

Abstract
Background -
Tetralogy of Fallot (TOF), the most common cyanotic heart defect in newborns, has evidence of multiple genetic contributing factors. Identifying variants that are clinically relevant is essential to understand patient-specific disease susceptibility and outcomes, and could contribute to delineating pathomechanisms.
Methods -
Using a clinically-driven strategy, we re-analyzed exome sequencing data from 811 probands with TOF, to identify rare loss-of-function and other likely pathogenic variants in genes associated with congenital heart disease (CHD).
Results -
We confirmed a major contribution of likely pathogenic variants in FLT4 (VEGFR3; n=14) and NOTCH1 (n=10), and identified 1–3 variants in each of 21 other genes, including ATRX, DLL4, EP300, GATA6, JAG1, NF1, PIK3CA, RAF1, RASA1, SMAD2, and TBX1. In addition, multiple loss-of-function variants provided support for three emerging CHD/TOF candidate genes: KDR (n=4), IQGAP1 (n=3), and GDF1 (n=8). In total, these variants were identified in 63 probands (7.8%). Using the 26 composite genes in a STRING protein interaction enrichment analysis revealed a biologically relevant network (p-value 3.3e-16), with VEGFR2 (KDR) and NOTCH1 representing central nodes. Variants associated with arrhythmias/sudden death and/or heart failure indicated factors that could influence long-term outcomes.
Conclusions -
The results are relevant to precision medicine for TOF. They suggest considerable clinical yield from genome-wide sequencing, with further evidence for KDR (VEGFR2) as a CHD/TOF gene, and for vascular endothelial growth factor (VEGF) and Notch signaling as mechanisms in human disease. Harnessing the genetic heterogeneity of single gene defects could inform etiopathogenesis and help prioritize novel candidate genes for TOF.
Journal Subject Terms: Genetics, Congenital Heart Disease, Developmental biology
Keywords: congenital heart disease, genetic variation, tetralogy of Fallot, vascular endothelial growth factor receptor, genomics
Introduction
Tetralogy of Fallot (TOF) affects about one in 3000 live births, and is the most common cyanotic heart defect in newborns1, 2. Initially described in 1671 by Danish anatomist Niels Stensen, the four components (pulmonary outflow tract obstruction, aorta overriding both ventricles, ventricular septal defect, and hypertrophy of the right ventricle) represent a single developmental anomaly3, 4. Further detailed anatomical documentation by Arthur Fallot, Maude Abbott, and others, laid the foundation first for palliative, and later corrective, surgical procedures4, 5. Advances in imaging, medical management, and surgeries across the lifespan have transformed TOF from a usually fatal pediatric condition to a chronic disease that is more prevalent in adults than children, with life expectancy into the seventh decade and beyond6–9.
The pursuit of determining the genetic underpinnings and recognizing how these may affect late outcomes in TOF, has proceeded in parallel with these clinical advances10–13. This research began with the recognition of multi-system syndromes in approximately 20% of patients, most commonly caused by 22q11.2 microdeletions, other copy number variants, or aneuploidies14, along with some single-gene defects15. Availability of newer genomic technologies, particularly genome-wide sequencing, has expanded gene discovery studies. The cumulative genetic evidence indicates a pattern of molecular etiology for TOF that is characterized by genetic heterogeneity and some distinction from congenital heart disease (CHD) as a whole14, 16–21. However, there has been relatively limited consideration of the clinical pathogenicity of genetic variants for TOF and translation of findings into the clinic22, 23.
Here, we re-analyzed data from the largest genome-wide sequencing dataset for TOF available, where an initial study had reported that deleterious variants in NOTCH1 and FLT4 surpassed statistical thresholds for genome-wide significance20, 24. By design, genome-wide variant burden analyses generally include abundant variants of uncertain significance, and lack power to detect the rare gene-disease associations which comprise the genetic heterogeneity of a disease and form the basis for defining clinically reportable genetic variants.
The objective of our study was to improve understanding of precisely such variants, i.e., those that are of greatest relevance to clinical practice for patients with TOF. We re-analyzed and re-annotated the raw sequence data files, and used American College of Medical Genetics (ACMG) interpretation guidelines25 to adjudicate variants in genes that were considered relevant for the congenital cardiac phenotype. This re-analysis identified 63 (7.8%) of 811 pediatric TOF probands to have pathogenic/likely pathogenic variants in known CHD genes (n = 49), or loss-of-function variants in emerging CHD/TOF candidate genes (n = 15; Figure 1). As expected given the difference in approach and methodology, few of these variants and genes were reported in the previous study20. The implicated genes encode proteins that functionally interact, indicating that the heterogeneous genetic architecture could inform mechanisms. Other pathogenic/likely pathogenic variants identified add to potential genetic implications for cardiovascular outcomes of TOF.
