; and M5 = Asp12Ala + Glu14Ala. All mutant dnaQ genes were synthesized and cloned on an integrative plasmid under control of the strong constitutive promoter ermE*p known to provide high-level gene expression in S. lividans. In addition, DnaQ926, an E. coli protein manifesting ‘mutator’ phenotype was also expressed in S. lividans TK24 under the same promoter after codon optimization and synthesis of the corresponding gene (30).
After intergeneric conjugation to S. lividans TK24, the recombinant plasmids expressing DnaQ mutants were site-specifically integrated into the chromosome. The wild-type copy of the dnaQ1 was subsequently deleted in recombinant strains expressing dnaQ1 mutants using gene replacement vector pYQS046 (see Supplementary Data, Supplementary Tables S1, S4). This was done in case DnaQ1 wild-type, which is expressed at a relatively high level (based on the RNAseq data) outcompetes DnaQ1 mutants in binding to the DnaE1 and DnaX. The deletion of dnaQ1 was done in the strains expressing the mutant versions of dnaQ because we considered it possible that complete absence of this subunit may destabilize the DNA PolIII complex, rendering the replication machinery non-functional. All constructed recombinant strains were tested for the frequency of spontaneous mutagenesis manifested by the appearance of rifampicin-resistant mutants (RifR) due to mutations in the β subunit of RNA polymerase (31). Surprisingly, frequencies of the RifR mutations in the recombinant strains did not differ from that of the control, S. lividans TK24 with integrated empty vector and carrying wild-type copy of dnaQ1 (data not shown). This result strongly suggested that DnaQ1 of S. lividans TK24, unlike its counterpart in E. coli, is not involved in proofreading during DNA replication in the former bacterium. To confirm this, we constructed S. lividans strains that did not express any DnaQ mutants. These strains harbored either dnaQ or dnaQ2 deletion constructed using gene replacement plasmids pYQS056 and pYQS057, respectively, or dnaQ3 insertionally inactivated via integration of a suicide vector pYQS058 (see Supplementary Data, Supplementary Tables S1 and S4). These mutants were also tested for the frequency of spontaneous mutagenesis, showing the same phenotype as the control (Figure 1). Seven of the eight spontaneous RifR mutants were verified by PCR amplification of the RNA polymerase subunit β gene fragment followed by sequencing, which revealed point mutations in line with those in a previous study (32), indicating that rifampicin resistance is a proper marker to evaluate spontaneous mutation frequency. Genome sequencing of S. lividans TK24 ΔdnaQ1::dnaQ1-M5 and TK24 ΔdnaQ2::dnaQ2-M5 did not show increased mutation frequency either (data not shown). Taking together, all these data allowed us to conclude that DNA polymerase III ϵ subunits are not involved in the proofreading of DNA replication in S. lividans.
Figure 1.
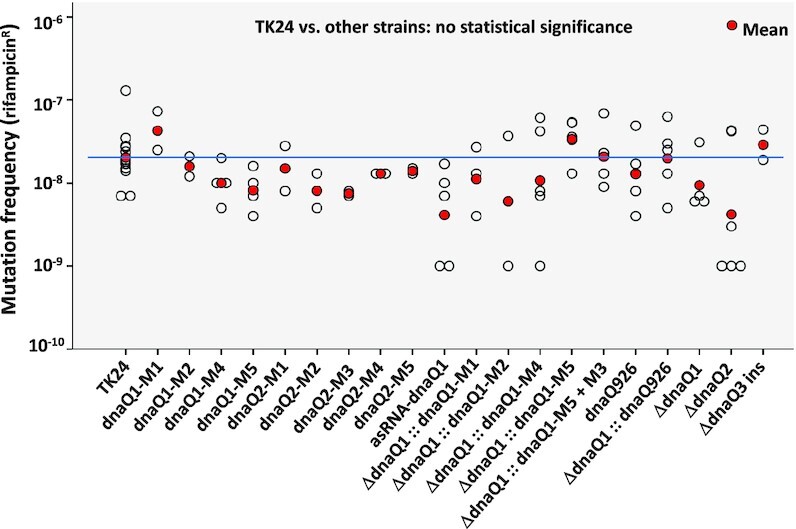
Spontaneous mutation frequency of recombinant S. lividans strains with manipulated dnaQ genes. ins – insertional inactivation via single crossover.
DnaQ1 and DnaQ2 are involved in SOS response in S. lividans
A previous study in E. coli has demonstrated that knock-out of dnaQ induces SOS response, which is manifested in the increase of mutation frequency upon exposure to DNA damaging agents (33). In another study, contradictory evidence was reported, claiming that a dnaQ knock-out mutant is deficient in the SOS response induced by nalidixic acid (34). In order to investigate the roles of DnaQ proteins in SOS response in S. lividans, spores of the wild type strain and dnaQ1-3 knockout mutants grown in the presence of a sub-inhibitory concentration of DNA damaging agent mitomycin C were collected and tested subsequently for the frequency of RifR mutations. As shown in Figure 2, S. lividans TK24 showed increased frequency of RifR mutations following exposure to mitomycin C (P = 0.001), which suggested a proper SOS response in this strain. The strains with dnaQ1 and dnaQ2 knock-outs, on the other hand, exhibited only moderately increased mutagenesis (not statistically significant), suggesting their deficiency in SOS response (Figure 2). After genetic complementation of the knockout mutants with corresponding dnaQ1 and dnaQ2 genes, the SOS response was restored. A similar trend was observed for the dnaQ3 knockout mutant, while the difference was not statistically significant. These findings confirm that DnaQ1 and DnaQ2 are both required for a proper SOS response in S. lividans, although their individual roles in this process remain unclear.
