

ABSTRACT
Chikungunya virus (CHIKV) is a mosquito-borne alphavirus that causes a debilitating febrile illness characterized by persistent muscle and joint pain. The widespread distribution of transmission-competent vectors, Aedes species mosquitoes, indicates the potential risk of large-scale epidemics with high attack rates that can severely impact public health globally. Despite this, currently, there are no antivirals available for the treatment of CHIKV infections. Thus, we aimed to identify potential drug candidates by screening a chemical library using a cytopathic effect-based high-throughput screening assay. As a result, we identified radicicol, a heat shock protein 90 (Hsp90) inhibitor that effectively suppressed CHIKV replication by blocking the synthesis of both positive- and negative-strand viral RNA as well as expression of viral proteins. Interestingly, selection for viral drug-resistant variants and mutational studies revealed nonstructural protein 2 (nsP2) as a putative molecular target of radicicol. Moreover, coimmunoprecipitation and in silico modeling analyses determined that G641D mutation in the methyltransferase (MT)-like domain of nsP2 is essential for its interaction with cytoplasmic Hsp90β chaperone. Our findings collectively support the potential application of radicicol as an anti-CHIKV agent. The detailed study of the underlying mechanism of action further contributes to our understanding of virus-host interactions for novel therapeutics against CHIKV infection.
KEYWORDS: Chikungunya virus, nsP2, radicicol, Hsp90, Hsp90 inhibitor, antiviral
INTRODUCTION
Chikungunya virus (CHIKV) is an arthropod-borne virus transmitted mainly by Aedes spp. mosquitoes that belongs to the genus Alphavirus in the family Togaviridae. It has a positive-sense, single-stranded RNA genome of approximately 12 kb in size (1). The genome reads in two open reading frames (ORF), encoding four nonstructural proteins (nsPs) and five structural proteins (2, 3). CHIKV was first identified during an outbreak in 1952 in present-day Tanzania. Since its discovery, CHIKV remained endemic in tropical areas of Africa, the Indian subcontinent, and Southeast Asia (4, 5). In 2013, CHIKV emerged in the Caribbean, causing more than one million suspected infections, followed by spreading into Central, South, and North America and worldwide (6–10). Its widespread distribution and transmission-competent vectors indicate the potential risk of large-scale epidemics with high attack rates that can severely impact public health globally. The CHIKV infection causes a febrile illness characterized by high fever, skin rash, long-lasting and debilitating polyarthralgia, and myalgia (8). In a large fraction of patients, joint and muscle pain can persist for several months to years, causing considerable suffering and economic loss (11). However, no specific treatment or preventive measures are currently available that can effectively control CHIKV outbreaks.
To address the urgent need for the identification and development of antivirals against CHIKV, we screened a library composed of highly purified compounds with defined chemical structures using a cytopathic effect (CPE)-based high-throughput screening (HTS) assay. Several hits selected from the primary screen were subjected to dose-response curve analysis to confirm the antiviral activity against CHIKV. As a result, we identified radicicol, a heat shock protein 90 (Hsp90) inhibitor, as a novel CHIKV inhibitor. The Hsp90s are highly abundant and conserved molecular chaperones essential for maintaining cellular homeostasis by promoting the folding, maturation, and activation of its client proteins and enabling them to carry out their biological functions (12–15). They are also necessary for the proper function of many multiprotein complexes (16–18).
The Hsp90 family chaperones have recently received significant attention because of their implication in viral life cycles and host stress responses (19, 20). In general, virus infection often induces high expression of intracellular stress-related proteins, including Hsp90, necessary for proper folding and assembly of viral proteins at various stages of the viral life cycle (12, 21). There is a growing body of evidence to suggest that Hsp90 is a crucial cellular factor associated with viral propagation, pathogenesis, or the host immune response (20, 22–25). The dependency of viruses on Hsp90 appears to be almost universal. The antiviral effects of Hsp90 inhibition have been confirmed for a broad range of both DNA and RNA viruses, including hepatitis B virus, human immunodeficiency virus type 1, herpes simplex virus 1, human cytomegalovirus, vesicular stomatitis virus, mumps virus, poliovirus, rubella virus, hepatitis C virus, Ebola virus, and influenza virus (12, 17, 26–34). Similarly, several studies have shown that Hsp90 is required for CHIKV replication, and the Hsp90 inhibitors, such as geldanamycin and its derivatives, can reduce CHIKV infection (35–37).
In the present study, we further investigated the antiviral properties of radicicol and its mechanism of action in CHIKV infection. Much of our effort focused on better understanding the significance of Hsp90 in the CHIKV life cycle by examining the molecular interaction of viral nsPs with Hsp90 family proteins. Our data revealed that radicicol targets the methyltransferase (MT)-like domain of nsP2, which physically interacts with a constitutively expressed cytoplasmic Hsp90β. Mutations of residues present in the predicted binding interface confer resistance to radicicol and contribute to the understanding of its mechanism of action and virus-host interactions.
RESULTS
Identification of radicicol as a novel inhibitor of CHIKV infection.
To identify antiviral compounds against CHIKV, we performed a CPE-based antiviral screening assay using a chemical library obtained from Korea Chemical Bank (Daejeon, Republic of Korea). The primary screen of 11,478 compounds (final concentration of 20 μM) identified 371 small molecules with anti-CHIKV activity above the cutoff (>34.8% inhibition), with a hit rate of 3.2%. The average Z′ score during the primary screen was 0.9 ± 0.05, suggesting that the assay was robust and reliable for detecting potential CHIKV inhibitors (see Fig. S1 in the supplemental material). Further confirmation of the antiviral activity of selected compounds against CHIKV infection was performed by testing them in an 8-point dose-response analysis (Fig. 1A and Table S1). The hit molecule, tanespimycin, a derivative of the antibiotic geldanamycin with known anti-CHIKV activity, was identified, confirming the validity of our screening system. Notably, radicicol, an Hsp90 inhibitor, showed the most prominent antiviral activity among hit molecules, with an estimated 50% effective concentration (EC50) of 0.04 ± 0.01 μM and a 50% cytotoxic concentration (CC50) of 6.44 ± 0.54 μM (Fig. 1B). Thus, radicicol was selected for further analysis based on its significant inhibition of CHIKV infection and its novel status as an antiviral. Next, to confirm its antiviral activity, we evaluated the effect of radicicol on the production of CHIKV infectious progeny virus by plaque assay. Interestingly, radicicol effectively reduced the infectious CHIKV titer in a dose-dependent manner with a maximal reduction of 3-log10 PFU/ml when cells were treated with 2 μM radicicol (Fig. 1C). Similarly, a plaque reduction assay also showed a dose-dependent inhibition of plaque formation in infected cells with the complete abrogation of plaques with 0.2 μM and 2 μM radicicol treatment (Fig. 1D). Taken together, the data confirmed that radicicol effectively inhibits in vitro CHIKV infection.
FIG 1.

Radicicol, an Hsp90 inhibitor, strongly inhibits CHIKV infection. (A) Flow chart of the compound screening process for the identification of CHIKV antivirals. (B) The antiviral activity of radicicol was determined by dose-response curve (DRC) analysis in CHIKV-infected Vero76 cells. Cells were mock-infected (triangles) or infected with CHIKV (circles) at an MOI of 0.1 in the presence of various concentrations of radicicol. At 48 hpi, cell viability was measured using the CellTiter 96 AQueous One Solution cell proliferation assay. (C and D) Reduction of CHIKV infection by radicicol in Vero76 cells as determined by measuring infectious viral titers from cell culture supernatant and formation of viral plaques. Data represent means (± standard deviation [SD]) of at least two independent experiments performed in duplicate. Statistical analyses were determined using paired Student’s t tests, and significant differences are indicated by * (P < 0.05).