Figure 1:
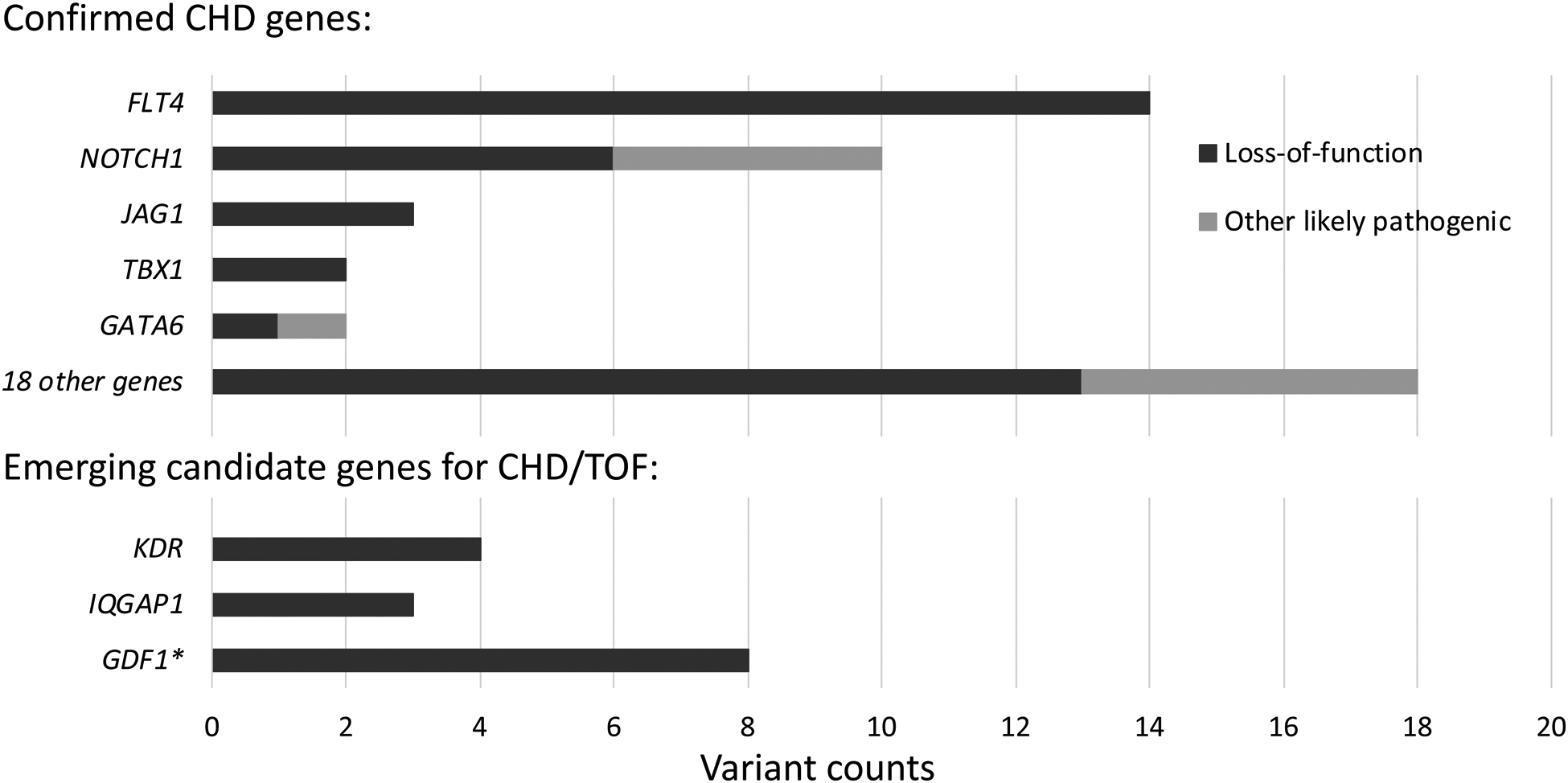
Clinically relevant variants associated with congenital heart disease (CHD), identified in exome sequencing data of n = 811 probands with tetralogy of Fallot (TOF). Likely pathogenic loss-of-function and missense variants meeting ACMG criteria for clinical relevance (pathogenic or likely pathogenic, n = 49) in 23 recognized CHD genes were identified in 48 probands. Loss-of-function variants (n = 15) in 3 emerging candidate genes for CHD/TOF were identified in another 15 probands. Variant and gene details are provided in Supplemental Tables II and III, respectively, and Figure 2 provides details of KDR variants.
* Note: Comprehensive assessment of gene GDF1 was not possible due to insufficient coverage of this locus with the exome sequencing data available.
Methods
Exome sequencing raw data (bam files) have been accessed through the European Genome-phenome Archive (EGA; https://www.ebi.ac.uk/ega), and can be made available to qualified researchers through accession number EGAS00001003302. As described here20, ethical approval had been obtained from the local institutional review boards at each participating centre prior to blood or saliva sample collection, and informed consent had been obtained from all subjects or their parents/legal guardians. The downloaded dataset was analyzed at The Centre for Applied Genomics (TCAG, The Hospital for Sick Children, Toronto, Canada) under a research protocol of the Hospital for Sick Children (REB# 0019980189). Full methods and quality metrics (Supplemental Table I) are available as supplemental data.
Results
Re-analyzing the exome sequencing data from 811 probands with TOF, we identified 48 probands with at least one pathogenic/likely pathogenic variant in a CHD-associated gene (48/811; 5.9%). Five CHD-associated genes had multiple pathogenic/likely pathogenic variants25 (Figure 1). These included 14 likely pathogenic loss-of-function variants in FLT4 (all loss-of-function), and 10 in NOTCH1 (6 loss-of-function, 4 missense) (Supplemental Table II). The prevalence for variants in these two genes was collectively 24/811 (3.0%). We also identified three likely pathogenic variants in JAG1 (OMIM-P 187500, 118450), and two each in TBX1 (OMIM-P 187500) and GATA6 (OMIM-P 187500, 600001) (Supplemental Table II). Consistent with the genetic heterogeneity of TOF, we identified 16 other individuals to have one pathogenic/likely pathogenic variant (11 loss-of-function, 5 missense) in 16 CHD genes: ARHGAP31, ATRX, CACNA1C, CHD7, CSNK2A1, DLL4, EP300, GATAD2B, KAT6A, LZTR1, NF1, NODAL, PIK3CA, RAF1, RASA1, and SMAD2, and one individual with loss-of-function variants in two genes, ASXL1 and PSMD12 (Supplemental Table II).
In a further 16 individuals in this TOF cohort, we identified loss-of-function variants in three emerging CHD candidate genes (KDR, IQGAP1, and GDF1), i.e., genes with substantial research evidence but as yet insufficient to clinically deem variants “likely pathogenic” (Figure 1, Supplemental Table III). For KDR (encoding VEGFR2), four individuals with TOF had high-confidence loss-of-function variants in KDR, compared with none in 6,201 controls from the 1000 genomes and MSSNG projects (Fisher’s exact test (FET): p = 1.8E-4), and compared with 9 in 76,156 controls from gnomAD (Chi-squared test with Yates’ correction (X2) = 83.46; p < 1E-5; observed/expected loss-of-function constraint score (o/e LOF) = 0.15). The significant findings in this large TOF cohort add to results of several recent studies, reporting rare loss-of-function variants in KDR in independent cohorts with TOF or other conotruncal defects18, 21, 26, which collectively provide evidence for KDR as a TOF/CHD gene (Figure 2).