Figure 2.
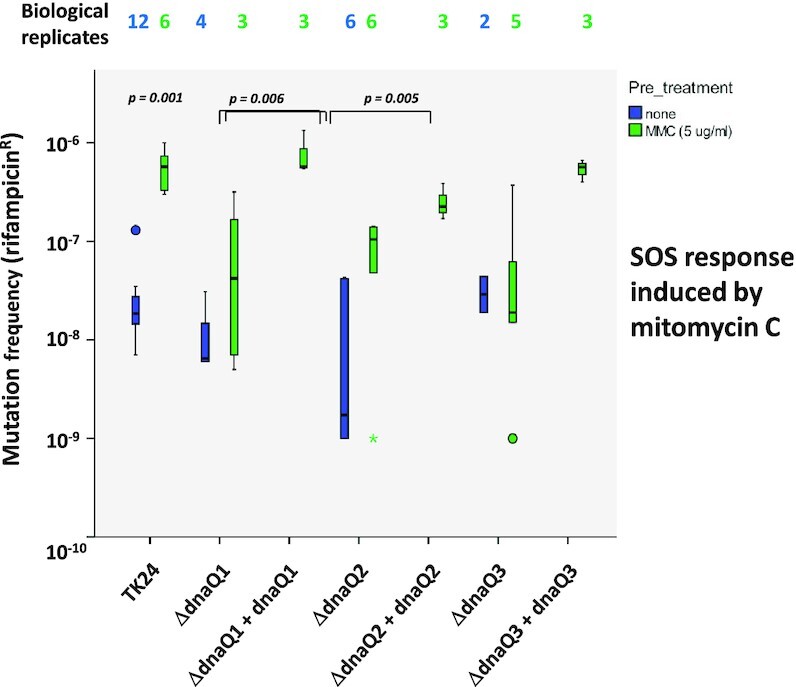
SOS response in dnaQ-deficient S. lividans strains treated with mitomycin C. Asterisk indicates an outlier.
Targeting DnaE1 for ‘mutator’ phenotype
A recent study on Mycobacterium tuberculosis showed that its DnaQ does not substantially contribute to replication fidelity, while DnaE1 itself is likely to be responsible for proofreading function during DNA replication, mediated by an intrinsic 3′-5′ exonuclease activity within its polymerase and histidinol phosphatase (PHP) domain (6).
Based on the alignment of amino acid sequences of S. lividans TK24 DnaE1and its mycobacterial counterpart, we designed two mutants that, according to the data reported for M. tuberculosis, could be affected in DNA replication proofreading. In these mutants, metal-coordinating residues in the PHP domain were altered (Asp19Asn in DnaE1_M1 and Glu69Gln in DnaE1_M2). Both mutants were expressed in S. lividans under control of the strong PA3 promoter (35) and recombinant strains tested for spontaneous mutagenesis. No differences in the frequency of RifR mutations were observed between the recombinant strains expressing these DnaE1 mutants and control strain with empty vector (data not shown).
The N-terminal part of DnaE1 (amino acids 8–194) encompasses the PHP domain, which was the primary target for mutagenesis in the first attempt to generate a ‘mutator’. The central part of DnaE1 (amino acids 316–756) represents catalytic polymerization domain DNA_pol3_alpha (36), while the C-terminal part contains two domains, HHH involved in non-specific DNA binding (37) and OB-fold implicated in binding single-stranded DNA (38).
In order to expand the options for achieving desired phenotype in S. lividans, the dnaE1 gene was dissected into several parts that would allow assembly aimed at combining mutations reported to result in ‘mutator’ phenotype (Figure 3). In particular, mutations were designed that targeted PHP, DNA_pol3_alpha and HHH domains in DnaE1. Gene parts were synthesized individually, and then assembled in various combinations under control of the strong synthetic promoter PA3 (35). All constructs were assembled on a low-copy plasmid pKC1218H (39) and introduced into S. lividans TK24. Transconjugants for each construct were tested for spontaneous mutagenesis frequency after one round of sporulation on the SFM medium supplemented with hygromycin by plating serial dilutions on the same medium supplemented with rifampicin. S. lividans TK24 carrying empty vector was used as a control. Recombinant strains expressing two of the DnaE1 mutants, M5 (D19N H44F) and M7 (D19N C154G D634E) exhibited considerable (up to ca 1000-fold) increase in spontaneous mutagenesis (data not shown). None of the other recombinant strains expressing DnaE1 mutants displayed spontaneous mutation frequency higher than the control strain S. lividans TK24 (pKC1218H). Since the strain overexpressing DnaE1 M7 mutant exhibited ca. 2-fold higher mutation frequency, it was chosen for further testing. The corresponding construct pMDE7 was again introduced into S. lividans TK24, and three independent transconjugants were tested for spontaneous mutagenesis as described above. All three displayed significant spontaneous mutagenesis, with frequencies ranging from 3 × 10–3 to 2 × 10–4, compared to 2 × 10–6 in the control. The reason for differences in spontaneous mutagenesis between the transconjugants remains unclear. The pMDE7 plasmid was also introduced into Streptomyces venezuelae NRRL B-65442 and Streptomyces albidoflavus (former Streptomyces albus) J1074, but examination of the transconjugants’ progeny for RifR mutants did not reveal enhanced mutagenesis (see Discussion).
Figure 3.
