

Abstract
Background:
Diabetes is a risk factor of heart failure and promotes cardiac dysfunction. Diabetic tissues are associated with NAD+ redox imbalance; however, the hypothesis that NAD+ redox imbalance causes diabetic cardiomyopathy (DCM) has not been tested. This investigation employed mouse models with altered NAD+ redox balance to test this hypothesis.
Methods:
Diabetic stress was induced in mice by streptozotocin. Cardiac function was measured by echocardiography. Heart and plasma samples were collected for biochemical, histological and molecular analyses. Two mouse models with altered NAD+ redox states (cardiac-specific knockout mice of Ndufs4, cKO; nicotinamide phosphoribosyltransferase transgenic mice, NAMPT) were used.
Results:
Diabetic stress caused cardiac dysfunction and lowered NAD+/NADH in wild-type mice. Mice with lowered cardiac NAD+/NADH without baseline dysfunction, cKO mice, were challenged with chronic diabetic stress. NAD+ redox imbalance in cKO hearts exacerbated systolic (FS: 27.6% vs 36.9% at 4-week, male cohort, P<0.05) and diastolic dysfunction (E’/A’: 0.99 vs 1.20, P<0.05) of diabetic mice in both sexes. Collagen levels and transcripts of fibrosis and extracellular matrix-dependent pathways did not show changes in diabetic cKO hearts, suggesting that the exacerbated cardiac dysfunction was due to cardiomyocyte dysfunction. NAD+ redox imbalance promoted superoxide dismutase 2 acetylation, protein oxidation, troponin-I S150 phosphorylation and impaired energetics in diabetic cKO hearts. Importantly, elevation of cardiac NAD+ levels by NAMPT normalized NAD+ redox balance, alleviated cardiac dysfunction (FS: 40.2% vs 24.8% in cKO:NAMPT vs cKO, P<0.05; E’/A’: 1.32 vs 1.04, P<0.05) and reversed pathogenic mechanisms in diabetic mice.
Conclusions:
Our results show that NAD+ redox imbalance to regulate acetylation and phosphorylation is a critical mediator of the progression of DCM, and suggest the therapeutic potential for DCM by harnessing NAD+ metabolism.
Introduction
Diabetes is characterized as a loss of glycemic control, and is a risk factor of heart failure. Epidemiological studies show a positive correlation of hyperglycemia with the increased risk of heart failure in both Type 1 and Type 2 diabetes patients. Diabetic cardiomyopathy (DCM) is defined as diabetes-associated cardiac dysfunction independent of coronary artery diseases or other confounding cardiovascular diseases 1. Mechanisms such as mitochondrial dysfunction and oxidative stress have been identified 2. However, how altered metabolism in diabetes, in particular NAD+-dependent pathways, leads to cardiac dysfunction is not fully understood.
NAD+-dependent pathways have emerged to play critical roles in metabolism-driven disease progression 3. NAD+ mediates electron transfer and redox reactions in metabolism. The oxidized form of NAD+ is converted to the reduced form (NADH) to mediate substrate catabolism via many oxidoreductase-dependent enzymes. The recycling of NADH to NAD+, and NAD+ redox balance (high NAD+/NADH ratio) are essential to maintain catabolism and ATP synthesis. The functions of NAD+ have been expanded with the discovery of NAD+-consuming enzymes, such as Sirtuins, a family of NAD+-dependent deacylases. Sirtuins remove acyl modifications on proteins by turning NAD+ into nicotinamide (NAM) and acyl-adenosine diphosphate ribose. Activation of Sirtuins is generally cytoprotective and inhibition of Sirtuins promotes pathogenesis in many metabolic diseases 3–5. The availability of NAD+ to Sirtuins, via changes in the NAD+/NADH or the NAD+ pool, is crucial to regulate mitochondrial and cellular functions. To replenish the loss of NAD+ consumed by Sirtuins, NAD+ is synthesized by several biosynthetic pathways. The NAD+ salvage pathway is the dominant pathway in a mouse heart 6: NAM generated from the reactions of Sirtuins is recycled to synthesize nicotinamide mononucleotide (NMN) by nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme of salvage pathway. NMN is then converted into NAD+ by nicotinamide mononucleotide adenylyltransferases (NMNATs). Elevating NAD+ levels by activation of the NAD+ biosynthetic pathways is therapeutic to multiple diseases 7–14. We previously showed that NAD+-dependent acetylation contributes to pressure overload-induced heart failure, and elevation of NAD+ levels alleviates the cardiac dysfunction 15, 16. Hyperglycemia is associated with decreased NAD+/NADH ratio, and promotes protein hyperacetylation and mitochondrial dysfunction in diabetic organs 10, 17-21. Although these data support a hypothesis that NAD+ redox imbalance in diabetic hearts promotes cardiac dysfunction, this hypothesis has not been directly tested.
We previously generated a mouse model of mitochondrial dysfunction by deleting Ndufs4, a complex I protein, only in the hearts (cardiac Ndufs4-KO, cKO) 15. Complex I deficiency in cKO mice leads to lowered cardiac NAD+/NADH, but surprisingly these mice have normal cardiac function and energetics under unstressed condition 15, 16. In this study, we challenged this mouse model of latent cardiac NAD+ redox imbalance with streptozotocin (STZ)-induced diabetic stress and examined DCM progression. We found that cKO mice showed exacerbated cardiac dysfunction in response to diabetic stress. Furthermore, elevation of cardiac NAD+ levels with cardiac-specific NAMPT transgenic mice (NAMPT) 22 ameliorated the cardiac dysfunction in both diabetic cKO and control mice. The exacerbated cardiac dysfunction in diabetic cKO was associated with increased levels of acetylation of antioxidant enzyme SOD2, oxidative stress, troponin I (TnI) S150 phosphorylation and impaired energetics. Importantly, NAMPT overexpression normalized NAD+/NADH ratio in diabetic cKO hearts and reversed these pathogenic mechanisms.
Research Design and Methods
Data supporting the findings of this study are available from the corresponding author upon reasonable request. Detailed animal care and experimental methods described below were available in Supplement.
Animal care and experiments
All animal procedures were approved by the IACUC at Oklahoma Medical Research Foundation and University of Washington.
Tissue harvest, processing and NAD+ quantification
Experiment details were available in Supplement.
Analysis of mRNA levels
Total RNA was extracted and quantified. To assess expression of genes related to fibrosis, cDNA samples were synthesized, and quantitative PCR reactions were performed using RT2 Profiler PCR Array for mouse extracellular matrix and adhesion molecules genes (Qiagen, Cat. No. 330231 ID: PAMM-013Z).
Western blotting
Pulverized cardiac tissues were homogenized in RIPA buffer (Sigma) with a protease, phosphatase and deacetylase inhibitor cocktail. Experiment details were available in Supplement.
Sample analyses by LC-MS/MS
Experiment details for metabolite analysis were available in Supplement. Briefly, samples were subjected to protein precipitation and metabolite extraction, and followed by centrifugation. The supernatants were collected and dried. The dried samples were reconstituted with 40% PBS/60% ACN. A pooled sample was used as the quality-control sample. The targeted LC-MS/MS method was used 23-25.