Figure 2:
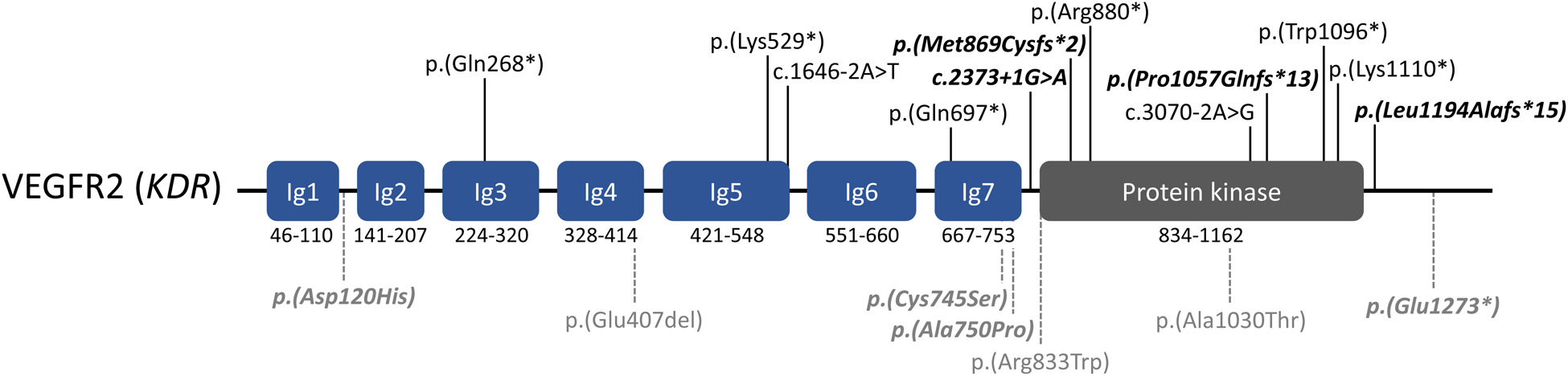
Variants identified in vascular endothelial growth factor receptor 2 (VEGFR2; KDR: NM_002253.2), in patients with TOF or other conotruncal defects. Rare loss-of-function variants (n = 12) are shown in black font (above, solid vertical lines). Rare variants of uncertain significance (in-frame deletion, missense variants, and stopgain variant in penultimate exon) are shown in gray font (below, dashed vertical lines). Variants from this study (Supplemental Tables III and VI) are in bold/italics. Variants reported in Jin et al.21: p.(Lys529*), c.1646-2A>T. Variants reported in Reuter et al.18, Figure 1: p.(Arg880*), p.(Trp1096*), p.(Glu407del), p.(Arg833Trp), p.(Ala1030Thr). Variants reported in Morton et al.26 (Note: some probands may overlap those reported by Jin et al.): p.(Gln268*), p.(Lys529*), c.1646-2A>T, p.(Gln697*), c.3070-2A>G, p.(Lys1110*).
For another vascular endothelial growth factor (VEGF) related gene, IQGAP1, with previous reports of loss-of-function variants in independent cohorts of TOF and other CHD18, 27, 28, we identified three individuals with loss-of-function variants. This was significantly enriched compared to 1000 genomes control data (3/811 vs. 1/2,504; FET: p = 4.8E-2), and to gnomAD (23/76,156; X2 = 18.29; p < 1E-5; o/e LOF = 0.19), but not when compared to control (parental) data from an autism cohort (3/811 vs. 5/3,697; FET: p = 1.6E-1). For GDF1, the pathogenicity of biallelic (homozygous or compound heterozygous) variants is well established [OMIM-P 208530]21, and heterozygous variants have also been suggested as risk factors for CHD29 [OMIM-P 613854]. In the current exome data, the prevalence of GDF1 heterozygous loss-of-function variants is likely to be underestimated due to insufficient coverage of this locus. Nonetheless, we identified 8 individuals to have such a variant, including an identical, heterozygous GDF1 stopgain variant p.Cys227* in 7 of the 706 unrelated probands having samples with adequate read depths (≥10x). In comparison, the carrier frequency of this variant was 6 of 3,697 European controls from MSSNG (FET: p = 1.9E-3) and 64 of 33,079 controls from gnomAD (X2 = 17.36; p < 1E-5), indicating significant enrichment of heterozygous loss-of-function variants in GDF1 in the TOF cohort studied.
Taken together, we identified 63 probands (7.8%) with n = 49 pathogenic/likely pathogenic variants in 23 known CHD genes (Supplemental Table II) or n = 15 loss-of-function variants in 3 emerging CHD/TOF candidate genes (Supplemental Table III) in these TOF exome data. We note that the majority of these variants (40/64, 62.5%) and genes (23/26, 88.5%) were not previously reported for this cohort20 (Supplemental Tables II, III).
We next considered the 23 established TOF genes and 3 candidate genes with significant findings as a group, in an in silico functional interaction analysis. This showed evidence for a highly interactive network of the encoded proteins (STRING interaction enrichment p value = 3.3E-16). VEGFR2 (KDR) and NOTCH1 were central nodes within this network map, each connecting directly with 11 other proteins. In all, 23 of the 26 genes (88.5%) form an interactive network, related to VEGFR2 (KDR) or NOTCH1 by ≤2 edges (Figure 3). Pathogenic/likely pathogenic loss-of-function variants from an independent cohort of 424 children with TOF21 supported, and slightly extended, this network, identifying NOTCH2 as an additional protein with multiple (five) interactions (Supplemental Table IV, Supplemental Figure I). All of the composite genes are expressed in human hearts (https://www.proteinatlas.org/), but at varying levels (Figure 3, Supplemental Figure I). A pathway enrichment map for the respective proteins is provided as supplemental material (Supplemental Figure II, Supplemental Table V).