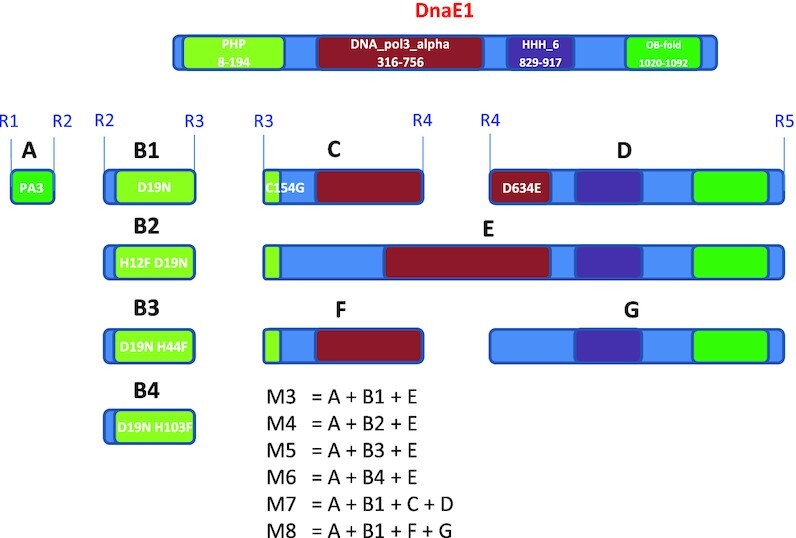
Dissected DnaE1 and a scheme for combinatorial assembly of mutants.
Coupling of the ‘mutator’ device with metabolite-specific biosensor
We have recently reported construction and testing of a biosensor based on a TetR-like repressor and indigoidine synthetase BpsA that can be used to detect production of a metabolite specified by the biosynthetic gene cluster encoding the abovementioned repressor (39). This biosensor was able to successfully detect production of coelimycin P1, a polyketide alkaloid which is normally not produced by S. lividans TK24, and has been reported from Streptomyces coleicolor M145 (40). Considering the fact that activation of a silent BGC or enhancement of its expression may require multiple mutations in the producer's genome (41,42), we decided to couple the newly constructed ‘mutator’ device with the coelimycin-specific biosensor.
The plasmid pMDE7 carrying the ‘mutator’ gene was introduced into the S. lividans TK24 (pYQS040) strain harboring the coelimycin biosensor, where the coelimycin cluster, cpk, was not expressed in the conditions used (Supplementary Table S5, Supplementary data). Three recombinant strains obtained after introduction of pMDE7 were propagated until sporulation on a solid medium with hygromycin to maintain the pMDE7 plasmid. The spores of each strain were plated in serial dilutions, and resulting colonies inspected for indigoidine production. In all three cases indigoidine-producing colonies appeared at a frequency of ca. 10–5, and four such colonies originating from three independent TK24 (pYQS040 + pMDE7) transconjugants were picked for further analyses, along with the control TK24 (pYQS040). The five selected strains were grown in liquid medium YEME, and culture extracts examined for the production of indigoidine by UV/Vis spectrophotometry and of coelimycin using high-resolution LC–MS. In addition, all extracts were analyzed for the presence of actinorhodin and undecylprodigiosin by LC–MS, and presence of the latter but not the former was confirmed in all extracts. Hence, we could exclude the possibility that blue pigment produced by the four strains was due to actinorhodin. In liquid cultures, all four TK24 (pYQS040+pMDE7) strains produced indigoidine, which could not be identified in the control strain. Surprisingly, coelimycin P1 could not be detected in any of the samples. However, by searching for compounds that produce common fragment ions with coelimycin P1, several potential congeners named coelimycin P3–P6 were identified (Supplementary Data, Supplementary Figures S1–S8). They were only present in trace amounts in the culture extract of the control strain but produced in much higher concentration in all four TK24 (pYQS040+pMDE7) recombinant strains (Supplementary Table S5, Supplementary data), which most likely explains the biosensor response. Detection of very low levels of coelimycin congeners in the control strain is not surprising, since S. lividans RedStrep1 (pYQS040), the derivative of TK24, was previously reported to produce traces of coelimycin P1 (39). These results provide further evidence for the activation of the cpk gene cluster in the biosensor-positive TK24 (pYQS040 + pMDE7) strains but also demonstrate our still very limited understanding of its biosynthesis products.
The data obtained strongly indicated that the spontaneous mutations introduced due to the error-prone replication activated the biosynthetic gene cluster for coelimycin, and the indigoidine biosensor reacted properly in the majority of the strains. We speculated that activation of the coelimycin BGC might have occurred due to one or more of the following: (i) inactivation of the repressor ScbR2; (ii) changes in scbR2 promoter region; (iii) changes in the promoter region of the transcriptional activator gene cpkN leading to its overexpression. To test these hypotheses, DNA fragments encompassing these regions were PCR-amplified from all 5 strains under study and sequenced. No mutations were identified in the amplified regions, suggesting that activation of coelimycin gene cluster was caused by mutations in other parts of S. lividans genome. On the other hand, continued functioning of the ‘mutator’ device could generate mutations within the cpk gene cluster, thus shutting down the coelimycin biosynthesis (see next subchapter).
Whole genome sequencing of indigoidine/coelimycin congener-producing strains
Although the error-prone replication most certainly will cause multiple point mutations in the genome, we made an attempt to identify them in all coelimycin-producing strains using genome sequencing in hope of finding a common pattern that may shed light on the reason for coelimycin BGC activation. Comparison of the reference strain carrying only the integrated plasmid pYQS040 and the four derivatives carrying the plasmid pMDE7 to the progenitor strain TK24 revealed a total of seven mutations found in all five (four missense and one synonymous exchanges in protein coding regions and two deletions in intergenic regions, Supplementary Table S6, Supplementary data). All these mutations were absent in the reference S. lividans TK24 genome, suggesting that they have accumulated during the laboratory maintenance and genetic manipulation of the latter leading to the strain TK 24 (pYQS040). Besides these ‘base’ mutations, the strains carrying plasmid pMDE7 all show many additional mutations that seem to be randomly distributed throughout the genomes (Figure 4), albeit the numbers varying wildly between the four strains: TK24-M7-BM1-1 with 96 additional mutations, TK24-M7-BM1-4 with 74, TK24-M7-BM2-9 with 66 and TK24-M7-BM3-3 with just 6. Only TK24-M7-BM1-1 and TK24-M7-BM1-4 share a significant number of mutations, 28 in total, which is unsurprising as they were derived from the same transconjugant.