Radicicol inhibits early stages of the CHIKV replication cycle.
Next, we conducted time-of-addition studies to evaluate step(s) of the CHIKV life cycle suppressed by radicicol. Virus attachment was performed at 4°C in the presence (cotreatment) or absence of radicicol for 1 h, followed by incubation at 37°C to allow virus internalization. After the temperature-shift, radicicol was added at 0, 1, 4, and 8 h postinfection (hpi) (posttreatment), and cell culture supernatants were collected at 24 hpi to measure infectious viral titers by plaque assay (Fig. 2A). As a result, a maximum of 3-log10 PFU/ml reduction in viral titers was observed when radicicol was added during virus adsorption and at 1 hpi. The infectious viral titers increased gradually when the compound was added at 4 h or 8 hpi, suggesting that the compound possibly interferes with initial postentry steps of the CHIKV life cycle, such as viral genome replication (Fig. 2B). To support this, the effect of radicicol on viral RNA synthesis was further evaluated in CHIKV-infected cells treated with radicicol for 6 and 12 hpi. The immunofluorescence analysis showed that radicicol suppresses CHIKV-induced double-stranded RNA (dsRNA) production. In dimethyl sulfoxide (DMSO)-treated control cells, dsRNA synthesis was detected at 6 hpi and it increased notably by 12 h after CHIKV infection. In contrast, CHIKV induced-dsRNA production was completely inhibited in radicicol-treated cells at 6 hpi and 12 hpi (Fig. 2C). Similarly, radicicol treatment caused a significant reduction in both negative- and positive-strand RNAs at 6 hpi, which was even more pronounced at 12 hpi, suggesting that radicicol inhibits a step of the CHIKV life cycle that occurs before the production of genomic and subgenomic RNAs (Fig. 2D).
FIG 2.

Radicicol inhibits CHIKV RNA genome replication. (A) Schematic representation of the time-of-addition experiment. (B) Vero cells were treated with 2 μM radicicol or 0.5% DMSO (vehicle control) during virus adsorption (co) and added to infected cells immediately after the temperature shift to allow synchronous entry and infection (0 h), at 1 h after entry (1 h), and at 4 h or 8 h postinfection. At 24 hpi, cell culture supernatants were collected for virus titration using a plaque assay. (C) Immunofluorescence assay for detection of dsRNA in radicicol-treated cells. Vero76 cells infected with CHIKV at an MOI of 10 were treated with 2 μM radicicol. At 6 and 12 hpi, cells were fixed, and the nuclei were counterstained with DAPI (blue). The viral RNA was analyzed by detecting dsRNA (green) under a fluorescence microscope. Scale bar = 100 μm. (D) RT-qPCR analysis of positive- and negative-strand CHIKV RNA genomes in radicicol-treated cells. Total RNA was extracted from CHIKV-infected cells treated with 2 μM radicicol at 6 and 12 hpi. Intracellular viral RNA levels (nsP3 gene) were normalized to β-actin mRNA. Data represent the means (±SD) of at least two independent experiments performed in duplicate. Statistical analyses were determined using paired Student’s t tests, and significant differences are indicated by * (P < 0.05).
Inhibition of Hsp90 by radicicol reduces expression of CHIKV nonstructural proteins.
To determine whether radicicol interferes with the functions of nsPs responsible for transcription of viral RNAs, the HEK293T cells infected with CHIKV were treated with increasing radicicol concentrations and subjected to Western blot analysis. As shown in Fig. 3A, the expression levels of viral proteins, nsP1, nsP2, and capsid, were significantly reduced with increasing concentrations of radicicol treatment, whereas the expression levels of the indicated Hsp90 family proteins remained unaltered. Next, the effect of radicicol on the expression of nsPs was evaluated with the treatment of HEK293T cells transiently expressing green fluorescent protein (GFP)-tagged nsP1, nsP2, nsP3, and nsP4 proteins. The cells expressing GFP were counted and analyzed using the IncuCyte live-cell imaging system. As a result, radicicol treatment caused a significant reduction of GFP-expression in cells transfected with nsP1-GFP plasmid compared to those of DMSO control cells (Fig. 3B). Expression of GFP alone as a control was not affected by radicicol, indicating the specificity of nsP1. Consistent with live-cell imaging results, Western blot analysis confirmed that expression of nsP1 was significantly reduced, while nsP2, nsP3, and nsP4 expression levels remained unaffected after treatment with radicicol (Fig. 3C). The results together suggested that radicicol inhibits viral replication specifically targeting CHIKV nsPs.
FIG 3.

Radicicol reduces expression of CHIKV nonstructural proteins. (A) HEK293T cells infected with CHIKV at an MOI of 1 were treated with increasing concentrations of radicicol. At 12 hpi, cell lysates were subjected to Western blot analysis using antibodies to nsP1, nsP2, capsid proteins, and Hsp90 family proteins (Hsp90α, Hsp90β, Grp94, and TRAP1). β-actin served as a loading control. (B) HEK293T cells were transfected with GFP-tagged empty control and CHIKV nonstructural protein-expressing plasmids and treated with DMSO or 2 μM radicicol. The GFP expression levels were monitored in real time using Incucyte live-cell analysis (Sartorius, Gottingen, Germany) for 24 h. The percentage of cells expressing GFP was analyzed and normalized to a percentage of object count per mm2 (OC) based on the cell confluence (normalized 100% cell confluence per mm2) pEGFP-empty-transfected cells. Data represent the means (±SD) of at least two independent experiments performed in duplicate. Statistical analyses were determined using paired Student’s t tests, and significant differences are indicated by * (P < 0.05). A representative image is shown. Scale bar = 400 μm. (C) HEK293T cells transfected with GFP-tagged nsPs were treated with DMSO or 2 μM radicicol. At 24 h posttransfection, the cell lysates were subjected to Western blot analysis with anti-GFP and anti-β-actin antibodies.
Hsp90β and TRAP1 are needed to support CHIKV replication.
Given that radicicol is an Hsp90 inhibitor, we evaluated the importance of Hsp90 function during CHIKV replication. As shown in Fig. 3A, radicicol treatment did not alter the expression of stress-inducible Hsp90α, cytoplasmic Hsp90β, glucose-regulated protein 94 (Grp94), or tumor necrosis factor receptor-associated protein 1 (TRAP1). To further identify the critical Hsp90 protein required during CHIKV replication, small interfering RNAs (siRNAs) were used to selectively silence the expression of HSP90α, HSP90β, Grp94, or TRAP1. Briefly, HEK293T cells were transfected with nontargeting (NT) siRNA or a pool of siRNAs targeting each member of the Hsp90 family for 48 h followed by CHIKV infection. At 12 hpi, cell lysates were collected, and viral protein and viral RNA levels were measured by Western blot analysis and reverse transcription-quantitative PCR (RT-qPCR), respectively. Interestingly, siRNA silencing of Hsp90β and TRAP1 transcripts resulted in a significant reduction of nsP1 and nsP2 expression, followed by Hsp90α silencing compared to those of the nontargeted siRNA-transfected controls (Fig. 4A). In contrast, nsP1 and nsP2 expression levels remained unaltered in Grp94 knockdown cells, suggesting that Grp94 is not involved in CHIKV replication (Fig. 4A). Similarly, the RT-qPCR analysis results further indicated that siRNA silencing of Hsp90β transcript showed the highest reduction in CHIKV RNA levels, followed by silencing of TRAP1 (Fig. 4B). In contrast, silencing of Hsp90α and Grp94 transcripts did not cause a decrease in CHIKV RNA levels. Taken together, siRNA knockdown data suggested that Hsp90β and TRAP1, and to a lesser extent, Hsp90α, but not Grp94, play essential roles in CHIKV replication.