Histology
Heart tissues were cut cross-sectionally at the mid-section and fixed with paraformaldehyde. Experiment details for histology were available in Supplement.
Statistical analysis
For comparisons involving two groups, unpaired 2-tailed t-tests were used. For analysis of sex-dependent changes, two-way ANOVA was used. All analyses were performed using GraphPad Prism 8.0. All data are expressed as mean ± SD, and a p<0.05 was considered significant. Metabolite levels were statistically analyzed by MetaboAnalyst 4.0. Adjusted P-value (FDR) cutoff for the dataset was set at 0.05. Raw data are available as supplementary materials. Heatmaps were generated by Morpheus.
Results
16 weeks of diabetic stress impaired cardiac function and lowered NAD+/NADH ratio
We subjected C57BL/6 wildtype mice (WT) to STZ-induced diabetic stress for 16 weeks, as systolic and diastolic dysfunction has been demonstrated in mice at 16 weeks after STZ treatment 26. STZ treatment in WT mice led to significant increase in fasting blood glucose levels (STZ mean glucose 640+/−64, vehicle glucose 103+/−15, P<0.01) at 16 weeks after treatment (Figure 1A). We showed that 16 weeks of diabetic stress in WT mice led to decline in systolic and diastolic function of the heart. Systolic function represented by fractional shortening (FS) decreased (STZ mean FS 33+/−7, vehicle mean FS 53+/−5, P<0.01) in STZ-treated diabetic mice (Figure 1B). E’/A’ ratio decreased (STZ mean E’/A’ 1.0+/−0.1, vehicle mean E’/A’ 1.5+/−0,1, P<0.01), e/E’ ratio and isovolumic relaxation time (IVRT) increased in diabetic mice, indicating diastolic dysfunction (Figure 1C-F). These declines in cardiac function were accompanied by a lowered NAD+/NADH ratio (STZ mean ratio 3.3+/−1.2, vehicle mean ratio 6.7+/−1.1, P<0.05, Figure 1G). An in vitro study has shown that the effects of STZ are not specific only to the beta cells, and that STZ treatment impairs contractile function of isolated rat cardiomyocytes 27. To determine if STZ injection causes acute toxicity in the heart, we examined the acute effects of STZ treatment on the heart. We observed that acute STZ treatment (1 day) did not affect cardiac function, NAD+ pool size, and fasting glucose levels (Supp. Figure I–A–D). STZ is rapidly metabolized and excreted by rodents 28. Therefore, the lowered NAD+/NADH ratio and cardiac dysfunction at 16 weeks after STZ treatment should be caused by chronic diabetic stress (Figure 1A), as observed in other diabetic tissues 10, 17-21, but not direct STZ-mediated toxicity to the heart. To determine if the decrease in NAD+/NADH ratio precedes cardiac dysfunction, WT mice were subjected to 2-week, STZ-induced diabetic stress. We found that NAD+/NADH ratio decreased in hearts after 2-week diabetic stress, while systolic function and diastolic function remained unchanged (Figure 1H-L).
Figure 1. 16-week diabetic stress leads to systolic and diastolic dysfunction and lowered NAD+/NADH ratio.
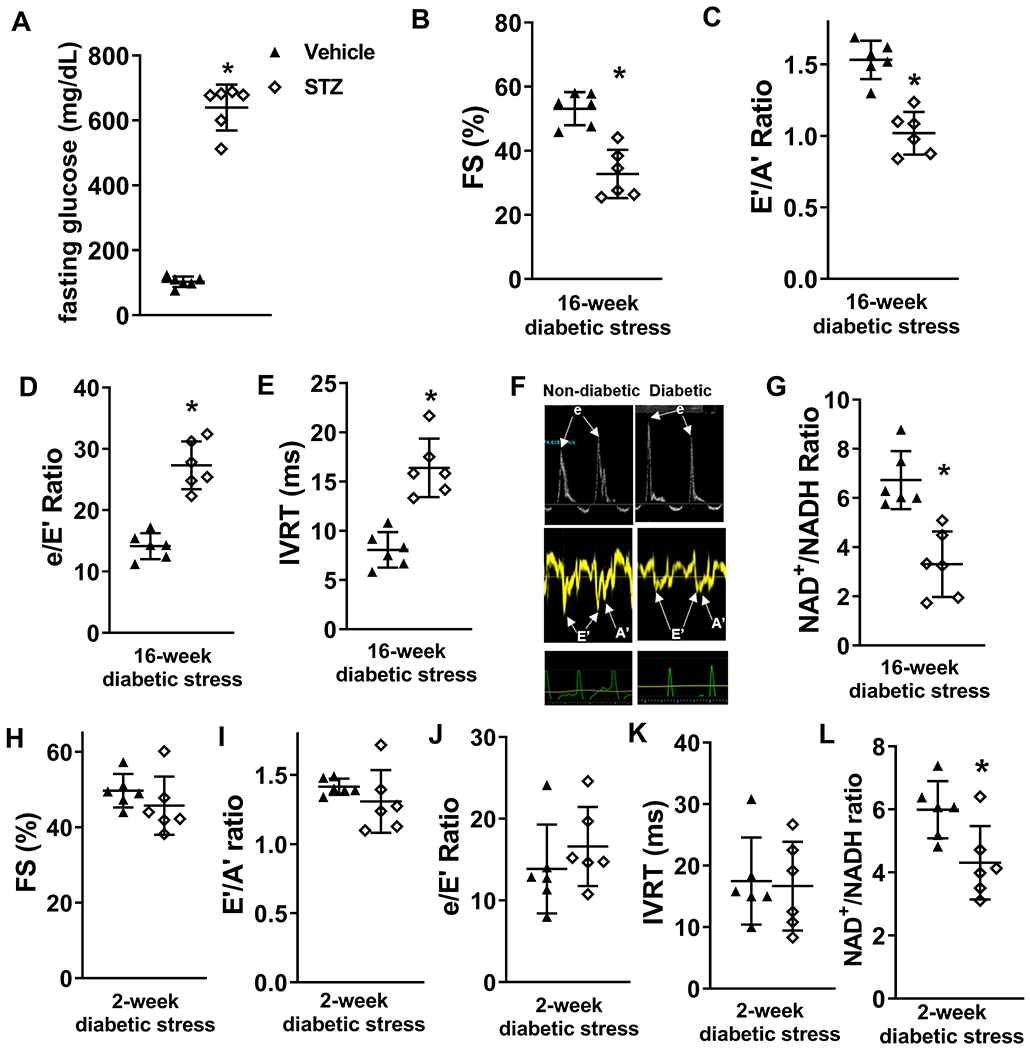
C57/BL6 wild type (WT) mice were treated with STZ to induce diabetes. Cardiac function was assessed with echocardiography 16 weeks after induction of diabetes. (A) Plasma glucose levels were measured 16 weeks after vehicle or STZ treatment. (B) Fractional shortening (FS), (C) E’/A’, (D) e/E’ ratio, and (E) isovolumic relaxation time (IVRT) were measured to evaluate systolic and diastolic functions. (F) Representative pulsed-wave (upper panels) and tissue (middle panels) doppler images and corresponding electrocardiogram (lower panels) images. (G) Cardiac NAD+/NADH ratio were measured. N=6. *: P<0.05 to vehicle. (H) FS, (I) E’/A’ ratio, (J) e/E’ ratio, (K) IVRT and (L) cardiac NAD+/NADH ratio were measured in mice 2-week after diabetic stress. N=6. *: P<0.05 to vehicle.