Figure 4.

Distribution of mutations in the genomes of the coelimycin-producing, biosensor-positive progeny of TK24 that carry pMDE7. The complete genome sequences of TK24-YQ040, TK24-M7-BM1-1, TK24-M7-BM1-4, TK24-M7-BM2-9 and TK24-M7-BM3-3 were established in a hybrid approach using Illumina and ONT data. The resulting genomes of the pMDE7-carrying strains were compared to that of TK24-YQ040 using snippy. Lines denote mutations, with green indicating synonymous replacements, blue missense mutations, red frameshifts & stops, and black presumably neutral mutations in intergenic regions. Lines above the chromosome bars (light grey) indicate insertions, with small triangles denoting the two inserted vectors pYQ040 (green) and pMDE7 (purple). cpk cluster is indicated as an orange bar.
By type, single nucleotide polymorphisms are the most common with 166 instances, while small deletions and insertions occur much less frequently with 19 and18 instances, respectively. Among the SNPs, 98 are transitions while 68 are transversions, indicating an almost equal rate of occurrence of the two types. Transversions to the complementary base are rare, though: only 2 out of the 68 transversions belong to this type. The insertions and most of the deletions consist of just one nucleotide (usually in short homopolymers), but three deletions are larger and result in the loss of 46, 74 and 177 nucleotides, respectively. Concerning the effect of the introduced mutations, 99 cause a missense mutation, 47 a synonymous replacement, 22 a frameshift, 4 a nonsense mutation and one an in-frame deletion of 59 amino acids. An additional 31 are intergenic, with 5 being in known regulatory structures, i.e., promoters (26).
A search for large scale mutations and rearrangements revealed some interesting findings: First, the inspection of the integrated plasmid pYQS040 revealed that in all 4 strains carrying the mutator plasmid pMDE7, the phiC31 integrase was disrupted by independent mutations: frame-shift introducing deletions in strains TK24-M7-BM1-1 and TK24-M7-BM2-9, a frame-shift causing insertion in strain TK24-M7-BM3-3 and a nonsense mutation in TK24-M7-BM1-4. Second, the plasmid pMDE7 was found integrated into the genome in all 4 TK24-M7 derivatives at the dnaE locus. Quite surprisingly, it is present at this location not in one copy, but as a concatemer of at least two to four copies. Due to its size of 9.7 kb it is impossible to discern the exact number of repeats in the genome (and whether there is any ‘free’ pMDE7 in the cells at all). If there is no free, self-replicating pMDE7 present, the coverage statistics of this region indicates that between 8 and 10 repeats are integrated in a row in the four strains. Closer inspection of the pMDE7 sequence in the four strains revealed as a possible cause for the non-replicative behavior of the plasmid a one nucleotide insertion in the SCP2* minimal replicon. In addition, it was found that the Cys154Gly point mutation in dnaE is absent in strain TK24-M7-BM3-3, which might explain the low number of mutations observed in this strain.
The TK24-M7-BM3-3 mutant, while apparently having only 6 additional mutations compared to the parent strain, produced the next highest levels of coelimycin congeners (Supplementary Table S5, Supplementary data). Three of the six mutations were located within coding regions, leading to inactivation of phiC31 integrase in the biosensor-harboring construct, SLIV_1507 coding for the ribosomal protein L10 and SLIV_22980 encoding putative sugar hydrolase. One mutation within coding region of SLIV_29345 (glucokinase) was synonymous, and hence would not cause a functional defect. Two other mutations were located within intergenic regions, namely G to A transition upstream of SLIV_03310 encoding a putative toxin immunity protein, and a 75-bp deletion in the putative regulatory region of SLIV_17715 encoding a FAD-binding oxidoreductase with C-terminal BBE domain. Interestingly, the same deletion was found in the genome of the TK24-M7-BM1-four mutant, which stems from a different original pMDE7 transconjugant.
In order to test whether the selected strains continue to accumulate mutations, we plated serial dilutions of spores obtained from the indigoidine-producing colonies of TK24-M7-BM1-1 and the TK24-M7-BM3-3 mutant. White, indigoidine non-producing colonies, appeared in both cases at a frequency of 3 × 10–3, suggesting that ‘mutator’ continues to function. Considering the size of the cpk cluster (ca. 70 kb), it is likely that this phenotype is due to the inactivation of the cpk biosynthetic genes or other genes involved in its regulation rather than the coelimycin-specific biosensor. Despite this, we were still able to detect coelimycin congeners in these mutants even after several sub-culturings that did not involve picking single indigoidine-producing colonies.