FIG 4.

Hsp90β is required for CHIKV replication and interacts with nsP2 protein. Gene silencing of Hsp90 family proteins, Hsp90α, Hsp90β, Grp94, and TRAP1. HEK293T cells were transfected with 100 nM nontargeting (NT) siRNA or siRNA Hsp90α, Hsp90β, Grp94, or TRAP1. At 48 h posttransfection, cells were infected with CHIKV at an MOI of 1. (A and B) After 12 hpi, cell lysates were collected and subjected to (A) Western blot and (B) RT-qPCR analyses. Statistical analyses were determined using paired Student’s t tests, and significant differences are indicated by * (P < 0.05). (C) Immunoprecipitation assay of GFP-tagged CHIKV-nonstructural proteins with Hsp90β. Briefly, GFP only (emptyGFP) and GFP-tagged CHIKV nsPs were expressed in HEK-293T cells for 24 h. At 24 h posttransfection, cell lysates were subjected to coimmunoprecipitation using 1 μg of anti-GFP antibodies. Western blot analysis was performed to determine the interaction between CHIKV nsPs and Hsp90β. Isotype control IgG was used as a negative control to pull down the protein complex.
To further determine the molecular mechanisms by which Hsp90s are involved in CHIKV replication, we assessed the interactions between HSP90 family proteins (Hsp90α, Hsp90β, TRAP1, and Grp94) and CHIKV nsPs. To do this, we evaluated whether Hsp90 proteins are pulled down together with overexpressed GFP-tagged CHIKV nsPs by coimmunoprecipitation (co-IP) assays. Interestingly, only Hsp90β was coimmunoprecipitated (co-IP) with CHIKV nsPs, whereas interaction of Hsp90α, TRAP1, or Grp94 with CHIKV nsPs was not detected (Fig. S2). Importantly, strong interactions of Hsp90β subunits with CHIKV nsP2, nsP3, and nsP4, but not with nsP1, protein were further confirmed by co-IP assays (Fig. 4C). The interactions between nsP2 and nsP4 with Hsp90β were more prominent than that of nsP3. In contrast to previous studies (35, 36), physical interactions between Hsp90α and CHIKV nsPs were not detected under our experimental conditions. GFP alone as a control did not show interaction with either Hsp90α or Hsp90β, indicating that the interaction of Hsp90β is specific to CHIKV nsPs.
Mutations in CHIKV nsP2 cause resistance to radicicol with decreased interaction with Hsp90β.
To gain deeper insight into the antiviral mechanism of radicicol and to facilitate identifying the drug’s putative target, we next pursued identification of the mutation(s) conferring resistance to radicicol. To obtain viruses resistant to radicicol, CHIKV was passaged in the presence of increasing concentrations of the drug, ranging from 0.06 μM to 5 μM. Although we used the same multiplicity of infection (MOI) of 0.1 in each passage, the viral titer gradually increased with several passages, indicating that drug-resistant variants were produced. Next, susceptibility changes to radicicol were assessed by an antiviral assay using viral cultures obtained from passages P0 to P15. As a result, radicicol-resistant variants arose along increasing numbers of passages in the presence of the compound, with a significant shift in EC50 values of approximately 30-fold higher in P15 compared to the parental virus (P0) (Fig. 5A). Subsequent sequencing of nonstructural genes of P15 viruses revealed two mutations, G641D and V740A, in the MT-like domain at the C terminus of nsP2, suggesting that this protein is involved in the radicicol antiviral mechanism (Fig. 5B and C). To verify the causal relationship between the mutations and interaction with Hsp90β, both G641D and V740A mutations were introduced into the wild-type (WT) GFP-tagged nsP2 expression plasmid, resulting in the mut_nsP2_P15 construct. Subsequently, we assessed the physical interaction between CHIKV nsP2 and Hsp90β by a co-IP assay as described previously. As shown in Fig. 5D, the amount of Hsp90β that coimmunoprecipitated with GFP-tagged mut_nsP2_P15 was significantly reduced compared to WT nsP2 protein, suggesting that the MT-like domain of nsP2 protein physically interacts with Hsp90β. Lastly, to further confirm the interaction between nsP2 and Hsp90β in the context of viral infection, we performed the reverse pulldowns using Hsp90β-specific antibody and immunoblotted with anti-CHIKV nsP1 and nsP2 polyclonal antibodies generated in-house (Fig. 5E). Indeed, the results showed that nsP2 coprecipitated with Hsp90β, while the control nsP1 did not coprecipitate with Hsp90β, thus validating the data obtained above from an overexpression system. Taken altogether, the resistant phenotype and genotype of mut_nsP2_P15 suggested that radicicol inhibits CHIKV replication through a specific antiviral mechanism that targets the MT-like domain of nsP2, which physically interacts with constitutively expressed cytoplasmic Hsp90β.
FIG 5.

Radicicol targets the MT-like domain of CHIKV nsP2, which interacts with Hsp90β. (A) The antiviral activity of radicicol against resistant virus strains selected by serial passages was determined. The dose-response curve (DRC) analysis was performed using Vero76 cells infected with radicicol-resistant variants from different passages in the presence of various concentrations of radicicol. At 48 hpi, cell viability was measured using the CellTiter 96 AQueous One Solution cell proliferation assay. (B) Amino acid sequence alignment of radicicol-resistant virus (CHIKV P15 radicicol). The residues substituted in the radicicol-resistant virus (G641D and V740A) are shown. (C) Schematic of the organization of the CHIKV genome encoding nsP1-4 (top) and the nsP2 protein. The residue substitutions are indicated. (D) Immunoprecipitation assay of GFP-tagged wild-type nsP2 (WT nsP2GFP) or GFP-tagged mutant (mut-nsP2GFP) nsP2 with Hsp90β. Briefly, GFP only (emptyGFP), WT nsPGFP, or mut-nsP2GFP were expressed in HEK-293T cells for 24 h. At 24 h posttransfection, cell lysates were subjected to coimmunoprecipitation using 2 μg of anti-GFP antibodies. Western blot analysis was performed to determine the interaction between CHIKV nsPs and Hsp90β. Isotype control IgG was used as a negative control to pull down the protein complex. (E) Immunoprecipitation assay of Hsp90β with CHIKV nsP1 or CHIKV nsP2 using virus-infected cell lysates. Briefly, HEK293T cells were mock infected or infected with CHIKV at an MOI of 2. At 24 h postinfection, cell lysates were subjected to coimmunoprecipitation using 10 μg of Hsp90β antibodies. Western blot analysis was performed to determine the interaction between Hsp90β and CHIKV nsP1 and nsP2. Isotype control mouse IgG2a was used as a negative control to pull down the protein complex.
Molecular modeling predicts that CHIKV nsP2 G641 has a higher binding affinity to Hsp90β.