Latent NAD+ redox imbalance exacerbated dysfunction of diabetic hearts
Although diabetic hearts displayed NAD+ redox imbalance and dysfunction, the causal role of the lowered NAD+/NADH ratio induced by diabetic stress to cardiac dysfunction has not been established. Therefore, we employed cardiac-specific Ndufs4-KO mice (cKO), a mouse model with lowered cardiac NAD+/NADH ratio, normal cardiac function and energetics, 15 to determine how NAD+ redox imbalance changes the progression of DCM. Male control and cKO mice were subjected to 8-week, STZ-induced diabetic stress (Figure 2A). STZ treatment led to insulin depletion, and the extent of insulin depletion was similar in control and cKO mice (Supp. Figure II–A). Fasting blood glucose levels of control and cKO mice at 8 weeks after STZ treatment were up-regulated to similar levels (Supp. Figure II-B). To assess the systemic metabolic changes induced by STZ treatment, we next analyzed plasma samples collected from these mice by quantitative metabolomics. Metabolic stress phenotypes, including increased circulating glucose and ceramide levels, were observed in STZ-induced diabetic control mice, when compared to non-diabetic control mice (Data Set 1). Of the 159 aqueous and 85 lipid metabolites surveyed, no significant changes in levels were observed between diabetic control and diabetic cKO mice (Supp. Figure II–C–D, FDR cutoff < 0.05, data available in Data Set 2). The results of the plasma metabolomic analyses suggest that cardiac-specific Ndufs4 deletion did not affect peripheral metabolism, and diabetic control and diabetic cKO hearts experienced similar metabolic stress induced by STZ. Despite the similar extent of metabolic stress, diabetic cKO mice exhibited reduced cardiac NAD+/NADH ratio compared to diabetic control mice (diabetic cKO mean ratio 3.7+/−0.7, diabetic control 7.3+/−0.8, P<0.01, Figure 2B). The diabetic stress promoted a decline in systolic function of control mice, which was exacerbated in cKO mice 2, 4, and 8 weeks after STZ injection (Figure 2C, Supp. Figure III–A, Supp. Table I). The diabetic cKO mice also exhibited exacerbated diastolic dysfunction represented by a lowered E’/A’ ratio, increased e/E’ ratio (diabetic cKO mean e/E’ 40.5+/−7.0, diabetic control, 24.5+/−6.9, P<0.05) and IVRT (diabetic cKO mean IVRT 22.6+/−4.6, diabetic control, 14.0+/−3.8, P<0.05), and an elevated myocardial performance index (MPI) compared to diabetic control mice (Figure 2D-G, Supp. Table I). Left ventricular dilation, cardiac hypertrophy and lung edema did not change in diabetic cKO hearts compared to diabetic controls (Figure 2H-J).
Figure 2. Latent NAD(H) redox imbalance in the heart exacerbates cardiac dysfunctions of diabetic male mice.
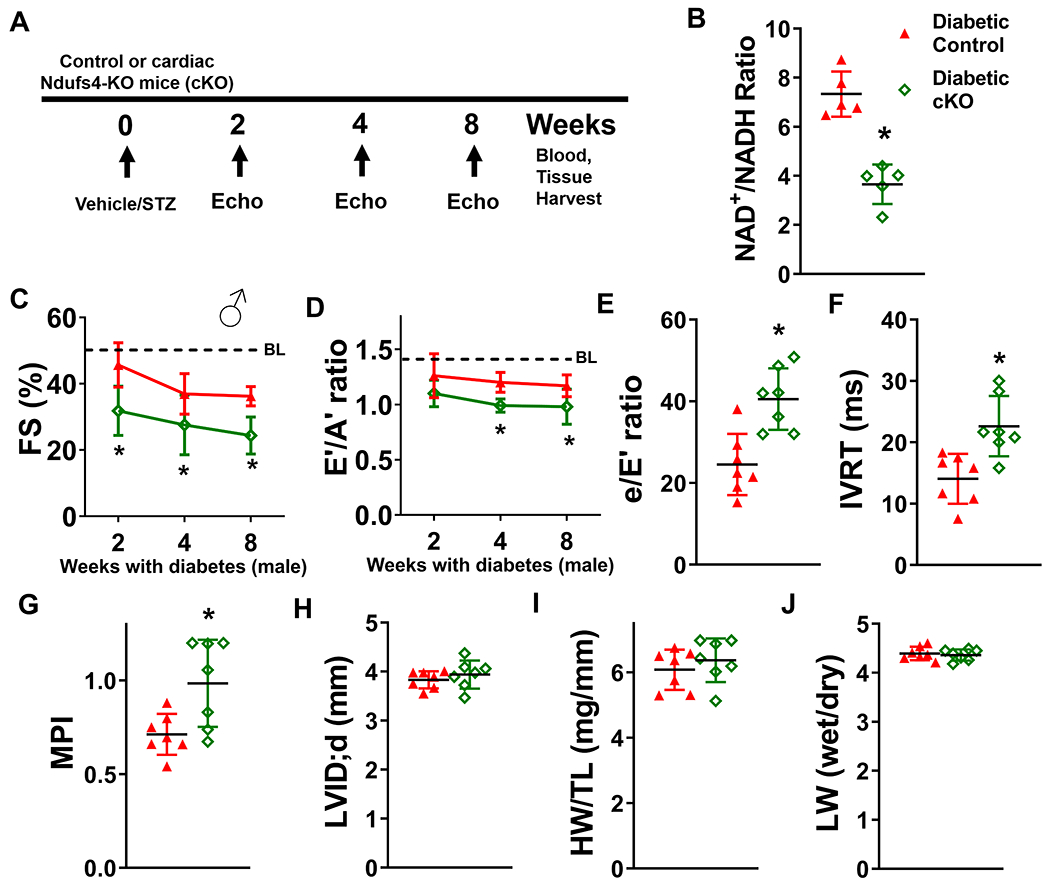
(A) Experimental plan. Control and cKO male mice were treated with STZ to induce diabetes. Cardiac function was measured at 2, 4, and 8 weeks after induction of diabetes. Blood and tissue samples were collected at the endpoint of the study. (B) Cardiac NAD+/NADH ratio was measured. N=5. Longitudinal changes in (C) fractional shortening (FS) and (D) E’/A’ ratio of diabetic control or cKO male mice were assessed. Dotted lines indicate average baseline (BL) values of non-diabetic control mice. Numerical values are available in Supplementary Table I. (E) e/E’ ratio, (F) isovolumic relaxation time (IVRT), (G) myocardial performance index (MPI) and (H) left ventricular internal dimension at diastole (LVID;d) were measured at 8 weeks after diabetes induction. (I) Cardiac hypertrophy (heart weight/tibia length; HW/TL) and (J) lung edema of diabetic control or cKO mice (lung wet weight/lung dry weight ratio; LW wet/dry) were determined at 8-week endpoint. N=7. *: P<0.05 to diabetic control mice.