DISCUSSION
Traditionally, strain development programs aimed at increasing the yield of secondary metabolites, in particular antibiotics, have been based on random mutagenesis of microbial strains by means of their exposure to UV- or X-radiation and various chemical mutagens (e.g. nitrosoguanidine) (43). This approach, although laborious and time-consuming, has led to the development of many high-producing strains that are currently being used to manufacture dozens of antibiotics and anti-cancer agents, e.g. erythromycin, nystatin and doxorubicin. With the onset of genomics, the strain development programs based on metabolic engineering started to emerge, sometimes generating truly spectacular outcomes, like in the case of anti-malarial drug artemisinin (44). However, regulation of secondary metabolism in the most prolific antibiotic producers, bacteria of the order Actinomycetales, is extremely complicated and each species appears to be unique in this respect. The latter makes rational metabolic engineering aimed at enhancement of antibiotic production or, as of late, activation of silent secondary metabolite biosynthesis gene clusters a formidable challenge. Genome sequencing of high-producing actinomycete mutants recently revealed different mutations that apparently contribute to the desired phenotype but vary significantly between the strains (41,42,45). Hence, it appears difficult to design a common strategy for metabolic engineering of various actinomycetes, and spontaneous mutagenesis coupled with smart selection strategy remains a viable alternative.
In this study, we targeted the replication machinery of S. lividans TK24, a model actinomycete bacterium, in order to generate ‘mutator’ phenotype allowing spontaneous mutagenesis without the use of radiation of chemical mutagens. Unlike in E. coli, ϵ subunits of DNA polymerase III were found to be dispensable for DNA replication and proofreading in S. lividans, much like in the distantly related actinobacterium M. tuberculosis (6). Both paralogs of this subunit turned out to be required for a proper SOS response, a complex mechanism promoting continuous replication of bacterial chromosome upon DNA damage, at the same time causing enhanced mutagenesis (46). The same phenomenon was described for the E. coli DnaQ protein, although the mechanism behind remains unclear (47). We then turned to the α subunit of DNA polymerase III as a target, but introduction of the single mutation D19N in the PHP domain of DnaE1analogous to that which led to the ‘mutator’ phenotype in M. tuberculosis had no effect in S. lividans. Only introduction of additional mutation in the same domain, or its combination with a mutation in the pol3 domain led to the desired phenotype. Interestingly, overexpression of the DnaE1-M7 mutant could elicit ‘mutator’ phenotype only in S. lividans, but not in S. venezuelae or S. albus. DnaE1 of S. lividans TK24 shares 91% and 92% amino acid sequence identity to the corresponding proteins of S. venezuelae and S. albidoflavus, respectively. It is plausible that the differences in amino acid sequences were sufficient to prevent proper interaction of the exogenous DnaQ1-M7 with the other endogenous DNA polymerase III subunits, thus preventing formation of a complex with deficient proofreading activity. Hence, we believe that introducing mutations D19N, C154G and D634E in DnaE1 proteins of any Streptomyces will have the same effect, yielding strain-specific ‘mutator’ devices.
Next, we decided to take advantage of the ‘mutator’ device pMDE7 based on engineered dnaE1-M7 gene for the purpose of activation of silent cpk gene cluster in S. lividans TK24. In order to monitor eventual cluster activation and production of coelimycin, we coupled the ‘mutator’ device with a coelimycin-specific biosensor. This allowed us to easily select strains that gave positive biosensor signal and turned out to produce coelimycin congeners, suggesting that a similar approach can be applied to many streptomycetes in order to reveal their true biosynthetic potential. It is notable that we could not detect formation of coelimycin P1, which would be expected to be the main product of cpk gene cluster in S. lividans. The biosynthetic pathway specified by the cpk gene cluster is remarkably complex and not well understood. The actual cpk product is hypothesized to be coelimycin A, a highly reactive bis-epoxide that is also assumed to possess antibacterial activity. However, this compound has never been isolated and characterized due to its instability. Coelimycin P1, which has no antibiotic activity, most likely originates from the non-enzymatic trapping of coelimycin A with N-acetylcysteine in the culture medium and subsequent oxidation (48). This hypothesis is supported by the detection of a different non-enzymatic trapping product with glutamate, named coelimycin P2, in a different culture medium (40). Furthermore, Juan Pablo Gomez-Escribano et al. reported that even the trapping product coelimycin P1 was not stable and ‘invariably suffered partial degradation during purification’ (40). Based on this information from literature, we are confident that coelimycin A is indeed produced by our mutant strains, but that different non-enzymatic trapping products and/or degradation products of this unstable species are formed either due to differences in the medium composition or sample preparation.
Sequencing of the genomes of selected coelimycin producers revealed a number of mutations that cannot be unequivocally associated with cpk cluster activation. These mutations need further comprehensive analyses, as they encompass not only point mutations, but also deletions and insertions in non-coding regions that may play an important regulatory role. It must also be considered that ‘background’ mutations identified in the parent strain harboring biosensor construct could have an influence on the coelimycin production phenotype only when combined with additional mutations introduced via error-prone DNA replication. It must be mentioned that, once the cluster product has been detected, it would be advantageous to shut down or eliminate the ‘mutator’ device from the mutant since it may negatively affect the production and/or the overall fitness of the strain. In this respect, we investigated the potential usefulness of two repressors, CymR and RolR, which respond to cumate and resorcinol, respectively, and have been reported to provide tight control of gene expression in Streptomyces (49). Both repressor genes under control of constitutive promoter were introduced into S. lividans TK24 carrying a plasmid with bpsA gene coupled to the respective operator. Although we could observe a clear repression of bpsA, induction could not be achieved with a wide range of inducer concentrations, possibly because of an efficient efflux of inducers form S. lividans. We plan to further explore other repressors for this purpose, or place the ‘mutator’ device on temperature-sensitive plasmid, e.g. pSG5 derivative (50).