Figure 3:

Functionally interacting proteins encoded by confirmed genes (n = 23) and emerging candidate genes (n = 3) for CHD/TOF identified in n = 811 individuals with TOF. Network analysis was performed using Cytoscape, STRING and the 26 genes identified from exome sequencing of this cohort (STRING interaction enrichment p value 3.3E-16; see text for details). Node sizes (circles) represent the connectivity (numbers of edges to other proteins). Node colors represent the interaction with VEGFR2/KDR (blue), NOTCH1 (yellow), or both (green). Edge widths represent the confidence (strength of data support for functional and physical protein associations, including textmining, experiments, databases, and co-expression). 23 of the 26 genes form an interactive network with VEGFR2 (gene KDR) and NOTCH1 as central nodes, each connecting directly with 11 other proteins.
Notably, we additionally identified rare nonsynonymous or predicted splice-altering variants of uncertain significance, according to ACMG interpretation guidelines25. This re-analysis classified the majority of NOTCH1 rare nonsynonymous missense and in-frame variants that were previously reported for this sample20 as were variants of uncertain significance (n = 21; Supplemental Table VI). Three of these variants – p.(Glu1294Lys), p.(Gly200Arg)30, and p.(Pro143Leu) – were identified in probands with other likely pathogenic variants: NOTCH1 p.(Gln1733*), NF1 c.5206-1G>C, and EP300 p.(Phe1595Val), respectively (Supplemental Table VI). There were other CHD-relevant genes or candidate genes with variants of uncertain significance, including loss-of-function variants in CHD4, ECE1, SMAD6, ZFPM1, PRKD1 and VEGFA (Supplemental Table VI). In some cases, clinical data or knowledge about the parental genotypes, which were inaccessible for this study, could have informed more accurate variant classifications.
We also report pathogenic/likely pathogenic variants for childhood-onset disorders, but with less established evidence at this time for clinical relevance to TOF/CHD (Supplemental Table VII). These include loss-of-function variants in POLR1A (2x), TCF12 (2x), APC, and GLI2 (Supplemental Table VII).
To further investigate the potential clinical utility of exome sequencing, we also interrogated the dataset for rare variants with potential clinical implications for cardiovascular management and outcome. In 16 (2.0%) of the 811 probands, we identified pathogenic/likely pathogenic variants meeting these criteria. There were 11 variants associated with cardiac hypertrophy, arrhythmia and sudden cardiac death: hypertrophic cardiomyopathy (MYBPC3 (3x), MYH7, MYL2, TNNI3), arrhythmogenic right ventricular dysplasia (DSP (2x), DSC2), Brugada syndrome (SCN5A), and dilated cardiomyopathy (DMD) (Supplemental Table VIII). There were also five variants in genes (LZTR1, RAF1, CACNA1C, NF1, RASA1) implicated in other conditions, including e.g., Noonan and Timothy syndromes31, 32, that in addition have been reported to be associated with CHD, thus were also considered in the above etiologic variant analysis (Supplemental Tables II, VIII).
Discussion
Precision medicine is an emerging concept that involves health management based on individual characteristics, including genetic disease susceptibilities and pharmacogenomics33, 34. Delineation of disease-causing variants and genes and their functional networks will advance both precision health initiatives and our understanding of the relevant molecular mechanisms. The rationale for re-analyzing the exome sequencing data from these 811 individuals with TOF was to identify sequence variants with sufficient evidence for pathogenicity according to consensus clinical guidelines25, that could thereby inform disease etiologies of TOF, and to detect rare and emerging gene-disease associations, unlikely to be identified in the previous study of these data20. We identified 49 pathogenic/likely pathogenic variants for TOF, plus 15 loss-of-function variants in emerging CHD/TOF candidate genes. The majority of variants (40/64, 62.5%) and genes (23/26, 88.5%) were not previously reported for this cohort20 (Supplemental Tables II, III). This demonstrates the novelty of our findings using a stringent clinical approach to variant assessment, and the benefits of re-analysis. Collectively, the results document both genetic and allelic heterogeneity in the pathogenesis of TOF across this large cohort. The findings may also help to define minimal clinical gene panels for TOF.
Novel results from this study also provide further evidence that haploinsufficiency of KDR (VEGFR2) contributes to risk for TOF. Rare KDR loss-of-function variants were previously reported in several cohorts with TOF and other conotruncal defects18, 21, 26 (Figure 2), and our results document highly significant enrichment in TOF compared to controls. None of these KDR variants were previously reported for this cohort20. Other recent studies report evidence that similar loss-of-function variants in KDR are also risk factors for pulmonary arterial hypertension, independent of any heart malformations35, 36. As for many CHD genes, this suggests pleiotropy for KDR requiring further study. There may also be clinical implications with respect to pulmonary hypertension in individuals with KDR-related TOF37.
For IQGAP1, loss-of-function variants were identified in this and in other CHD cohorts18, 27, 28 (including several de novo variants). However, we found a similar prevalence of variants in parents of probands with autism, used here as a control sample. Besides its essential role in VEGF receptor signaling, IQGAP1 regulates and integrates other cellular processes, including neuronal functions38. We consider IQGAP1 a promising candidate gene for CHD, but further statistical support and phenotypic characterization will be needed.
Taken together, pathogenic/likely pathogenic variants in known CHD genes and variants in candidate genes were identified in a total of 26 genes: ARHGAP31, ASXL1, ATRX, CACNA1C, CHD7, CSNK2A1, DLL4, EP300, FLT4, GATAD2B, GATA6, GDF1, JAG1, IQGAP1, KAT6A, KDR, LZTR1, NF1, NODAL, NOTCH1, PIK3CA, PSMD12, RAF1, RASA1, SMAD2, and TBX1. The results further indicated that the encoded proteins form highly inter-connected networks of functional interaction. This suggests that the genetic heterogeneity identified through human disease studies may help to inform overlapping or unifying molecular pathomechanisms for TOF. Notably, VEGFR2 (encoded by KDR) and NOTCH1 form central nodes in this interaction network, supporting and extending evidence that the developing right outflow tract is vulnerable to VEGF/Notch dysregulation39–42. VEGF signaling was recently reported to be the top canonical pathway associated with de novo variants in conotruncal defects28, and low VEGF expression was linked to TOF risk in a historical family study43. Delineating the relevant protein networks and associated pathomechanisms will help to rank novel candidate genes and could inform potential therapeutic targets. For example, loss-of-function variants in TCF12 (Supplemental Table IV) were recently reported in multiple individuals with unexplained CHD26, and the encoded protein functionally interacts with three confirmed TOF-associated proteins (NOTCH1, SMAD2, EP300).