We performed the same diabetogenic protocol (Figure 2A) on female control and cKO mice. Similarly, diabetic stress induced systolic and diastolic dysfunction in female diabetic control mice, which was significantly worsened in female diabetic cKO mice (Figure 3A-C, Supp. Figure 3B, Supp. Table I). IVRT, MPI, left ventricular dilation, cardiac hypertrophy, and lung edema were not different between female diabetic control and cKO mice (Figure 3D-H). To determine if cardiac-specific Ndufs4 deletion mediates any sex-dependent changes on DCM progression, we compared the effects of STZ on cardiac function and geometry of male and female diabetic control and cKO mice. Although Ndufs4 deletion only significantly worsened IVRT and MPI in male diabetic cKO mice but in not female diabetic cKO mice, there were no significant interactions between sex and genotype by two-way ANOVA (Supp. Table II). Overall, the results suggest that NAD+ redox imbalance is a positive regulator for the progression of DCM in both sexes.
Figure 3. NAD(H) redox imbalance in the heart also exacerbates cardiac dysfunctions of diabetic female mice.
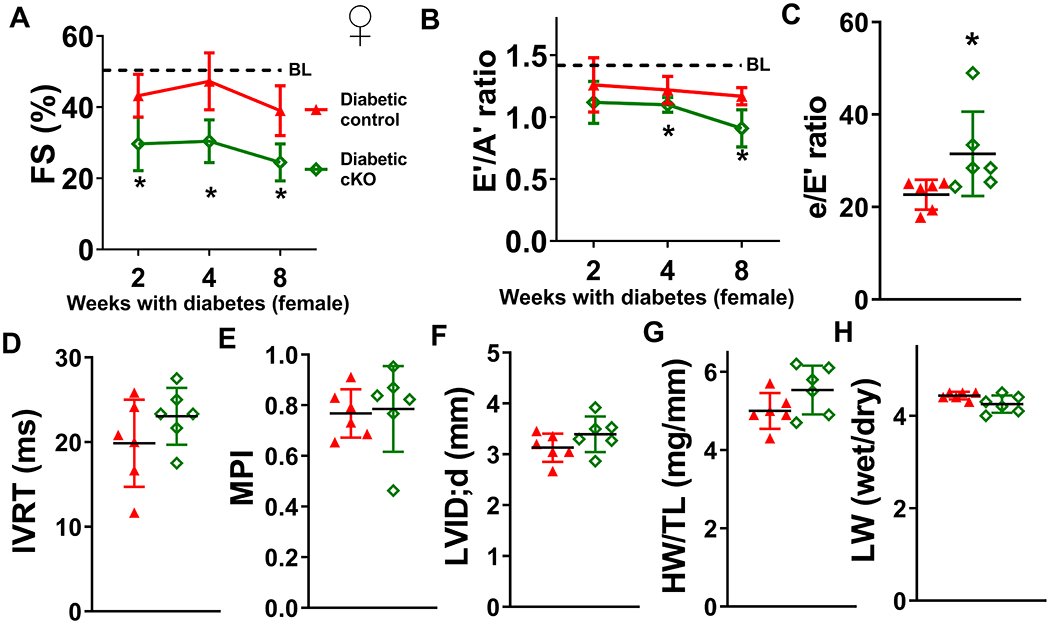
Control and cKO female mice were treated with STZ to induce diabetes. Cardiac function was assessed at 2, 4, and 8 weeks after induction of diabetes. (A) Longitudinal changes in fractional shortening (FS) and (B) E’/A’ ratio of diabetic control or cKO female mice were measured. Dotted lines indicate average baseline (BL) values of non-diabetic control mice. Numerical values are available in Supplementary Table I. (C) e/E’ ratio, (D) IVRT, (E) MPI, (F) LVID;d, (G) HW/TL, and (H) LW wet/dry ratio of diabetic control or cKO female mice were determined at 8-week endpoint. N=6. *: P<0.05 to diabetic control mice.
Tissue fibrosis was not associated with exacerbated DCM induced by NAD+ redox imbalance
Diabetic hearts are often associated with increased fibrosis, which contributes to cardiac dysfunction, especially diastolic dysfunction 29. We therefore quantified tissue collagen contents of mid-sectioned heart slices by trichrome staining. We observed similar fibrosis levels (~4%) in diabetic control and diabetic cKO hearts (Figure 4A). There was also no difference in cardiomyocyte size between the two groups (Figure 4B). Consistent with the fibrosis levels, expression of extracellular matrix (ECM)-related genes were similar between diabetic control and diabetic cKO hearts: transcript levels of Adamts proteinases, integrins, laminins, matrix metalloproteinases and collagen subtypes (Figure 4C-D, Supp. Figure IV–A–C) were not different. These results suggest that altered ECM environment and fibrosis do not account for the exacerbated dysfunction, but cardiomyocyte dysfunction may be the culprit.
Figure 4. NAD+ redox imbalance exacerbates diabetic cardiomyopathy, independent on tissue fibrosis.
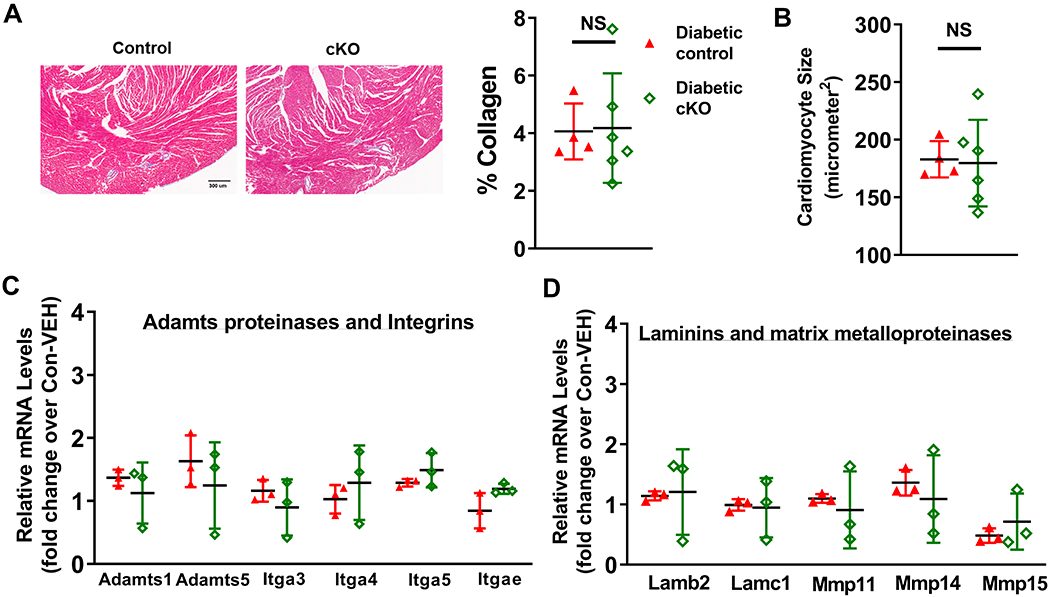
(A) Collagen levels of diabetic mouse hearts were quantified by trichrome staining. (B) Cardiomyocyte sizes of these hearts were quantified. N=4-6. Transcript expression levels of fibrosis-related genes were quantified by qPCR analyses. (C) Adamts proteinases and integrins, and (D) laminins and MMPs in diabetic control and diabetic cKO male hearts were measured. N=3.