To explore the structural basis of CHIKV nsP2 and Hsp90 interaction, we performed protein-protein docking and molecular dynamics (MD) simulations. As shown in Fig. 6A, WT CHIKV-nsP2 (wtCHIKV-nsP2) binds to Hsp90 complexed with radicicol around the ligand-binding entrance. The binding mode of wtCHIKV-nsP2 with Hsp90 remained similar over 500 ns of simulation time relative to the starting structure, suggesting its preferable stable binding mode (Fig. S3). In contrast, mutant CHIKV-nsP2 (mutCHIKV-nsP2; G641D and V740A) showed conformational changes after the systems reached equilibrium within 200 ns MD simulations (Fig. 6B). Furthermore, the MD simulations indicated that mutCHIKV-nsP2 was no longer tightly bound to Hsp90 complexed with radicicol resulting from steric repulsion between the D641 residue of mutCHIKV-nsP2 and the D54 residue of Hsp90. These data correlated with the reduced interaction between the mut-nsPGFP and Hsp90β shown earlier by coimmunoprecipitation assay (Fig. 5D). Taken together, the wtCHIKV-nsP2 with the G641 residue exhibited relatively higher complex stability with Hsp90 than the mutCHIKV-nsP2 throughout MD simulation, suggesting the importance of this residue for the complex binding.
FIG 6.
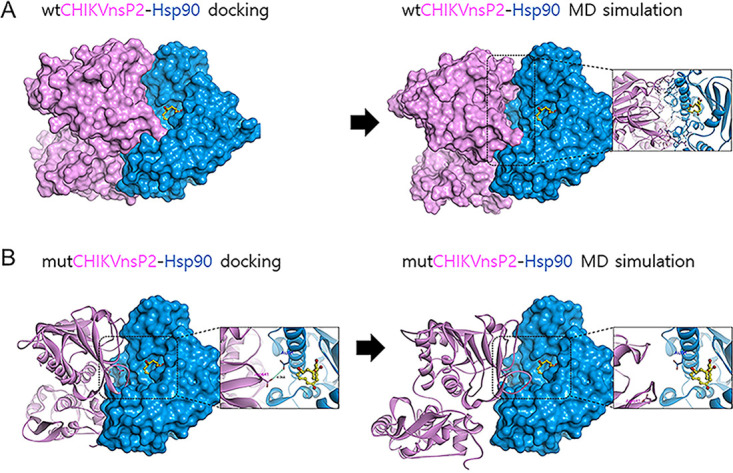
Proposed CHIKV nsP2-Hsp90 complex models. (A and B) The superimposition of protein-protein docking modes of (A) wtCHIKV nsP2-Hsp90 and (B) mutCHIKV nsP2-Hsp90 over 500 ns of simulation time. Wild-type CHIKV nsP2 exhibits more stable conformation than mutant CHIKV nsP2. CHIKVnsP2, pink ribbon and surface model; Hsp90, blue ribbon and surface model; Asp641 in mutCHIKVnsP2, pink stick model; Asp54 in Hsp90, blue stick model; radicicol, yellow ball and stick model.
DISCUSSION
In this study, we demonstrated the antiviral activity of radicicol against CHIKV infection, identified by screening a chemical library using a CPE-based HTS assay. Our data revealed that radicicol effectively inhibits CHIKV replication and the production of infectious viral particles in a dose-dependent manner. Radicicol is an antifungal macrolactone antibiotic, originally isolated from the fungus Monosporium bonorden, specifically targeting Hsp90 chaperone proteins (38). Similar to benzoquinone ansamycins, such as geldanamycin, it competitively binds to the N-terminal ATP/ADP-binding pocket region of Hsp90 proteins, preventing its conformational change that renders the chaperone functionally inactive (39, 40).
Here, we aimed to characterize the antiviral mechanism of radicicol associated with Hsp90-mediated regulation of CHIKV replication. Given a significant reduction of viral RNA levels affecting the synthesis of both positive- and negative-strand RNAs upon radicicol treatment (Fig. 2C and D), we focused our studies on the nonstructural proteins of CHIKV. Using siRNA-mediated gene silencing, we confirmed the dependency on Hsp90 family proteins for CHIKV replication. We observed that the depletion of Hsp90β and TRAP1 resulted in a significantly decreased viral RNA and protein levels, suggesting that these Hsp90s are essential for efficient CHIKV replication. The dependency on Hsp90β for CHIKV replication was further recapitulated by co-IP assays showing direct interactions between Hsp90β and nsP2, nsP3, and nsP4. However, these results were somewhat different from previous studies that reported interactions of nsP2 (35), nsP3, and nsP4 (36) proteins with the counterpart isoform Hsp90α, rather than Hsp90β. The discrepant results could be due to different CHIKV strains, cell types, or experimental conditions employed in each study. We cannot entirely rule out the possibility that the N-terminal tags on the recombinant nsPs used in our co-IP experiments might have influenced the properties of the proteins. Furthermore, radicicol-resistant variants and the mutational study revealed two mutations, G641D and V740A, located in the C-terminal domain of nsP2 that conferred resistance to radicicol, which are particularly important for binding to its chaperone, further supporting the interaction of nsP2 with Hsp90β (Fig. 5D). Consistent with the results obtained from the overexpression system, the interaction between nsP2 and Hsp90β was also confirmed during viral infection by coimmunoprecipitation (Fig. 5E). In addition, protein-protein docking analysis coupled with MD simulation predicted a stable conformation of the nsP2 and Hsp90 complex, whereas the D641 of the mutant nsP2 exhibited a longer distance with Hsp90β D54 via steric repulsion, suggesting the critical role of amino acid position 641 for its interaction with Hsp90β. In contrast, nsP2 V740 residue was predicted to be positioned on the opposite side of the Hsp90β binding interface. The significance of V740A mutation in CHIKV replication is unclear; however, our data together provide additional support to the overall importance of the nsP2 MT-like domain in viral replication and host-cell interaction (41–45). Interestingly, Stapleford et al. reported that the nsP2 G641D variant, induced by RNA mutagens (e.g., ribavirin and 5-fluorouracil [5-FU]), possesses increased replication fidelity and, together with nsP4, further augments replication complex fidelity, supporting the functional linkage of these two proteins (46). It would be interesting to test whether radicicol suppresses CHIKV replication by adding selective pressure for intrinsically higher-fidelity RdRp, similar to RNA mutagens such as ribavirin (47). Given that viral polymerases are the largest class of identified Hsp90 viral client proteins (12), it would be possible that inhibition of Hsp90 function also induces changes in stability or physical interactions of the nsP4 with the viral RNA genome and/or other nsPs in the replication complex, which compromises efficient viral genome replication. However, the molecular basis of nsP4 recognition by Hsp90 remains to be fully understood.
Interestingly, our data consistently showed that radicicol treatment induces a significant reduction of nsP1, more than nsP2, in virus-infected cells as well as in cells overexpressing individual nsP proteins. Similarly, silencing of Hsp90α, Hsp90β, or TRAP1 mRNAs also reduced the expression of nsP1 protein to a great extent, showing that it could serve as a target of Hsp90-mediated antiviral effect induced by radicicol. Notably, our co-IP assay showed no physical interaction between nsP1 and any tested Hsp90 proteins (Fig. S2), suggesting that nsP1 is not an Hsp90 client protein. Previously, Das et al. indicated that Hsp90-associated Akt protein is phosphorylated during CHIKV infection, which activates the mammalian target of rapamycin (mTOR). In contrast, geldanamycin treatment significantly reduced the amount of phosphorylated Akt and mTOR in infected cells (35). Thus, we speculate that radicicol-mediated Hsp90 inhibition may also suppress mTOR activation, which induces the autophagy lysosomal pathway, resulting in increased degradation of intracellular molecules via macroautophagy and/or microautophagy in bulk, in particular, the molecules that are not associated with chaperones (48, 49). However, the mechanism by which nsP1 is degraded after treatment with radicicol warrants further investigation.