Genome mining aimed at the discovery of new drugs currently mostly relies on biosynthetic cluster activation by means of overexpression of positive regulators, inactivation of repressors, promoter exchange, as well as heterologous expression that may be combined with the above (51). The advantage of the using a ‘mutator’ device is that it acts globally, thus affecting the genes that may indirectly influence production of desired metabolite, which is then detected by a specific biosensor. Other genome mining approaches target specific genes in the BGCs of interest, and do not affect global regulators or other, not yet characterized genes, that may contribute to cluster activation. Biosensors may not be easily constructed without significant efforts for biosynthetic gene clusters lacking metabolite-responsive repressors, although this is also possible (52). However, many BGCs in Streptomyces encode various types of repressors known to be ligand-responsive, such as those belonging to the TetR, MarR, PadR and LysR families (53–55). We have recently sequenced genomes of 13 Streptomyces isolated from the rhizosphere of Edelweiß (56), and surveyed the genomes for the presence of unique BGCs that we would like to target for activation (unpublished data). In total, we identified 79 such clusters, and 32 of those contained one or more genes encoding repressor(s) belonging to the families mentioned above. Hence, we believe that this new approach to mutator/biosensor-guided activation of silent or poorly expressed BGCs in Streptomyces, and perhaps other bacteria, represents a valuable contribution to the genome mining for novel natural products.
DATA AVAILABILITY
All DNA sequencing data is available via NCBI/ANE/DDBJ linked under BioProject PRJNA699418, the complete genome sequence of S. lividans TK24-YQS040 is accessible via BioProject PRJNA713129.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Jan Kormanec of Ustav Molekularnej Biologie Slovenskej Akadémie Vied, for kindly providing indicator BpsA plasmid pBPSA and Anna Fabisikova from the Mass Spectrometry Center of the Faculty of Chemistry, University of Vienna, for her assistance with LC–MS data acquisition. We are also thankful to Johanna Höller for her help in performing some experiments.
Contributor Information
Olga N Sekurova, Department of Pharmaceutical Sciences, Division of Pharmacognosy, University of Vienna, Vienna, Austria.
Yi-Qian Sun, Department of Biotechnology and Food Science, Norwegian University of Science and Technology (NTNU), Trondheim, Norway; Department of Clinical and Molecular Medicine, Norwegian University of Science and Technology (NTNU), Trondheim, Norway.
Martin Zehl, Department of Analytical Chemistry, Faculty of Chemistry, University of Vienna, Austria.
Christian Rückert, Center for Biotechnology (CeBiTec), Universität Bielefeld, Bielefeld, Germany.
Anna Stich, Department of Pharmaceutical Sciences, Division of Pharmacognosy, University of Vienna, Vienna, Austria.
Tobias Busche, Center for Biotechnology (CeBiTec), Universität Bielefeld, Bielefeld, Germany.
Jörn Kalinowski, Center for Biotechnology (CeBiTec), Universität Bielefeld, Bielefeld, Germany.
Sergey B Zotchev, Department of Pharmaceutical Sciences, Division of Pharmacognosy, University of Vienna, Vienna, Austria.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Commission under the 7th Framework Program through the ‘Collaborative Project’ action ‘STREPSYNTH’ [613877]; University of Vienna; Austrian Science Fund (FWF) [P 30986-B32]. Funding for open access charge: Universität Wien.
Conflict of interest statement. None declared.
REFERENCES
- 1.Denamur E., Matic I.. Evolution of mutation rates in bacteria. Mol. Microbiol. 2006; 60:820–827. [DOI] [PubMed] [Google Scholar]
- 2.Shevelev I.V., Hubscher U.. The 3′ 5′ exonucleases. Nat. Rev. Mol. Cell. Biol. 2002; 3:364–376. [DOI] [PubMed] [Google Scholar]
- 3.Fijalkowska I.J., Schaaper R.M.. Mutants in the Exo I motif of Escherichia coli dnaQ: defective proofreading and inviability due to error catastrophe. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:2856–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Itakura M., Tabata K., Eda S., Mitsui H., Murakami K., Yasuda J., Minamisawa K.. Generation of Bradyrhizobium japonicum mutants with increased N2O reductase activity by selection after introduction of a mutated dnaQ gene. Appl. Environ. Microbiol. 2008; 74:7258–7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whatley Z., Kreuzer K.N.. Mutations that separate the functions of the proofreading subunit of the Escherichia coli replicase. G3. 2015; 5:1301–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rock J.M., Lang U.F., Chase M.R., Ford C.B., Gerrick E.R., Gawande R., Coscolla M., Gagneux S., Fortune S.M., Lamers M.H.. DNA replication fidelity in Mycobacterium tuberculosis is mediated by an ancestral prokaryotic proofreader. Nat. Genet. 2015; 47:677–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maki H., Mo J.Y., Sekiguchi M.. A strong mutator effect caused by an amino acid change in the alpha subunit of DNA polymerase III of Escherichia coli. J. Biol. Chem. 1991; 266:5055–5061. [PubMed] [Google Scholar]
- 8.Atanasov A.G., Zotchev S.B., Dirsch V.M.International Natural Product Sciences Taskforce and Supuran, C.T. . Natural products in drug discovery: advances and opportunities. Nat. Rev. Drug. Discov. 2021; 20:200–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flett F., de Mello Jungmann-Campello D., Mersinias V., Koh S.L., Godden R., Smith C.P.. A ‘gram-negative-type’ DNA polymerase III is essential for replication of the linear chromosome of Streptomyces coelicolor A3(2). Mol. Microbiol. 1999; 31:949–958. [DOI] [PubMed] [Google Scholar]