Elevated SOD2 acetylation promoted oxidative stress in diabetic cKO hearts
NAD+ is required for deacetylation by Sirtuins, and NAD+ redox imbalance has been associated with protein hyperacetylation. Therefore, we explored the roles of NAD+ redox imbalance in regulating protein acetylation in diabetic hearts. Consistent with the lowered NAD+/NADH ratio, global protein acetylation increased in diabetic cKO hearts compared to diabetic controls (Figure 5A, Supp. Figure IV–D). Specifically, we observed that acetylation levels of superoxide dismutase 2 at lysine-68 (SOD2-K68Ac) were elevated in diabetic cKO hearts (diabetic cKO mean SOD2-K68Ac levels 2.8+/−0.6, diabetic control 1.2+/−0.2, P<0.01, Figure 5B, Supp. Figure IV–E). SOD2-K68Ac is known to suppress its antioxidant activity 30, suggesting that diabetic cKO hearts could be more prone to oxidative stress. Consistently, levels of protein carbonylation, an irreversible protein oxidative modification used as a marker of oxidative stress, were elevated in diabetic cKO hearts (Figure 5C, Supp. Figure IV–F). Transcript levels of NADPH oxidase (Nox) isoforms, key ROS generating proteins, were similar in diabetic control and diabetic cKO hearts (Figure 5D). These results suggest that NAD+ redox imbalance promotes oxidative stress through SOD2 acetylation in diabetic hearts.
Figure 5. NAD+ redox imbalance regulated SOD2 acetylation, protein oxidation and myofilament protein phosphorylation.
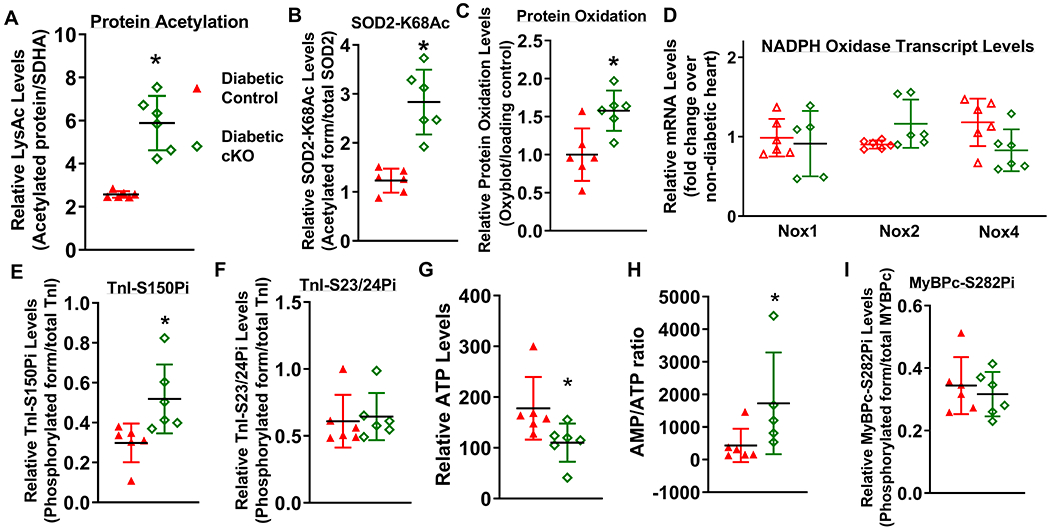
(A) Global lysine acetylation levels of protein extracts from diabetic control or diabetic cKO hearts were assessed by Western blot. (B) Acetylation levels of SOD2 at lysine-68 were assessed by Western blot analysis. (C) Protein oxidation levels of diabetic mouse hearts were determined by Oxyblot analysis. (D) Relative mRNA levels of pro-oxidant genes (Nox1, Nox2 and Nox4) were measured by qPCR. Phosphorylation levels of (E) TnI at Serine 150 (TnI-S150Pi), (F) TnI-S23/24Pi, (G) ATP levels and (H) AMP/ATP ratio were measured. Phosphorylation levels of (I) MyBPc at Serine 282 (MyBPc-S282Pi) were measured. N=6. *: P<0.05 to diabetic control mice.
NAD+ redox imbalance regulated TnI phosphorylation
We then examined if phosphorylation of myofilament proteins, which control cardiomyocyte contraction and relaxation, were altered by NAD+ redox imbalance in diabetic hearts. Phosphorylation states of troponin I (TnI) and myosin binding protein c (MyBPc) can modulate contractile and relaxation properties of the myocardium in diseased hearts 31, 32. TnI is an inhibitory subunit of troponin, and phosphorylation of TnI at S150 and at S23/24 (TnI-S150Pi and TnI-S23/24Pi) has been shown to coordinately regulate myofilament calcium sensitivity as an adaptive response in ischemic hearts 33, 34. Diabetic cKO hearts displayed increased levels of TnI-S150Pi compared to diabetic controls, while levels of TnI-S23/24Pi were not different (diabetic cKO mean TnI-S150Pi levels 0.52+/−0.16, diabetic control 0.29+/−0.09, P<0.05, Figure 5E-F, Supp. Figure IV–G–H). As TnI-S150Pi prolongs calcium dissociation 33, these results suggest that NAD+ redox imbalance induces TnI-S150 phosphorylation to exacerbate diastolic dysfunction in DCM (Figure 2D-F). AMPK, a key sensor of cellular energy states, has been shown to phosphorylates TnI-S150 35. AMPK-T172 phosphorylation is known to activate AMPK and AMPK-S485 phosphorylation is known to suppress AMPK activity 36. Phosphorylation levels at both sites were not different between diabetic control and diabetic cKO hearts (Supp. Figure IV–I–J) and AMPK activity assay detected similar AMPK activities in lysates of diabetic control and diabetic cKO hearts (Supp. Figure IV–K). In addition to the regulation by phosphorylation, AMPK is also allosterically activated by AMP binding, which is competitively inhibited by ATP 36, 37. Despite normal energetics in unstressed cKO hearts 15, we found that ATP levels decreased and AMP/ATP ratio increased in diabetic cKO hearts, compared to diabetic controls (Figure 5G,H). AMP and ADP levels trended to increase in diabetic cKO hearts (Supp. Figure IV–L,M). These results indicated lowered cellular energy status in diabetic cKO hearts compared to diabetic controls. The impaired energetics in diabetic cKO hearts potentially mediates AMPK activation allosterically and leads to increased phosphorylation of TnI-S150.