In conclusion, we described radicicol, an Hsp90 inhibitor, as a novel antiviral against CHIKV infection and described the importance of Hsp90s activity for efficient CHIKV replication. We extended this work to demonstrate the molecular interaction of CHIKV nsPs with Hsp90 family proteins, identifying the interaction between nsP2 MT-like domain and Hsp90β. Due to the lack of an infectious cDNA clone of the CHIKV strain used in this study, we could not further confirm the significance of G641D and V740A mutations for binding to Hsp90 protein by reverse genetics. Nevertheless, this work supports the vital role of Hsp90 during CHIKV infection and offers insights into the alternative future development of potential therapeutics to treat the disease. Future work in our laboratory will further investigate the role of mutations in the Hsp90-mediated regulation of CHIKV replication and the regulation of associated host cell response for an in-depth understanding of CHIKV biology, replication, and infection.
MATERIALS AND METHODS
Cells and virus.
African green monkey Vero76 cells (ATCC CRL-1587) and human embryonic kidney HEK293T cells (ATCC CRL-3216) were maintained in minimum essential medium (MEM; HyClone, Utah, USA) and Dulbecco’s modified Eagle’s medium (DMEM; HyClone) supplemented with 5% fetal bovine serum (FBS; Atlas Biologicals, USA) at 37°C in a 5% CO2 incubator. The CHIKV isolate (CHIKV/NCCP43132) obtained from the National Culture Collection for Pathogens, Korea Disease Control and Prevention Agency (Osong, Republic of Korea) was amplified, and viral titers were measured by plaque assay on Vero76 cells.
Chemicals and antibodies.
A chemical library composed of 11,478 structurally diverse and biologically active compounds was obtained from Korea Chemical Bank at Korea Research Institute of Chemical Technology (Daejeon, Republic of Korea). Chloroquine and radicicol were purchased from Sigma-Aldrich (USA). All chemical compounds were dissolved in 100% dimethyl sulfoxide (DMSO; Sigma-Aldrich). Mouse monoclonal antibodies to GFP, Hsp90α, and Hsp90β (Abcam, Cambridge, UK), anti-dsRNA J2 (Scicons, Budapest, Hungary), β-actin (Thermo Fisher Scientific, MA, USA), and anti-CHIKV capsid from mouse ascites (ATCC) were used. Rabbit polyclonal antibodies to GFP (Abcam), Grp94 and TRAP1 (GeneTex, California, USA), and anti-Hsp90β and anti-GFP rabbit monoclonal antibodies (Cell Signaling Technology, Massachusetts, USA) were used. The rabbit anti-CHIKV-nsP1 and -nsP2 polyclonal sera were raised and purified in-house, as described previously (50). Anti-mouse and anti-rabbit Alexa Fluor 488 (AF488) and horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Thermo Fisher Scientific (Invitrogen, California, USA).
Plasmids.
The plasmids encoding recombinant viral proteins were generated following molecular cloning techniques. The CHIKV nsP1, nsP2, nsP3, and nsP4 genes were PCR amplified from tissue culture fluid-derived CHIKV genomic RNA using the primer pairs listed in Table S2. The individual PCR products were cloned into pEGFPC1 vector (Takra Bio USA, Inc., California, USA), resulting in pEGFP-nsP1, -nsP2, -nsP3, and nsP4 expression plasmids with an N-terminal enhanced GFP (eGFP)-tag. The plasmids were verified by DNA sequencing (Bioneer, Daejeon, Republic of Korea). The CHIKV nsP2 gene containing G641D and V740A mutations was synthesized and cloned into a pEGFP-C1 plasmid. The sequence of pEGFP encoding the nsP2 mutant gene was verified by DNA sequencing (Bioneer).
Primary antiviral screening assay.
A CPE-inhibition assay using a chemical library was performed to screen inhibitors of CHIKV as previously described with some modifications (51). Briefly, Vero76 cells were seeded in 96-well plates at a density of 2 × 104 cells/well. After overnight incubation, equal volumes of CHIKV inoculum (multiplicity of infection [MOI], 0.1) and 20 μM compound dilutions were added and incubated at 37°C in a 5% CO2 incubator. At 48 h postinfection (hpi), cell viability was measured using the CellTiter 96 AQueous One Solution cell proliferation assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Absorbance at 490 nm was measured using a Synergy H1 multimode microplate reader (BioTek, Winooski, VT, USA). Mock-infected cells treated with 0.5% DMSO (cell control) and 100 μM chloroquine-treated cells were used as controls. The antiviral activity of hit compounds was determined from the dose-response curve (DRC). The 50% effective concentration (EC50) and the compound-specific toxicity (50% cytotoxic concentration [CC50]) were calculated with Prism 8 (GraphPad Software, California, USA) using the nonlinear regression formula as follows: log (inhibitor) versus response-variable slope (4 parameters) model.
Plaque reduction assay.
Confluent Vero76 cell monolayers in 12-well tissue culture plates were infected with CHIKV at an MOI of 0.002 in the absence or presence of radicicol. The virus was adsorbed for 1 h at 37°C, and the cell monolayers were washed twice and maintained in an overlay medium (MEM containing 0.75% carboxymethylcellulose) containing radicicol. After incubation for 24 h at 37°C, the cell monolayers were fixed and stained with crystal violet solution for plaque counting.
Time-of-addition assay.
A day before infection, Vero76 cells were seeded in 12-well tissue culture plates at a density of 2 × 105 cells/well. The next day, confluent cell monolayers were inoculated with CHIKV at an MOI of 1 for 1 h on ice. The unadsorbed virus was removed by washing twice with ice-cold phosphate-buffered saline (PBS) and replenished with a fresh culture medium. Subsequently, plates were shifted to 37°C to allow synchronous entry and infection. Cell monolayers were treated with 2 μM radicicol from adsorption (virus attachment/binding) and onward or were added at specific time points during 37°C incubation. To assess the antiviral activity of radicicol, the amount of infectious viral particles in the cell culture supernatants after 24 hpi was measured by plaque assay expressed as PFU/ml.
Reverse transcription-quantitative PCR (RT-qPCR).
For quantification of intracellular mRNA and viral RNA, total cellular RNA was extracted using the RNeasy minikit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The positive- and negative-strand viral RNAs were reverse-transcribed, followed by amplification with nsP3-F/nsP3-R and Tag-F/nsP3-R primers, respectively, as described previously (52), using the SuperScript III first-strand synthesis system (Invitrogen) and SsoFast EvaGreen supermix (Bio-Rad, California, USA). The cycling conditions were 95°C for 5 min, followed by 40 cycles of 95°C for 15 s and 60°C for 30 s. For quantification of Hsp90α, Hsp90β, Grp94, and TRAP1 mRNA expression, the RT-qPCR was performed using specific primer pairs for each gene (Table S1) and the QuantiTect SYBR Green RT-PCR kit (Qiagen). The cycling condition was 50°C for 30 min and 95°C for 15 min, followed by 35 cycles of 94°C for 15 s, 56°C for 30 s, and 72°C for 30 s. The β-actin gene (housekeeping gene) and the QuantStudio 6 Flex real-time PCR system (Applied Biosystems, California, USA) were used in this study. Specificity verification of the amplified PCR products was confirmed by melt curve analysis.