- 10.Tsai H.H., Shu H.W., Yang C.C., Chen C.W.. Translesion-synthesis DNA polymerases participate in replication of the telomeres in Streptomyces. Nucleic Acids Res. 2012; 40:1118–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flett F., Mersinias V., Smith C.P.. High efficiency intergeneric conjugal transfer of plasmid DNA from Escherichia coli to methyl DNA-restricting streptomycetes. FEMS Microbiol. Lett. 1997; 155:223–229. [DOI] [PubMed] [Google Scholar]
- 12.Knirschova R., Novakova R., Mingyar E., Bekeova C., Homerova D., Kormanec J.. Utilization of a reporter system based on the blue pigment indigoidine biosynthetic gene bpsA for detection of promoter activity and deletion of genes in Streptomyces. J. Microbiol. Meth. 2015; 113:1–3. [DOI] [PubMed] [Google Scholar]
- 13.Sambrook J., Fritsch E., Maniatis T.. Molecular Cloning: A Laboratory Manual. 1989; 2nd ednCold Spring Harbor: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 14.Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A. 3rd, Smith H.O.. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009; 6:343–345. [DOI] [PubMed] [Google Scholar]
- 15.Hu H., Zhang Q., Ochi K.. Activation of antibiotic biosynthesis by specified mutations in the rpoB gene (encoding the RNA polymerase beta subunit) of Streptomyces lividans. J. Bacteriol. 2002; 184:3984–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsolis K.C., Hamed M.B., Simoens K., Koepff J., Busche T., Rückert C., Oldiges M., Kalinowski J., Anné J., Kormanec J.et al.. Secretome dynamics in a Gram-positive bacterial model. Mol. Cell Proteomics. 2019; 18:423–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hilker R., Stadermann K.B., Schwengers O., Anisiforov E., Jaenicke S., Weisshaar B., Zimmermann T., Goesmann A.. ReadXplorer 2-detailed read mapping analysis and visualization from one single source. Bioinformatics. 2016; 32:3702–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koren S., Walenz B.P., Berlin K., Miller J.R., Bergman N.H., Phillippy A.M.. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017; 27:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walker B.J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., Cuomo C.A., Zeng Q., Wortman J., Young S.K., Earl A.M.. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014; 9:e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H.Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013; arXiv doi:26 May 2013, preprint: not peer reviewedhttps://arxiv.org/pdf/1303.3997v2.
- 21.Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Margulies M., Egholm M., Altman W.E., Attiya S., Bader J.S., Bemben L.A., Berka J., Braverman M.S., Chen Y.J., Chen Z.et al.. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005; 437:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gordon D., Green P.. Consed: a graphical editor for next-generation sequencing. Bioinformatics. 2013; 29:2936–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown A.S., Calcott M.J., Collins V.M., Owen J.G., Ackerley D.F.. The indigoidine synthetase BpsA provides a colorimetric ATP assay that can be adapted to quantify the substrate preferences of other NRPS enzymes. Biotechnol. Lett. 2020; 42:2665–2671. [DOI] [PubMed] [Google Scholar]
- 25.Oberhofer M., Wackerlig J., Zehl M., Büyük H., Cao J.J., Prado-Roller A., Urban E., Zotchev S.B.. Endophytic Akanthomyces sp. LN303 from Edelweiss produces emestrin and two new 2-hydroxy-4 pyridone alkaloids. ACS Omega. 2021; 6:2184–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Droste J., Ruckert C., Kalinowski J., Hamed M.B., Anne J., Simoens K., Bernaerts K., Economou A., Busche T.. Extensive reannotation of the genome of the model streptomycete Streptomyces lividans TK24 based on transcriptome and proteome information. Front. Microbiol. 2021; 12:744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reinhardt H.C., Yaffe M.B.. Phospho-Ser/Thr-binding domains: navigating the cell cycle and DNA damage response. Nat. Rev. Mol. Cell. Biol. 2013; 14:563–580. [DOI] [PubMed] [Google Scholar]
- 28.Echols H., Lu C., Burgers P.M.. Mutator strains of Escherichia coli, mutD and dnaQ, with defective exonucleolytic editing by DNA polymerase III holoenzyme. Proc. Natl. Acad. Sci. U.S.A. 1983; 80:2189–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takano K., Nakabeppu Y., Maki H., Horiuchi T., Sekiguchi M.. Structure and function of dnaQ and mutD mutators of Escherichia coli. Mol. Gen. Genet. 1986; 205:9–13. [DOI] [PubMed] [Google Scholar]
- 30.Fijalkowska I. J., Schaaper R.M.. Mutants in the Exo I motif of Escherichia coli dnaQ: defective proofreading and inviability due to error catastrophe. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:2856–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Floss H.G., Yu T.W.. Rifamycin-mode of action, resistance, and biosynthesis. Chem. Rev. 2005; 10:621–632. [DOI] [PubMed] [Google Scholar]
- 32.Hu H., Zhang Q., Ochi K.. Activation of antibiotic biosynthesis by specified mutations in the rpoB gene (encoding the RNA polymerase beta subunit) of Streptomyces lividans. J. Bacteriol. 2002; 184:3984–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slater S.C., Lifsics M.R., O’Donnell M., Maurer R.. holE, the gene coding for the theta subunit of DNA polymerase III of Escherichiacoli: characterization of a holE mutant and comparison with a dnaQ (epsilon-subunit) mutant. J. Bacteriol. 1994; 176:815–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pohlhaus J.R., Long D.T., O’Reilly E., Kreuzer K.N.. The epsilon subunit of DNA polymerase III Is involved in the nalidixic acid-induced SOS response in Escherichia coli. J. Bacteriol. 2008; 190:5239–5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Siegl T., Tokovenko B., Myronovskyi M., Luzhetskyy A.. Design, construction and characterisation of a synthetic promoter library for fine-tuned gene expression in actinomycetes. Metab. Eng. 2013; 19:98–106. [DOI] [PubMed] [Google Scholar]