MyBPc is a sarcomeric protein that interacts with myosin and actin, and phosphorylation of MyBPc fine-tunes actin-myosin cross-bridge cycling to regulate cardiac contraction and relaxation. MyBPc S282 phosphorylation (MyBPc-S282Pi) is critical for normal cardiac function, and reduced MyBPc-S282Pi has been observed in failing hearts 32. Levels of MyBPc-S282Pi were unchanged in diabetic cKO hearts (Figure 5I, Supp. Figure IV–N), suggesting that it is not responsible for the worsened cardiac function.
Elevation of NAD+ levels ameliorated dysfunction of diabetic hearts
To validate the causal role of cardiac NAD+ redox imbalance in DCM, we examined the effects of elevation of cardiac NAD+ levels by cardiac NAMPT over-expression on DCM development. Transgenic mice expressing NAMPT in the heart (NAMPT) were crossed with cKO or control mice 16. The same diabetogenic protocol was used on these mice (Figure 6A). Over-expression of NAMPT in the heart increased NAD+/NADH ratio and NAD+ pool in diabetic cKO hearts, and improved systolic and diastolic dysfunction (Figure 6B-D, Supp. Figure V–A). These improvements occurred despite the similar extents of hyperglycemia in cKO and cKO:NAMPT mice (Supp. Figure V–B). The body weight changes at 8 weeks after diabetes induction were similar (Supp. Figure V–C). Heart rate, left ventricular dilation, hypertrophy, and lung edema were also unchanged, while IVRT and MPI were improved by cardiac-specific NAMPT over-expression (Supp. Figure V–D–I). Importantly, NAMPT over-expression lowered the levels of SOD2-K68Ac (diabetic cKO:NAMPT mean SOD2-K68Ac levels 0.54+/−0.06, diabetic cKO 1.07+/−0.12, P<0.01) and TnI-S150Pi (diabetic cKO:NAMPT mean TnI-S150Pi levels 0.41+/−0.21, diabetic cKO 0.83+/−0.26, P<0.05) in diabetic cKO hearts, while levels of TnI-S23/24Pi were similar (Figure 6E-G, Supp. Figure V–J–L). To determine if preventing NAD+ redox imbalance also ameliorates DCM progression in control mice, we compared the impacts of diabetic stress on cardiac function of cardiac-specific NAMPT transgenic mice and their control littermates. Diabetic stress induced systolic and diastolic dysfunction in control mice, which was ameliorated by cardiac NAMPT overexpression (Figure 6H,I). NAMPT expression did not change ATP levels, AMP/ATP ratio, AMPK activity, AMPK-T172Pi and AMPK-S485Pi levels (Supp. Figure V–M–Q). At the same time, levels of TnI-S150Pi were not different between diabetic NAMPT compared to diabetic controls (Supp. Figure V–R). The extent of cardiac dysfunction in diabetic controls was relatively modest compared to that of diabetic cKO mice and diabetic control hearts did not display depleted ATP levels (Supp. Figure V–S). These results suggest that the energetic stress-induced AMPK activation and TnI-S150 phosphorylation pathogenic axis was not activated in diabetic control hearts, and was not sensitive to NAMPT expression in this condition. Overall, the data from this report collectively support the causal roles of NAD+ redox imbalance in the progression of DCM.
Figure 6. Elevation of cardiac NAD+ levels alleviates diabetic cardiomyopathy in cKO and control mice.
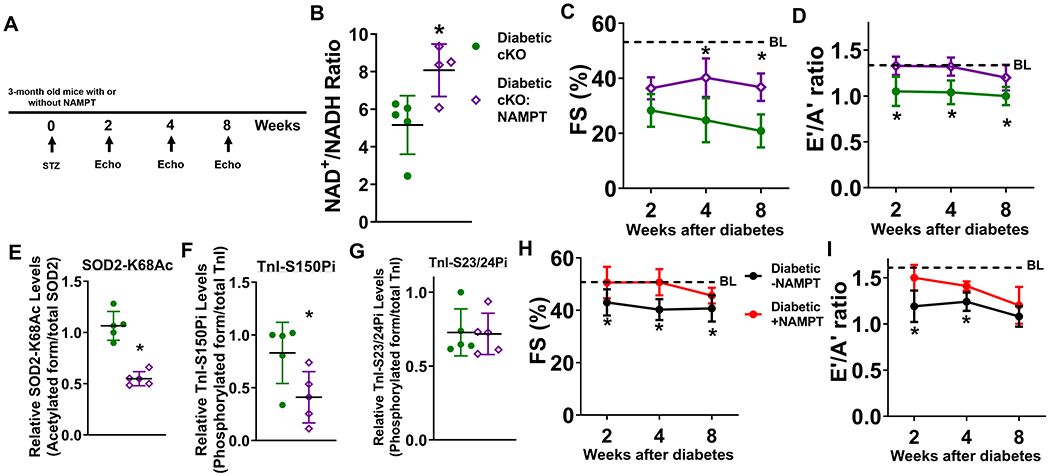
(A) Experimental plan. STZ was administered to male cKO and cKO:NAMPT mice to induce diabetes. Cardiac function of cKO and cKO:NAMPT mice was measured at 2, 4, and 8 weeks after induction of diabetes. (B) Cardiac NAD+/NADH ratio were measured. N=4-5. (C) FS and (D) E’/A’ ratio were assessed longitudinally. N=6. Dotted lines indicate average baseline (BL) values of non-diabetic control mice. Numerical values are available in Supplementary Table I. Levels of (E) SOD2-K68Ac, (F) TnI-S150Pi, and (G) TnI-S23/24Pi of indicated hearts were measured by Western blots. N=5. (H) FS and (I) E’/A’ ratio were measured longitudinally in control mice with or without NAMPT expression in the hearts after diabetes induction. N=5. *: P<0.05 to diabetic cKO mice or diabetic-NAMPT mice.
Discussion
Diabetes increases the risk of heart failure and causes DCM. Although pathogenic mechanisms such as lipotoxicity, oxidative stress and mitochondrial dysfunction have been demonstrated in diabetic hearts 2, the mechanisms by which diabetes-induced metabolic stresses lead to cardiac dysfunction are not fully understood. Hyperglycemia and dyslipidemia are the hallmarks of diabetes, and diabetes alters cardiac metabolism such as substrate utilization 2. NAD+ is an important redox cofactor for glucose and lipid oxidation. Changes in NAD+ metabolism such as NAD+ redox imbalance emerges as critical mediators of disease pathogeneses 15, 16. NAD+ redox imbalance has long been observed in tissues from diabetic mice 10, 17-19, but the hypothesis that diabetes-driven NAD+ redox imbalance promotes DCM remains to be tested. In this study, we demonstrated the causal role of NAD+ redox imbalance in promoting DCM (Figure 7). The key findings in supporting this notion are that 1) cardiac NAD+/NADH ratio decreases and precedes cardiac dysfunction after diabetic stress; 2) a pre-existing, lowered cardiac NAD+/NADH ratio exacerbates the progression of DCM, which is alleviated by elevating NAD+ levels; and 3) NAD+ redox imbalance mediates dysfunction of diabetic hearts in part by increased oxidative stress, impaired energetics and TnI-S150 phosphorylation, but not by enhanced cardiac fibrosis.
