
Figure 3.
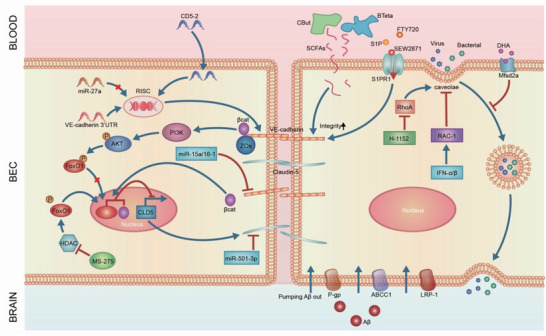
Restoration of BBB integrity as a therapeutic target. 1) Adherens and tight junction molecules. VE‐cadherin is the major adherens junction molecule. VE‐cadherin adhesion prevents FOXO1 accumulation in the nucleus to regulate claudin‐5 expression. Loss of VE‐cadherin leads to FOXO1 and β‐catenin translocation to nucleus, leading to repression of claudin‐5 transcription. CD5‐2, a target site blocker, binds to the binding site of miR‐27a to prevent its regulation of VE‐cadherin, leading to specific upregulation in VE‐cadherin expression and downstream claudin‐5. Claudin‐5 and ZO‐1 are major tight junction molecules. Deletion or inhibition of microRNAs, which target claudin‐5, restores the BBB. MS‐275, an inhibitor of HDAC, suppresses FOXO1 activity to rescue claudin‐5 expression. SCFAs, produced in the colon by bacteria, regulate occludin and claudin‐5 in the BBB in mice with depression. Treatment with bacteria such as C. tyrobutyricum (CBut) or B. thetaiotaomicron (BTeta) may serve as a new strategy to rescue BBB integrity. 2) Transcytosis. Bacterial and viral pathogens employ caveolae‐mediated transcytosis to escape lysosomal degradation and cross the BBB, leading to BBB breakdown. Inhibition of RhoA by its inhibitor H‐1152 or activation of Rac‐1 by interferons (IFN), including IFN‐α or IFN‐β, significantly reduced transcytosis across brain endothelial cells. Mfsd2a is a key molecule that inhibits transcytosis in the BBB. The lipid composition of brain endothelial cells and Msfd2a serve as key players in the regulation of transcytosis and may constitute targets to modulate transcytosis in the BBB for therapeutic purposes. 3) Efflux transporters. Lower efflux transporter expression and activity results in abnormal BBB function, contributing to neurological disease. In AD, LRP‐1 and ABC efflux transporter like P‐gp and ABCC1 are deficient, causing insufficient Aβ clearance and AD pathologies. Pharmacological rescue of these efflux transporters has been shown to alleviate AD in mice.