Immunofluorescence assay.
An immunofluorescence assay (IFA) was performed to evaluate the effect of radicicol on the double-stranded RNA (dsRNA) in infected Vero76 cells. Briefly, Vero76 cells were infected with CHIKV at an MOI of 10 in the presence of 2 μM radicicol or DMSO at 37°C. After 6 hpi and 12 hpi, fixed cells were incubated with anti-dsRNA IgG2 for 1 h at room temperature, followed by incubation with AF488-conjugated goat anti-mouse IgG antibodies for 1 h at room temperature. Nuclei were counterstained using mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, California, USA). Image analysis was performed using a confocal microscope (Zeiss LSM 710; Zeiss, Oberkochen, Germany).
Plasmid transfection.
HEK293T cells were transfected with pEGFP-nsP1, pEGFP-nsP2, pEGFP-nsP3, pEGFP-nsP4, or control plasmid (pEGFP-C1) using Lipofectamine 2000 (Thermo Fisher Scientific) and treated with 2 μM radicicol or DMSO for 24 h. Expression levels of GFP-tagged CHIKV nsPs were monitored in real time using Incucyte live-cell analysis (Sartorius, Gottingen, Germany). At 24 h posttransfection, cell counts expressing GFP were analyzed and normalized to a percentage of object count per mm2 (OC) based on the cell confluence (normalized 100% cell confluence per mm2) and pEGFP-empty-transfected cells. The expression levels of GFP-tagged nsPs from radicicol-treated cells were also evaluated by Western blot analysis.
Western blot analysis.
Sample lysates were prepared using M-PER buffer (Thermo Fisher Scientific), and the protein concentration was measured using a bicinchoninic acid (BCA) assay (Pierce BCA protein assay kit; Thermo Fisher Scientific). Subsequently, equal amounts of samples were resolved by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene difluoride (PVDF) membrane (Merk Millipore, MA, USA). Immunoblotting was performed using primary antibodies and secondary antibodies conjugated with horseradish peroxidase (HRP). Anti-β-actin antibody was used to detect β-actin, a loading control. The detection was performed using a chemiluminescent substrate SuperSignal West Pico PLUS (Thermo Fisher Scientific) and LAS-3000 imager (Fuji, Tokyo, Japan).
Gene knockdown using siRNA.
The siRNA complexes targeting Hsp90AA1 (Hsp90α), Hsp90AB1 (Hsp90β), Hsp90B1 (Grp94), and TRAP1 (TRAP1) mRNAs and the control siRNA were purchased from Bioneer. siRNA-mediated gene knockdown was performed using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s instructions. Efficient knockdown of the targets was monitored by Western blot analysis at 48 h after siRNA transfection.
Coimmunoprecipitation.
HEK293T cells were transfected with pEGFP-nsP1, pEGFP-nsP2, pEGFP-nsP3, pEGFP-nsP4, pEGFP-nsP2mut, or control pEGFP-C1 plasmids as described above. At 24 h posttransfection, cells were lysed using Pierce IP lysis/wash buffer, and the cell lysates were clarified by centrifugation at 13,000 rpm for 10 min at 4°C. For the pulldown of protein samples, 900 μg of total protein of each precleared supernatant was incubated with 1 μg of polyclonal anti-GFP antibody (Abcam) or normal rabbit IgG (Cell Signaling Technology) at 4°C overnight with constant mixing using an end-over-end rotor. For reverse pulldowns using virus-infected cells lysates, HEK293T cells were mock infected or infected with CHIKV at an MOI of 2. After 12 hpi, cell lysates were prepared as described above. For immunoprecipitation of protein samples, 900 μg of total protein was incubated with 10 μg of monoclonal Hsp90β (Abcam) or mouse IgG2a isotype control antibody (Cell Signaling Technology) at 4°C overnight. The immune complexes formed were pulled out using Pierce protein A/G magnetic beads (Pierce classic magnetic IP/co-IP kit; Thermo Fisher Scientific). Finally, beads were collected using a magnetic stand and washed twice with Pierce IP lysis/wash buffer, followed by elution of immune complexes in molecular-grade nuclease-free water. Eluted samples were analyzed by SDS-PAGE and Western blotting assays.
Resistance selection and phenotyping of resistant CHIKV variants.
For generation of radicicol-resistant viruses, wild-type CHIKV (P0) was inoculated at an MOI of 0.1 in Vero76 cells in the presence of increasing concentrations of radicicol, ranging from 0.06 μM to 5 μM, in a 12-well tissue culture plate. Then, 3 days postinfection, the supernatants from cultures with >50% cytopathic effect under drug pressure were used to infect new cell monolayers in the presence of increasing concentrations of radicicol. The procedure was repeated for 15 consecutive passages until a drug-resistant virus was obtained. In parallel, the DMSO-treated virus was passaged along and used as a reference. The virus titers from each passage were determined by plaque assay. The resistance phenotype was determined by antiviral assay with increasing concentrations of radicicol (DRC) as described above. The genotype of each viral passage was determined by sequencing the P1234 region of CHIKV variant genomes.
Protein-protein docking, modeling, and molecular dynamics simulations.
Rigid-body protein-protein docking between CHIKV nsP2 and Hsp90 was simulated using the ZDOCK protocol in the Discovery Studio 2020 (Dassault Systèmes, California, USA). The crystal structures of CHIKV nsP2 (PDB ID: 3RTK) and Hsp90 bound to radicicol (PDB ID: 4EGK) were obtained from the Protein Data Bank (https://www.rcsb.org), and two protein structures were assigned the Chemistry at Harvard Macromolecular Mechanics (CHARMM) force field for protein-protein docking procedures. The top clustered poses of docking models between CHIKV nsP2 and Hsp90 were chosen by visual inspection with high ZDOCK score from the top 2,000 poses. Ten high-scoring and eye-selection docking models were selected by ZDOCK analysis. The mutant of CHIKV nsP2 was generated using the Build Mutant protocol in Discovery Studio 2020. G641D and V740A were set for selected wild-type protein-protein docking models by ZDOCK, and the optimization level was set to high; other parameters were default settings. For further refinement of each ZDOCK pose, molecular dynamics (MD) simulations were performed using Desmond v6.4 with OPLS3e force field in Schrodinger Suite v2020-4 (Schrödinger LLC, New York, USA). The System Builder was used for solvation employing predefined TIP3P water molecules in an orthorhombic box with dimensions of 12 Å by 12 Å by 12 Å, and the overall complex was neutralized by adding Cl− counterions. The NaCl concentration was 0.15 mol/liter. Production of MD simulations at 500 ns in length was carried out under periodic boundary conditions in the NPT ensemble at normal temperatures (300 K) and pressure (101,325 pascals). Recording intervals of 1.2 and 500 ps were used for energy calculation and trajectory analysis. The final proposed wtCHIKV nsP2-Hsp90 and mutCHIKV nsP2-Hsp90 docking models for the equilibrium state of the MD simulations were represented using Discovery Studio 2020.