Even with overlapping molecular functions, however, the phenotypic spectrum (pleiotropy) can vary largely not only from one gene to another, and one variant to another, but for individuals with the same variant within and between families. Most of the identified pathogenic/likely pathogenic variants were in CHD genes associated with multisystemic genetic disorders (Supplemental Table II), as may be expected for confirmed genes in early stages of clinical interpretation. Clinical genetic testing results may thus, in certain cases, flag the potential for an increased risk for comorbidities including neurodevelopmental delays. On the other hand, pathogenic variants historically identified through syndromic phenotypes may have cardiac phenotypes without classic extracardiac expression14. For most individual genetic predispositions however, the extent of the disease spectrum is yet unknown. Delineating genotype-associated clinical traits, and understanding their penetrance, will be essential for genetic counselling, familial risk assessments, and informing outcome.
Identifying the genetic etiologies of TOF can improve clinical management, by providing information on outcomes and risks related to the variant, in addition to those related to the cardiac anatomical severity and other clinical parameters13, 44, 45. After the surgical repair of TOF, heart failure and arrhythmias are leading causes of morbidity, impaired quality of life, and mortality. Genetic factors that can affect the molecular and structural properties of the heart and vasculature may play a role. In this study, 16 (2.0%) of 811 probands were identified to have pathogenic/likely pathogenic variants that could affect cardiac surveillance and management recommendations (Supplemental Table VIII). Longitudinal clinical data will be needed to characterize adverse or favourable outcomes of patient populations that include such genetic variant data, in order to identify predictive markers and to inform preventive and therapeutic interventions, as part of precision medicine.
Advantages and limitations
We analyzed genetic risk variants in the largest available exome dataset of individuals with TOF. We prioritized variants with sufficient evidence for pathogenicity, according to consensus clinical guidelines25, in order to increase the “signal-to-noise ratio” in the reporting of single gene defects. In contrast to primarily statistical approaches, such as that previously applied to this cohort20, this clinical approach enabled us to capitalize on the expected genetic and allelic heterogeneity of pathogenic variants in TOF. For example, 18 genes were identified with only one pathogenic/likely pathogenic variant each in this cohort. The lack of variant segregation data and the inaccessibility of individual clinical information, such as anatomical subtypes, disease progression, associated features, or age for this cohort20, however limited the interpretation of the findings. Our analyses were also restricted to rare small sequence variants and insertions/deletions in regions targeted by exome sequencing, typically involving ~95% of exonic regions. The exome data available did not allow us to assess structural aberrations, such as rare copy number variants, despite their established contribution to CHD14 (individuals with typical 22q11.2 deletions were however excluded20). All variants reported here were identified in the heterozygous state, passed internal quality metrics, and were visually validated in the aligned sequencing reads, however we could not confirm their accuracy by Sanger sequencing.
Conclusions
We studied the largest published exome sequencing dataset of patients with TOF, interpreting variants from the perspective of clinical pathogenicity. The identified genetic results add evidence for a major contribution of VEGF/Notch dysregulation, including KDR/VEGFR2, and provide novel findings for functionally interacting protein networks relevant to the pathomechanism of TOF. We anticipate that clinical genomic sequencing, especially where capability of detecting structural variants is included, will become an essential component for assessing risks and outcomes in patients with CHD22. Re-analysis of existing datasets is warranted, and valuable, especially as our knowledge to identify and interpret disease-related variants continuously evolves.
Supplementary Material
Acknowledgments:
Sequence data has been accessed through the European Genome-phenome Archive, which is hosted by the EBI and the CRG, under accession number EGAS00001003302 (https://ega-archive.org). We thank Page et al.20 for allowing us access to the data, and the patients and their families for participation in this research effort. We thank the staff at The Centre for Applied Genomics (TCAG), a node of CGEn, for support in data analysis.
Sources of Funding:
S.W.S. is funded by the Glaxo Smith Kline-CIHR Chair in Genome Sciences at the University of Toronto and The Hospital for Sick Children. A.S.B. holds the Dalglish Chair in 22q11.2 Deletion Syndrome at the University Health Network and University of Toronto. B.D.K. is supported by a British Heart Foundation Personal Chair.
Nonstandard Abbreviations and Acronyms:
- TOF
Tetralogy of Fallot
- CHD
Congenital heart disease
- ACMG
American College of Medical Genetics
- EGA
European Genome-phenome Archive
Footnotes
Publisher's Disclaimer: This article is published in its accepted form; it has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation: Genomic and Precision Medicine involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final published version.
Disclosures: S.W.S. serves on the Scientific Advisory Committees of Population Diagnostics and Deep Genomics, and is a Highly Cited Academic Advisor to the King Abdulaziz University. The other authors declare no conflicts of interest.