Figure 7. Graphical summary.
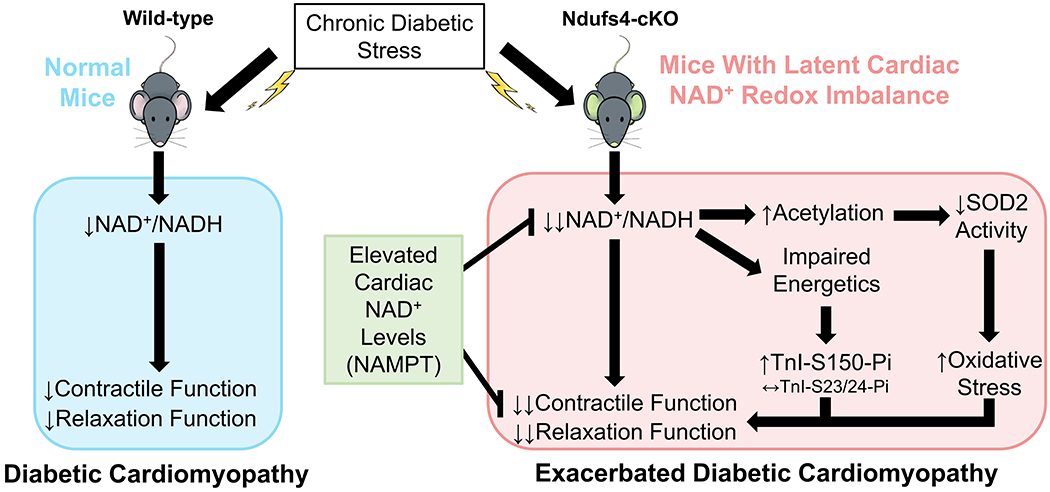
This study revealed the causal roles of NAD+ redox imbalance and cardiac dysfunction in hearts under chronic diabetic stress. In a mouse model with latent NAD+ redox imbalance (Ndufs4-cKO), we observed exacerbation of cardiac dysfunction in responses to chronic diabetic stress. The exacerbated contractile and relaxation dysfunction was mediated by protein hyperacetylation, in particular SOD2 acetylation which led to increased oxidative stress, and increased phosphorylation of TnI-S150 triggered by impaired energetics. Importantly, cardiac-specific elevation of NAD+ levels ameliorated diabetic cardiomyopathy and reversed pathogenic mechanisms, supporting the roles of NAD+ redox imbalance in the progression of diabetic cardiomyopathy.
Diabetic stress is associated with lowered NAD+/NADH ratio, also known as pseudohypoxia, in various tissues including hearts 18, 38. Functional decline in diabetic hearts has also been reported in parallel 2. However, whether NAD+ redox imbalance causes DCM has not been directly tested. We found that 16-week diabetic stress lowered cardiac NAD+/NADH ratio, accompanied by systolic and diastolic dysfunction. We also showed that the decline in cardiac NAD+/NADH ratio occurred before cardiac dysfunction. Importantly, we observed that a mouse model with lowered cardiac NAD+/NADH ratio (due to Ndufs4 deficiency in hearts) exhibits exacerbated DCM, and that cardiac dysfunction in diabetic cKO or control hearts can both be rescued by restoration of NAD+/NADH ratio (Figure 2,3,6). This is the first direct evidence to support the causal role of NAD+ redox imbalance in the progression of DCM. A recent report showed the role of hepatic NAD+ reductive stress (low NAD+/NADH ratio) in regulating changes in diabetes-induced metabolite levels (alpha-hydroxybutyrate) and insulin resistance in vivo 39, supporting the pathogenic role of NAD+ redox imbalance in diabetes and its complications. We previously demonstrated that NAD+ redox imbalance is a critical mediator in pressure overload-induced heart failure 16. Whether diabetes and hypertension have additive or synergistic effects on cardiac NAD+ redox imbalance to promote cardiac dysfunction remains to be determined.
While the causal role of NAD+ redox imbalance for DCM is well-supported by our current data, the mechanism by which diabetic stress induces NAD+ redox imbalance is not well-established. Previous studies have provided evidence that hyperglycemia and dyslipidemia promote NAD+ redox imbalance 21, 40. Mitochondrial dysfunction and impaired NAD+-dependent malate aspartate shuttle (MAS) have been shown as important mediators of the lowered NAD+/NADH ratio 21, 41, 42. MAS delivers cytosolic NADH to electron transport chain for ATP generation. Interestingly, MAS activity is suppressed in hypertrophied hearts in a NAD+ redox balance-dependent manner 16, 43, 44. Whether MAS activity decreases in diabetic hearts and leads to NAD+ redox imbalance requires further investigations.
Fibrosis is a hallmark of heart disease of different etiologies, especially in diabetic hearts 29, and we did observe a slight increase in fibrosis in STZ-treated diabetic hearts (Supp. Figure IV–O). However, we did not observe increased fibrosis associated with exacerbated dysfunction of diabetic cKO hearts. The similar extents of fibrosis in diabetic control and diabetic cKO hearts suggest that other mechanisms, e.g. cardiomyocyte dysfunction, is responsible for the exacerbated DCM. Our results suggest that the exacerbation can be mediated by cardiomyocyte dysfunction induced by elevated SOD2 acetylation and oxidative stress in diabetic cKO hearts. Other NAD+-sensitive protein acetylation, e.g. sensitized permeability transition pore by acetylation, may play a role in DCM progression 16.
Our data showed that diabetic cKO hearts displayed elevated TnI-S150Pi, exacerbated diastolic dysfunction and prolonged IVRT, which are all restored by NAMPT over-expression. AMPK-dependent TnI-S150Pi increases myofilament calcium sensitivity and prolongs calcium dissociation 31, 33, 34, while TnI-S23/24Pi decreases calcium sensitivity and accelerates calcium dissociation 31. Combination of TnI-S150Pi and TnI-S23/24Pi retains calcium sensitivity and accelerates calcium dissociation, playing an adaptive role during ischemic injury 31. Increased TnI-S150Pi and unchanged TnI-S23/24Pi in diabetic cKO hearts would suggest that NAD+ redox imbalance promotes TnI-S150Pi to play a maladaptive role in diastolic dysfunction. Although unstressed cKO hearts have normal energetics 15, we detected impaired energetics, indicated by depleted ATP levels and increased AMP/ATP ratio, in diabetic cKO hearts (Figure 5G,H). AMP binding activates AMPK via both enhancing T172 phosphorylation and allosteric activation independent of T172 phosphorylation, while ATP competitively inhibits AMP binding to AMPK 37, 45. The increased AMP/ATP ratio and unchanged T172 phosphorylation in diabetic cKO suggest that the impaired energetics potentially leads to allosteric activation of AMPK to promote TnI-S150Pi. Although we measured AMPK activities of heart lysates, the assay did not preserve the endogenous energetic environment and the effect of increased AMP/ATP ratio on allosteric activation of AMPK in diabetic cKO hearts in vivo cannot be determined. Our results suggest that NAD+ redox imbalance predisposes cKO hearts to diabetic cardiomyopathy by promoting energetic stress-dependent AMPK activation and TnI-S150Pi. Oxidative stress has also been shown to activate AMPK 46. We showed that diabetic cKO hearts exhibit increased oxidative stress, however, whether increased oxidative stress in cKO hearts contributes to AMPK activation requires further investigation.