Statistical analysis.
Statistically significant differences were determined by a one-tailed Student’s t test. The P < 0.005 value was considered statistically significant.
ACKNOWLEDGMENTS
The chemical library used in this study was kindly provided by the Korea Chemical Bank (www.chembank.org) of the Korea Research Institute of Chemical Technology (KRICT), Daejeon, Republic of Korea.
This research was supported by KRICT intramural funding (grant numbers KK2032-20 and KK1803-E00) and Incheon National University Research Grant, 2016. Y.Y.G. was supported by a start-up fund from the City University of Hong Kong, Hong Kong SAR, China.
Footnotes
Supplemental material is available online only.
Contributor Information
Jung-Yong Yeh, Email: yehjy@inu.ac.kr.
Yun Young Go, Email: yunygo@cityu.edu.hk.
REFERENCES
- 1.Strauss JH, Strauss EG. 1994. The alphaviruses: gene expression, replication, and evolution. Microbiol Rev 58:491–562. doi: 10.1128/MR.58.3.491-562.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li L, Jose J, Xiang Y, Kuhn RJ, Rossmann MG. 2010. Structural changes of envelope proteins during alphavirus fusion. Nature 468:705–708. doi: 10.1038/nature09546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Voss JE, Vaney MC, Duquerroy S, Vonrhein C, Girard-Blanc C, Crublet E, Thompson A, Bricogne G, Rey FA. 2010. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 468:709–712. doi: 10.1038/nature09555. [DOI] [PubMed] [Google Scholar]
- 4.Lumsden WH. 1955. An epidemic of virus disease in Southern Province, Tanganyika Territory, in 1952–53. II. General description and epidemiology. Trans R Soc Trop Med Hyg 49:33–57. doi: 10.1016/0035-9203(55)90081-x. [DOI] [PubMed] [Google Scholar]
- 5.Sudeep AB, Parashar D. 2008. Chikungunya: an overview. J Biosci 33:443–449. doi: 10.1007/s12038-008-0063-2. [DOI] [PubMed] [Google Scholar]
- 6.Fischer M, Staples JE, Arboviral Diseases Branch NCfE, Zoonotic Infectious Diseases CDC . 2014. Notes from the field: Chikungunya virus spreads in the Americas—Caribbean and South America, 2013–2014. MMWR Morb Mortal Wkly Rep 63:500–501. [PMC free article] [PubMed] [Google Scholar]
- 7.Leparc-Goffart I, Nougairede A, Cassadou S, Prat C, de Lamballerie X. 2014. Chikungunya in the Americas. Lancet 383:514. doi: 10.1016/S0140-6736(14)60185-9. [DOI] [PubMed] [Google Scholar]
- 8.Weaver SC, Forrester NL. 2015. Chikungunya: evolutionary history and recent epidemic spread. Antiviral Res 120:32–39. doi: 10.1016/j.antiviral.2015.04.016. [DOI] [PubMed] [Google Scholar]
- 9.Robinson MC. 1955. An epidemic of virus disease in Southern Province, Tanganyika Territory, in 1952–53. I. Clinical features. Trans R Soc Trop Med Hyg 49:28–32. doi: 10.1016/0035-9203(55)90080-8. [DOI] [PubMed] [Google Scholar]
- 10.Ross RW. 1956. The Newala epidemic. III. The virus: isolation, pathogenic properties and relationship to the epidemic. J Hyg (Lond) 54:177–191. doi: 10.1017/s0022172400044442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burt FJ, Rolph MS, Rulli NE, Mahalingam S, Heise MT. 2012. Chikungunya: a re-emerging virus. Lancet 379:662–671. doi: 10.1016/S0140-6736(11)60281-X. [DOI] [PubMed] [Google Scholar]
- 12.Geller R, Taguwa S, Frydman J. 2012. Broad action of Hsp90 as a host chaperone required for viral replication. Biochim Biophys Acta 1823:698–706. doi: 10.1016/j.bbamcr.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peterson LB, Blagg BS. 2009. To fold or not to fold: modulation and consequences of Hsp90 inhibition. Future Med Chem 1:267–283. doi: 10.4155/fmc.09.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zuehlke A, Johnson JL. 2010. Hsp90 and co-chaperones twist the functions of diverse client proteins. Biopolymers 93:211–217. doi: 10.1002/bip.21292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiosis G, Vilenchik M, Kim J, Solit D. 2004. Hsp90: the vulnerable chaperone. Drug Discov Today 9:881–888. doi: 10.1016/S1359-6446(04)03245-3. [DOI] [PubMed] [Google Scholar]
- 16.Makhnevych T, Houry WA. 2012. The role of Hsp90 in protein complex assembly. Biochim Biophys Acta 1823:674–682. doi: 10.1016/j.bbamcr.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 17.Naito T, Momose F, Kawaguchi A, Nagata K. 2007. Involvement of Hsp90 in assembly and nuclear import of influenza virus RNA polymerase subunits. J Virol 81:1339–1349. doi: 10.1128/JVI.01917-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prodromou C. 2012. The ‘active life’ of Hsp90 complexes. Biochim Biophys Acta 1823:614–623. doi: 10.1016/j.bbamcr.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shim HY, Quan X, Yi YS, Jung G. 2011. Heat shock protein 90 facilitates formation of the HBV capsid via interacting with the HBV core protein dimers. Virology 410:161–169. doi: 10.1016/j.virol.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 20.Ju HQ, Xiang YF, Xin BJ, Pei Y, Lu JX, Wang QL, Xia M, Qian CW, Ren Z, Wang SY, Wang YF, Xing GW. 2011. Synthesis and in vitro anti-HSV-1 activity of a novel Hsp90 inhibitor BJ-B11. Bioorg Med Chem Lett 21:1675–1677. doi: 10.1016/j.bmcl.2011.01.098. [DOI] [PubMed] [Google Scholar]
- 21.Nagy PD, Wang RY, Pogany J, Hafren A, Makinen K. 2011. Emerging picture of host chaperone and cyclophilin roles in RNA virus replication. Virology 411:374–382. doi: 10.1016/j.virol.2010.12.061. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Jin F, Wang R, Li F, Wu Y, Kitazato K, Wang Y. 2017. HSP90: a promising broad-spectrum antiviral drug target. Arch Virol 162:3269–3282. doi: 10.1007/s00705-017-3511-1. [DOI] [PubMed] [Google Scholar]
- 23.Nakagawa S, Umehara T, Matsuda C, Kuge S, Sudoh M, Kohara M. 2007. Hsp90 inhibitors suppress HCV replication in replicon cells and humanized liver mice. Biochem Biophys Res Commun 353:882–888. doi: 10.1016/j.bbrc.2006.12.117. [DOI] [PubMed] [Google Scholar]
- 24.Chase G, Deng T, Fodor E, Leung BW, Mayer D, Schwemmle M, Brownlee G. 2008. Hsp90 inhibitors reduce influenza virus replication in cell culture. Virology 377:431–439. doi: 10.1016/j.virol.2008.04.040. [DOI] [PubMed] [Google Scholar]
- 25.Geller R, Andino R, Frydman J. 2013. Hsp90 inhibitors exhibit resistance-free antiviral activity against respiratory syncytial virus. PLoS One 8:e56762. doi: 10.1371/journal.pone.0056762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sakata M, Katoh H, Otsuki N, Okamoto K, Nakatsu Y, Lim CK, Saijo M, Takeda M, Mori Y. 2019. Heat shock protein 90 ensures the integrity of rubella virus p150 protein and supports viral replication. J Virol 93:e01142-19. doi: 10.1128/JVI.01142-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katoh H, Kubota T, Nakatsu Y, Tahara M, Kidokoro M, Takeda M. 2017. Heat shock protein 90 ensures efficient mumps virus replication by assisting with viral polymerase complex formation. J Virol 91:e02220-16. doi: 10.1128/JVI.02220-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu J, Anselmo D. 2000. In vitro reconstitution of a functional duck hepatitis B virus reverse transcriptase: posttranslational activation by Hsp90. J Virol 74:11447–11455. doi: 10.1128/jvi.74.24.11447-11455.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Momose F, Naito T, Yano K, Sugimoto S, Morikawa Y, Nagata K. 2002. Identification of Hsp90 as a stimulatory host factor involved in influenza virus RNA synthesis. J Biol Chem 277:45306–45314. doi: 10.1074/jbc.M206822200. [DOI] [PubMed] [Google Scholar]