References:
- 1.Liu Y, Chen S, Zuhlke L, Black GC, Choy MK, Li N, Keavney BD. Global birth prevalence of congenital heart defects 1970–2017: updated systematic review and meta-analysis of 260 studies. Int J Epidemiol. 2019;48:455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bailliard F, Anderson RH. Tetralogy of Fallot. Orphanet J Rare Dis. 2009;4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diaz-Frias J, Guillaume M. Tetralogy of Fallot StatPearls; Treasure Island (FL); 2020. [PubMed] [Google Scholar]
- 4.Neill CA, Clark EB. Tetralogy of Fallot. The first 300 years. Tex Heart Inst J. 1994;21:272–9. [PMC free article] [PubMed] [Google Scholar]
- 5.Gelb BD. History of Our Understanding of the Causes of Congenital Heart Disease. Circ Cardiovasc Genet. 2015;8:529–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stefanescu Schmidt AC, DeFaria Yeh D, Tabtabai S, Kennedy KF, Yeh RW, Bhatt AB. National Trends in Hospitalizations of Adults With Tetralogy of Fallot. Am J Cardiol. 2016;118:906–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agarwal A, Thombley R, Broberg CS, Harris IS, Foster E, Mahadevan VS, John A, Vittinghoff E, Marcus GM, Dudley RA. Age- and Lesion-Related Comorbidity Burden Among US Adults With Congenital Heart Disease: A Population-Based Study. J Am Heart Assoc. 2019;8:e013450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marelli AJ, Ionescu-Ittu R, Mackie AS, Guo L, Dendukuri N, Kaouache M. Lifetime prevalence of congenital heart disease in the general population from 2000 to 2010. Circulation. 2014;130:749–56. [DOI] [PubMed] [Google Scholar]
- 9.Diller GP, Kempny A, Alonso-Gonzalez R, Swan L, Uebing A, Li W, Babu-Narayan S, Wort SJ, Dimopoulos K, Gatzoulis MA. Survival Prospects and Circumstances of Death in Contemporary Adult Congenital Heart Disease Patients Under Follow-Up at a Large Tertiary Centre. Circulation. 2015;132:2118–25. [DOI] [PubMed] [Google Scholar]
- 10.Lahiri S, Gil W, Daria S, Joshua G, Parul J, Redmond B, Elizabeth W. Genetic abnormalities/syndromes significantly impact perioperative outcomes of conotruncal heart defects. Ann Pediatr Cardiol. 2020;13:38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Formigari R, Michielon G, Digilio MC, Piacentini G, Carotti A, Giardini A, Di Donato RM, Marino B. Genetic syndromes and congenital heart defects: how is surgical management affected? Eur J Cardiothorac Surg. 2009;35:606–14. [DOI] [PubMed] [Google Scholar]
- 12.Russell MW, Chung WK, Kaltman JR, Miller TA. Advances in the Understanding of the Genetic Determinants of Congenital Heart Disease and Their Impact on Clinical Outcomes. J Am Heart Assoc. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Mil S, Heung T, Malecki S, Van L, Chang J, Breetvelt E, Wald R, Oechslin E, Silversides C, Bassett AS. Impact of a 22q11.2 Microdeletion on Adult All-Cause Mortality in Tetralogy of Fallot Patients. Can J Cardiol. 2020;36:1091–1097. [DOI] [PubMed] [Google Scholar]
- 14.Costain G, Silversides CK, Bassett AS. The importance of copy number variation in congenital heart disease. NPJ Genom Med. 2016;1:16031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morgenthau A, Frishman WH. Genetic origins of tetralogy of Fallot. Cardiol Rev. 2018;26:86–92. [DOI] [PubMed] [Google Scholar]
- 16.Lahm H, Schon P, Doppler S, Dressen M, Cleuziou J, Deutsch MA, Ewert P, Lange R, Krane M. Tetralogy of Fallot and Hypoplastic Left Heart Syndrome - Complex Clinical Phenotypes Meet Complex Genetic Networks. Curr Genomics. 2015;16:141–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Silversides CK, Lionel AC, Costain G, Merico D, Migita O, Liu B, Yuen T, Rickaby J, Thiruvahindrapuram B, Marshall CR, et al. Rare copy number variations in adults with tetralogy of Fallot implicate novel risk gene pathways. PLoS Genet. 2012;8:e1002843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reuter MS, Jobling R, Chaturvedi RR, Manshaei R, Costain G, Heung T, Curtis M, Hosseini SM, Liston E, Lowther C, et al. Haploinsufficiency of vascular endothelial growth factor related signaling genes is associated with tetralogy of Fallot. Genet Med. 2019;21:1001–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manshaei R, Merico D, Reuter MS, Engchuan W, Mojarad BA, Chaturvedi R, Heung T, Pellecchia G, Zarrei M, Nalpathamkalam T, et al. Genes and Pathways Implicated in Tetralogy of Fallot Revealed by Ultra-Rare Variant Burden Analysis in 231 Genome Sequences. Front Genet. 2020;11:957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Page DJ, Miossec MJ, Williams SG, Monaghan RM, Fotiou E, Cordell HJ, Sutcliffe L, Topf A, Bourgey M, Bourque G, et al. Whole Exome Sequencing Reveals the Major Genetic Contributors to Nonsyndromic Tetralogy of Fallot. Circ Res. 2019;124:553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, Zeng X, Qi H, Chang W, Sierant MC, et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. 2017;49:1593–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reuter MS, Chaturvedi RR, Liston E, Manshaei R, Aul RB, Bowdin S, Cohn I, Curtis M, Dhir P, Hayeems RZ, et al. The Cardiac Genome Clinic: implementing genome sequencing in pediatric heart disease. Genet Med. 2020;22:1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jia Y, Louw JJ, Breckpot J, Callewaert B, Barrea C, Sznajer Y, Gewillig M, Souche E, Dehaspe L, Vermeesch JR, et al. The diagnostic value of next generation sequencing in familial nonsyndromic congenital heart defects. Am J Med Genet A. 2015;167A:1822–9. [DOI] [PubMed] [Google Scholar]