- 36.Aravind L., Koonin E.V.. Phosphoesterase domains associated with DNA polymerases of diverse origins. Nucleic Acids Res. 1998; 26:3746–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doherty A.J., Serpell L.C., Ponting C.P.. The helix-hairpin-helix DNA-binding motif: a structural basis for non-sequence-specific recognition of DNA. Nucleic Acids Res. 1996; 24:2488–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bochkarev A., Pfuetzner R.A., Edwards A.M., Frappier L.. Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature. 1997; 385:176–181. [DOI] [PubMed] [Google Scholar]
- 39.Sun Y.Q., Busche T., Rückert C., Paulus C., Rebets Y., Novakova R., Kalinowski J., Luzhetskyy A., Kormanec J., Sekurova O.N., Zotchev S.B.. Development of a biosensor concept to detect the production of cluster-specific secondary metabolites. ACS Synth. Biol. 2017; 6:1026–1033. [DOI] [PubMed] [Google Scholar]
- 40.Gomez-Escribano J.P., Song L., Fox D.J., Yeo V., Bibb M.J., Challis G.L.. Structure and biosynthesis of the unusual polyketide alkaloid coelimycin P1, a metabolic product of the cpk gene cluster of Streptomyces coelicolor M145. Chem. Sci. 2012; 3:2716–2720. [Google Scholar]
- 41.Peano C., Damiano F., Forcato M., Pietrelli A., Palumbo C., Corti G., Siculella L., Fuligni F., Tagliazucchi G.M., De Benedetto G.E.et al.. Comparative genomics revealed key molecular targets to rapidly convert a reference rifamycin-producing bacterial strain into an overproducer by genetic engineering. Metab. Eng. 2014; 26:1–16. [DOI] [PubMed] [Google Scholar]
- 42.Peano C., Talà A., Corti G., Pasanisi D., Durante M., Mita G., Bicciato S., De Bellis G., Alifano P.. Comparative genomics and transcriptional profiles of Saccharopolyspora erythraea NRRL 2338 and a classically improved erythromycin over-producing strain. Microb. Cell Fact. 2012; 11:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simpson I.N., Caten C.E.. Recurrent mutation and selection for increased penicillin titre in Aspergillus nidulans. J. Gen. Microbiol. 1979; 113:209–217. [DOI] [PubMed] [Google Scholar]
- 44.Paddon C.J., Keasling J.D.. Semi-synthetic artemisinin: a model for the use of synthetic biology in pharmaceutical development. Nat. Rev. Microbiol. 2014; 12:355–367. [DOI] [PubMed] [Google Scholar]
- 45.Wang R., Kong F., Wu H., Hou B., Kang Y., Cao Y., Duan S., Ye J., Zhang H.. Complete genome sequence of high-yield strain S. lincolnensis B48 and identification of crucial mutations contributing to lincomycin overproduction. Synth. Syst. Biotechnol. 2020; 5:37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maslowska K.H., Makiela-Dzbenska K., Fijalkowska I.J.. The SOS system: a complex and tightly regulated response to DNA damage. Environ. Mol. Mutagen. 2019; 60:368–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pohlhaus J.R., Long D.T., O’Reilly E., Kreuzer K.N.. The epsilon subunit of DNA polymerase III Is involved in the nalidixic acid-induced SOS response in Escherichia coli. J. Bacteriol. 2008; 190:5239–5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Challis G.L.Exploitation of the Streptomyces coelicolor A3(2) genome sequence for discovery of new natural products and biosynthetic pathways. J. Ind. Microbiol. Biotechnol. 2014; 41:219–232. [DOI] [PubMed] [Google Scholar]
- 49.Horbal L., Fedorenko V., Luzhetskyy A.. Novel and tightly regulated resorcinol and cumate-inducible expression systems for Streptomyces and other actinobacteria. Appl. Microbiol. Biotechnol. 2014; 98:8641–8655. [DOI] [PubMed] [Google Scholar]
- 50.Muth G.The pSG5-based thermosensitive vector family for genome editing and gene expression in actinomycetes. Appl. Microbiol. Biotechnol. 2018; 102:9067–9080. [DOI] [PubMed] [Google Scholar]
- 51.Rutledge P.J., Challis G.L.. Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat. Rev.Microbiol. 2015; 13:509–523. [DOI] [PubMed] [Google Scholar]
- 52.Zhang. J., Jensen M.K., Keasling J.D.. Development of biosensors and their application in metabolic engineering. Curr. Opin. Chem. Biol. 2015; 28:1–8. [DOI] [PubMed] [Google Scholar]
- 53.Will W.R., Fang F.C.. The evolution of MarR family transcription factors as counter-silencers in regulatory networks. Curr. Opin. Microbiol. 2020; 55:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hauf S., Möller L., Fuchs S., Halbedel S.. PadR-type repressors controlling production of a non-canonical FtsW/RodA homologue and other trans-membrane proteins. Sci. Rep. 2019; 9:10023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fernandez-López R., Ruiz R., de la Cruz F., Moncalián G.. Transcription factor-based biosensors enlightened by the analyte. Front. Microbiol. 2015; 6:648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oberhofer M., Hess J., Leutgeb M., Gössnitzer F., Rattei T., Wawrosch C., Zotchev S.B.. Exploring actinobacteria associated with rhizosphere and endosphere of the native alpine medicinal plant Leontopodium nivale subspecies alpinum. Front. Microbiol. 2019; 10:2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All DNA sequencing data is available via NCBI/ANE/DDBJ linked under BioProject PRJNA699418, the complete genome sequence of S. lividans TK24-YQS040 is accessible via BioProject PRJNA713129.