- 30.Smith DR, McCarthy S, Chrovian A, Olinger G, Stossel A, Geisbert TW, Hensley LE, Connor JH. 2010. Inhibition of heat-shock protein 90 reduces Ebola virus replication. Antiviral Res 87:187–194. doi: 10.1016/j.antiviral.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Waxman L, Whitney M, Pollok BA, Kuo LC, Darke PL. 2001. Host cell factor requirement for hepatitis C virus enzyme maturation. Proc Natl Acad Sci U S A 98:13931–13935. doi: 10.1073/pnas.241510898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ujino S, Yamaguchi S, Shimotohno K, Takaku H. 2009. Heat-shock protein 90 is essential for stabilization of the hepatitis C virus nonstructural protein NS3. J Biol Chem 284:6841–6846. doi: 10.1074/jbc.M806452200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anderson I, Low JS, Weston S, Weinberger M, Zhyvoloup A, Labokha AA, Corazza G, Kitson RA, Moody CJ, Marcello A, Fassati A. 2014. Heat shock protein 90 controls HIV-1 reactivation from latency. Proc Natl Acad Sci U S A 111:E1528–E1537. doi: 10.1073/pnas.1320178111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geller R, Vignuzzi M, Andino R, Frydman J. 2007. Evolutionary constraints on chaperone-mediated folding provide an antiviral approach refractory to development of drug resistance. Genes Dev 21:195–205. doi: 10.1101/gad.1505307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Das I, Basantray I, Mamidi P, Nayak TK, B MP, Chattopadhyay S, Chattopadhyay S. 2014. Heat shock protein 90 positively regulates Chikungunya virus replication by stabilizing viral non-structural protein nsP2 during infection. PLoS One 9:e100531. doi: 10.1371/journal.pone.0100531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rathore AP, Haystead T, Das PK, Merits A, Ng ML, Vasudevan SG. 2014. Chikungunya virus nsP3 & nsP4 interacts with HSP-90 to promote virus replication: HSP-90 inhibitors reduce CHIKV infection and inflammation in vivo. Antiviral Res 103:7–16. doi: 10.1016/j.antiviral.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 37.Nayak TK, Mamidi P, Kumar A, Singh LP, Sahoo SS, Chattopadhyay S, Chattopadhyay S. 2017. Regulation of viral replication, apoptosis and pro-inflammatory responses by 17-AAG during Chikungunya virus infection in macrophages. Viruses 9:3. doi: 10.3390/v9010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delmotte P, Delmotte-Plaque J. 1953. A new antifungal substance of fungal origin. Nature 171:344. doi: 10.1038/171344a0. [DOI] [PubMed] [Google Scholar]
- 39.Pearl LH, Prodromou C. 2006. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu Rev Biochem 75:271–294. doi: 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- 40.Schulte TW, Akinaga S, Soga S, Sullivan W, Stensgard B, Toft D, Neckers LM. 1998. Antibiotic radicicol binds to the N-terminal domain of Hsp90 and shares important biologic activities with geldanamycin. Cell Stress 3:100–108. doi:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garmashova N, Gorchakov R, Frolova E, Frolov I. 2006. Sindbis virus nonstructural protein nsP2 is cytotoxic and inhibits cellular transcription. J Virol 80:5686–5696. doi: 10.1128/JVI.02739-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frolov I, Agapov E, HoffmanTA, Jr, Pragai BM, Lippa M, Schlesinger S, Rice CM. 1999. Selection of RNA replicons capable of persistent noncytopathic replication in mammalian cells. J Virol 73:3854–3865. doi: 10.1128/JVI.73.5.3854-3865.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dryga SA, Dryga OA, Schlesinger S. 1997. Identification of mutations in a Sindbis virus variant able to establish persistent infection in BHK cells: the importance of a mutation in the nsP2 gene. Virology 228:74–83. doi: 10.1006/viro.1996.8364. [DOI] [PubMed] [Google Scholar]
- 44.Weiss B, Rosenthal R, Schlesinger S. 1980. Establishment and maintenance of persistent infection by Sindbis virus in BHK cells. J Virol 33:463–474. doi: 10.1128/JVI.33.1.463-474.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frolov I, Schlesinger S. 1994. Comparison of the effects of Sindbis virus and Sindbis virus replicons on host cell protein synthesis and cytopathogenicity in BHK cells. J Virol 68:1721–1727. doi: 10.1128/JVI.68.3.1721-1727.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stapleford KA, Rozen-Gagnon K, Das PK, Saul S, Poirier EZ, Blanc H, Vidalain PO, Merits A, Vignuzzi M. 2015. Viral polymerase-helicase complexes regulate replication fidelity to overcome intracellular nucleotide depletion. J Virol 89:11233–11244. doi: 10.1128/JVI.01553-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cameron CE, Castro C. 2001. The mechanism of action of ribavirin: lethal mutagenesis of RNA virus genomes mediated by the viral RNA-dependent RNA polymerase. Curr Opin Infect Dis 14:757–764. doi: 10.1097/00001432-200112000-00015. [DOI] [PubMed] [Google Scholar]
- 48.Kaushik S, Cuervo AM. 2018. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol 19:365–381. doi: 10.1038/s41580-018-0001-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmeisser K, Parker JA. 2019. Pleiotropic effects of mTOR and autophagy during development and aging. Front Cell Dev Biol 7:192. doi: 10.3389/fcell.2019.00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar A, Mamidi P, Das I, Nayak TK, Kumar S, Chhatai J, Chattopadhyay S, Suryawanshi AR, Chattopadhyay S. 2014. A novel 2006 Indian outbreak strain of Chikungunya virus exhibits different pattern of infection as compared to prototype strain. PLoS One 9:e85714. doi: 10.1371/journal.pone.0085714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shin JS, Jung E, Kim M, Baric RS, Go YY. 2018. Saracatinib inhibits Middle East respiratory syndrome-coronavirus replication in vitro. Viruses 10:283. doi: 10.3390/v10060283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lani R, Hassandarvish P, Shu MH, Phoon WH, Chu JJ, Higgs S, Vanlandingham D, Abu Bakar S, Zandi K. 2016. Antiviral activity of selected flavonoids against Chikungunya virus. Antiviral Res 133:50–61. doi: 10.1016/j.antiviral.2016.07.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental tables and figures. Download AAC.00135-21-s0001.pdf, PDF file, 0.8 MB (843KB, pdf)