- 24.Fotiou E, Williams S, Martin-Geary A, Robertson DL, Tenin G, Hentges KE, Keavney B. Integration of Large-Scale Genomic Data Sources With Evolutionary History Reveals Novel Genetic Loci for Congenital Heart Disease. Circ Genom Precis Med. 2019;12:442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morton SU, Shimamura A, Newburger PE, Opotowsky AR, Quiat D, Pereira AC, Jin SC, Gurvitz M, Brueckner M, Chung WK, et al. Association of Damaging Variants in Genes With Increased Cancer Risk Among Patients With Congenital Heart Disease. JAMA Cardiol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petrovski S, Aggarwal V, Giordano JL, Stosic M, Wou K, Bier L, Spiegel E, Brennan K, Stong N, Jobanputra V, et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet. 2019;393:758–767. [DOI] [PubMed] [Google Scholar]
- 28.Sevim Bayrak C, Zhang P, Tristani-Firouzi M, Gelb BD, Itan Y. De novo variants in exomes of congenital heart disease patients identify risk genes and pathways. Genome Med. 2020;12:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karkera JD, Lee JS, Roessler E, Banerjee-Basu S, Ouspenskaia MV, Mez J, Goldmuntz E, Bowers P, Towbin J, Belmont JW, et al. Loss-of-function mutations in growth differentiation factor-1 (GDF1) are associated with congenital heart defects in humans. Am J Hum Genet. 2007;81:987–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blue GM, Kirk EP, Giannoulatou E, Dunwoodie SL, Ho JW, Hilton DC, White SM, Sholler GF, Harvey RP, Winlaw DS. Targeted next-generation sequencing identifies pathogenic variants in familial congenital heart disease. J Am Coll Cardiol. 2014;64:2498–506. [DOI] [PubMed] [Google Scholar]
- 31.Allanson JE, Roberts AE. Noonan Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K and Amemiya A, eds. GeneReviews((R)) Seattle (WA); 1993. [Google Scholar]
- 32.Napolitano C, Splawski I, Timothy KW, Bloise R, Priori SG. Timothy Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K and Amemiya A, eds. GeneReviews((R)) Seattle (WA); 1993. [Google Scholar]
- 33.Tada H, Fujino N, Nomura A, Nakanishi C, Hayashi K, Takamura M, Kawashiri MA. Personalized medicine for cardiovascular diseases. J Hum Genet. 2021;66:67–74. [DOI] [PubMed] [Google Scholar]
- 34.Dainis AM, Ashley EA. Cardiovascular Precision Medicine in the Genomics Era. JACC Basic Transl Sci. 2018;3:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eyries M, Montani D, Girerd B, Favrolt N, Riou M, Faivre L, Manaud G, Perros F, Graf S, Morrell NW, et al. Familial pulmonary arterial hypertension by KDR heterozygous loss of function. Eur Respir J. 2020;55. [DOI] [PubMed] [Google Scholar]
- 36.Swietlik EM, Greene D, Zhu N, Megy K, Cogliano M, Rajaram S, Pandya D, Tilly T, Lutz KA, Welch CCL, et al. Bayesian Inference Associates Rare KDR Variants with Specific Phenotypes in Pulmonary Arterial Hypertension. Circ Genom Precis Med. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dimopoulos K, Condliffe R, Tulloh RMR, Clift P, Alonso-Gonzalez R, Bedair R, Chung NAY, Coghlan G, Fitzsimmons S, Frigiola A, et al. Echocardiographic Screening for Pulmonary Hypertension in Congenital Heart Disease: JACC Review Topic of the Week. J Am Coll Cardiol. 2018;72:2778–2788. [DOI] [PubMed] [Google Scholar]
- 38.White CD, Erdemir HH, Sacks DB. IQGAP1 and its binding proteins control diverse biological functions. Cell Signal. 2012;24:826–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van den Akker NM, Caolo V, Molin DG. Cellular decisions in cardiac outflow tract and coronary development: an act by VEGF and NOTCH. Differentiation. 2012;84:62–78. [DOI] [PubMed] [Google Scholar]
- 40.Jain R, Rentschler S, Epstein JA. Notch and cardiac outflow tract development. Ann N Y Acad Sci. 2010;1188:184–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luxan G, D’Amato G, MacGrogan D, de la Pompa JL. Endocardial Notch Signaling in Cardiac Development and Disease. Circ Res. 2016;118:e1–e18. [DOI] [PubMed] [Google Scholar]
- 42.van den Akker NM, Molin DG, Peters PP, Maas S, Wisse LJ, van Brempt R, van Munsteren CJ, Bartelings MM, Poelmann RE, Carmeliet P, et al. Tetralogy of fallot and alterations in vascular endothelial growth factor-A signaling and notch signaling in mouse embryos solely expressing the VEGF120 isoform. Circ Res. 2007;100:842–9. [DOI] [PubMed] [Google Scholar]
- 43.Lambrechts D, Devriendt K, Driscoll DA, Goldmuntz E, Gewillig M, Vlietinck R, Collen D, Carmeliet P. Low expression VEGF haplotype increases the risk for tetralogy of Fallot: a family based association study. J Med Genet. 2005;42:519–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Michielon G, Marino B, Formigari R, Gargiulo G, Picchio F, Digilio MC, Anaclerio S, Oricchio G, Sanders SP, Di Donato RM. Genetic syndromes and outcome after surgical correction of tetralogy of Fallot. Ann Thorac Surg. 2006;81:968–75. [DOI] [PubMed] [Google Scholar]
- 45.Villafane J, Feinstein JA, Jenkins KJ, Vincent RN, Walsh EP, Dubin AM, Geva T, Towbin JA, Cohen MS, Fraser C, et al. Hot topics in tetralogy of Fallot. J Am Coll Cardiol. 2013;62:2155–66. [DOI] [PubMed] [Google Scholar]
- 46.Lappalainen I, Almeida-King J, Kumanduri V, Senf A, Spalding JD, Ur-Rehman S, Saunders G, Kandasamy J, Caccamo M, Leinonen R, et al. The European Genome-phenome Archive of human data consented for biomedical research. Nat Genet. 2015;47:692–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cordell HJ, Topf A, Mamasoula C, Postma AV, Bentham J, Zelenika D, Heath S, Blue G, Cosgrove C, Granados Riveron J, et al. Genome-wide association study identifies loci on 12q24 and 13q32 associated with tetralogy of Fallot. Hum Mol Genet. 2013;22:1473–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11 10 1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, Sidiropoulos K, Cook J, Gillespie M, Haw R, et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020;48:D498–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Allen HL, Flanagan SE, Shaw-Smith C, De Franco E, Akerman I, Caswell R, International Pancreatic Agenesis C, Ferrer J, Hattersley AT, Ellard S. GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat Genet. 2011;44:20–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pierson TM, Otero MG, Grand K, Choi A, Graham JM Jr., Young JI, Mackay JP. The NuRD complex and macrocephaly associated neurodevelopmental disorders. Am J Med Genet C Semin Med Genet. 2019;181:548–556. [DOI] [PubMed] [Google Scholar]
- 60.Mirzaa G, Conway R, Graham JM Jr., Dobyns WB. PIK3CA-Related Segmental Overgrowth. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K and Amemiya A, eds. GeneReviews((R)) Seattle (WA); 1993. [Google Scholar]
- 61.Bayrak-Toydemir P, Stevenson DA. Capillary Malformation-Arteriovenous Malformation Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K and Amemiya A, eds. GeneReviews((R)) Seattle (WA); 1993. [PubMed] [Google Scholar]
- 62.Mohapatra B, Casey B, Li H, Ho-Dawson T, Smith L, Fernbach SD, Molinari L, Niesh SR, Jefferies JL, Craigen WJ, et al. Identification and functional characterization of NODAL rare variants in heterotaxy and isolated cardiovascular malformations. Hum Mol Genet. 2009;18:861–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.De Luca A, Sarkozy A, Consoli F, Ferese R, Guida V, Dentici ML, Mingarelli R, Bellacchio E, Tuo G, Limongelli G, et al. Familial transposition of the great arteries caused by multiple mutations in laterality genes. Heart. 2010;96:673–7. [DOI] [PubMed] [Google Scholar]
- 64.Granadillo JL, Chung WK, Hecht L, Corsten-Janssen N, Wegner D, Nij Bijvank SWA, Toler TL, Pineda-Alvarez DE, Douglas G, Murphy JJ, et al. Variable cardiovascular phenotypes associated with SMAD2 pathogenic variants. Hum Mutat. 2018;39:1875–1884. [DOI] [PubMed] [Google Scholar]
- 65.Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, Romano-Adesman A, Bjornson RD, Breitbart RE, Brown KK, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498:220–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meester JAN, Sukalo M, Schroder KC, Schanze D, Baynam G, Borck G, Bramswig NC, Duman D, Gilbert-Dussardier B, Holder-Espinasse M, et al. Elucidating the genetic architecture of Adams-Oliver syndrome in a large European cohort. Hum Mutat. 2018;39:1246–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chinton J, Huckstadt V, Mucciolo M, Lepri F, Novelli A, Gravina LP, Obregon MG. Providing more evidence on LZTR1 variants in Noonan syndrome patients. Am J Med Genet A. 2020;182:409–414. [DOI] [PubMed] [Google Scholar]
- 68.Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, Vertino-Bell A, Smaoui N, Neidich J, Monaghan KG, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2016;18:696–704. [DOI] [PubMed] [Google Scholar]
- 69.Weiss K, Lazar HP, Kurolap A, Martinez AF, Paperna T, Cohen L, Smeland MF, Whalen S, Heide S, Keren B, et al. The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis. Genet Med. 2020;22:389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fahed AC, Gelb BD, Seidman JG, Seidman CE. Genetics of congenital heart disease: the glass half empty. Circ Res. 2013;112:707–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stanczak P, Witecka J, Szydlo A, Gutmajster E, Lisik M, Augusciak-Duma A, Tarnowski M, Czekaj T, Czekaj H, Sieron AL. Mutations in mammalian tolloid-like 1 gene detected in adult patients with ASD. Eur J Hum Genet. 2009;17:344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zatyka M, Priestley M, Ladusans EJ, Fryer AE, Mason J, Latif F, Maher ER. Analysis of CRELD1 as a candidate 3p25 atrioventicular septal defect locus (AVSD2). Clin Genet. 2005;67:526–8. [DOI] [PubMed] [Google Scholar]
- 73.Wild A, Kalff-Suske M, Vortkamp A, Bornholdt D, Konig R, Grzeschik KH. Point mutations in human GLI3 cause Greig syndrome. Hum Mol Genet. 1997;6:1979–84. [DOI] [PubMed] [Google Scholar]
- 74.Thienpont B, Zhang L, Postma AV, Breckpot J, Tranchevent LC, Van Loo P, Mollgard K, Tommerup N, Bache I, Tumer Z, et al. Haploinsufficiency of TAB2 causes congenital heart defects in humans. Am J Hum Genet. 2010;86:839–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kirk EP, Sunde M, Costa MW, Rankin SA, Wolstein O, Castro ML, Butler TL, Hyun C, Guo G, Otway R, et al. Mutations in cardiac T-box factor gene TBX20 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. Am J Hum Genet. 2007;81:280–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Intarak N, Theerapanon T, Thaweesapphithak S, Suphapeetiporn K, Porntaveetus T, Shotelersuk V. Genotype-phenotype correlation and expansion of orodental anomalies in LTBP3-related disorders. Mol Genet Genomics. 2019;294:773–787. [DOI] [PubMed] [Google Scholar]
- 77.Walton RZ, Bruce AE, Olivey HE, Najib K, Johnson V, Earley JU, Ho RK, Svensson EC. Fog1 is required for cardiac looping in zebrafish. Dev Biol. 2006;289:482–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Werner P, Latney B, Deardorff MA, Goldmuntz E. MESP1 Mutations in Patients with Congenital Heart Defects. Hum Mutat. 2016;37:308–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lahm H, Deutsch MA, Dressen M, Doppler S, Werner A, Horer J, Cleuziou J, Schreiber C, Bohm J, Laugwitz KL, et al. Mutational analysis of the human MESP1 gene in patients with congenital heart disease reveals a highly variable sequence in exon 1. Eur J Med Genet. 2013;56:591–8. [DOI] [PubMed] [Google Scholar]
- 80.Weaver KN, Watt KE, Hufnagel RB, Navajas Acedo J, Linscott LL, Sund KL, Bender PL, Konig R, Lourenco CM, Hehr U, et al. Acrofacial Dysostosis, Cincinnati Type, a Mandibulofacial Dysostosis Syndrome with Limb Anomalies, Is Caused by POLR1A Dysfunction. Am J Hum Genet. 2015;96:765–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Monreal AW, Zonana J, Ferguson B. Identification of a new splice form of the EDA1 gene permits detection of nearly all X-linked hypohidrotic ectodermal dysplasia mutations. Am J Hum Genet. 1998;63:380–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet. 2013;381:333–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.