We showed that cardiac NAMPT over-expression improved cardiac function in diabetic cKO mice, supporting that NAD+ redox imbalance is responsible for the exacerbated DCM progression. The observation that cardiac NAMPT over-expression also improved cardiac function in diabetic control mice without Ndufs4 deletion further supports a causal role of NAD+ redox imbalance in DCM progression. NAMPT over-expression in cardiomyocytes has been shown to promote hypertrophy and inflammation via secreted form of NAMPT (eNAMPT) 47. In this study, we observed no change in eNAMPT levels in plasma of mice with or without NAMPT over-expression (Supp. Figure V–T). Therefore, eNAMPT and its role in cardiac hypertrophy and inflammation did not play a role in the interpretation of this study. It is conceivable that disease mechanisms of DCM involve other NAD+-dependent pathways beyond NAD+ redox imbalance and protein acetylation reported here. NAD+ metabolism involves metabolites and enzymes in the NAD+ consumption, synthesis and redox pathways, coordinating cellular NAD+ homeostasis 48. Our study employed cardiac-specific perturbations of NAD+ redox balance (cKO and NAMPT) and demonstrated the roles of cardiac NAD+ redox imbalance in DCM. However, we cannot rule out the contributions of NAD+-derived metabolites and systemic changes in NAD+ metabolism in promoting DCM progression. Metabolites in the NAD+ salvage pathway, e.g. nicotinamide mononucleotide or nicotinamide riboside, are prime agents for pharmacologic elevation of NAD+ levels as therapeutics for heart disease 16. In addition, the benefit of activation of NAD+ synthesis by harnessing the de novo pathway and the role of NMRK2 up-regulation in cardiomyopathy have been reported 49. Therefore, further assessments of changes in NAD+ metabolism in DCM are warranted, and will likely identify new targets for therapy. A limitation of this study is that we employed a STZ-induced Type 1 diabetic model, and the roles of NAD+ redox imbalance in Type 1 DCM progression cannot be generalized to cardiomyopathy induced by Type 2 diabetes or aging. These risk factors have been associated with abnormal NAD+ metabolism; however, specific mechanisms that alter NAD+ metabolism (redox state, synthesis or consumption) in these conditions and their roles in cardiac dysfunction require further investigations.
This study establishes the causal role of NAD+ redox imbalance in DCM. Our results support that NAD+ redox imbalance mediates cardiac dysfunction in DCM in both sexes. This highlights the importance to identify adaptive and maladaptive mechanisms triggered by NAD+ redox imbalance in DCM. Our pre-clinical data also demonstrate the potential benefit of expanding the NAD+ pool as a therapy for DCM.
Supplementary Material
What is new?
Latent NAD+ redox imbalance in the heart exacerbates systolic and diastolic dysfunction in responses to chronic diabetes.
NAD+ redox imbalance elevates global protein and SOD2 acetylation, which promotes oxidative stress in diabetic hearts.
NAD+ redox imbalance promotes TnI-S150 phosphorylation and diastolic dysfunction via impaired energetics and oxidative stress in diabetic hearts.
Elevation of cardiac NAD+ levels mitigates NAD+ redox imbalance, dysfunction in diabetic hearts, and reversed SOD2- and TnI-dependent pathogenic mechanisms.
What are the clinical implications?
Our results suggest that elevation of NAD+ levels is therapeutic to heart failure progression attributable to chronic diabetes.
Our findings support the rationale to further characterize NAD+-dependent mechanisms to identify new therapeutic targets of heart failure.
Acknowledgements
CFL designed and performed the experiments, interpreted the data, and wrote the manuscript. YAC performed experiments and wrote the manuscript. ADC, XS, HG, and CML performed the experiments and analyzed the data. CFL is the guarantor of the study and has full access to all the data in the study and takes responsibility for the integrity of the data and accuracy of the data analysis.
RT and JS kindly provided mouse models. Dr. Richard Palmiter provided Ndufs4-flox mice via a MTA. All authors have reviewed and edited the manuscript.
Sources of Funding
This work has been supported in part by research funds from the NIH (1P20GM139763-01), American Heart Association (17SDG33330003), from a recruitment grant and a pilot grant of the Presbyterian Health Foundation of Oklahoma, and from a research grant of the Oklahoma Center for Adult Stem Cell Research, a program of TSET (all to CFL).
This work has also been supported by the NIH, K99AG051735 and R00AG051735 (both to YAC). NW BioTrust and NWBioSpecimen, are supported by the National Cancer Institute grant P30 CA015704, Institute of Translational Health Sciences grant UL1 TR000423, the University of Washington School of Medicine and Department of Pathology.
Nonstandard abbreviations or acronyms
- NAD+
nicotinamide adenine dinucleotide
- NADH
reduced nicotinamide adenine dinucleotide
- NAM
nicotinamide
- NMN
nicotinamide mononucleotide
- NAMPT
nicotinamide phosphoribosyltransferase
- NMNAT
nicotinamide mononucleotide adenylyltransferase
- ATP
adenosine triphosphate
- ADP
adenosine diphosphate
- AMP
adenosine monophosphate
- DCM
diabetic cardiomyopathy
- STZ
streptozotocin
- FS
fractional shortening
- IVRT
isovolumic relaxation time
- MPI
myocardial performance index
- SOD2
superoxide dismutase 2
- TnI
troponin I
- MyBPc
myosin binding protein c
- ECM
extracellular matrix
- Nox
NADPH oxidase
- AMPK
AMP-activated protein kinase
- MAS
malate aspartate shuttle
Footnotes
Disclosures
No financial or non-financial competing interest is disclosed associated with the investigation in this manuscript by all authors.
References
- 1.Boudina S and Abel ED. Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord. 2010;11:31–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ritchie RH and Abel ED. Basic Mechanisms of Diabetic Heart Disease. Circ Res. 2020;126:1501–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Imai S and Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24:464–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guarente L Calorie restriction and sirtuins revisited. Genes Dev. 2013;27:2072–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choudhary C, Weinert BT, Nishida Y, Verdin E and Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nature reviews Molecular cell biology. 2014;15:536–50. [DOI] [PubMed] [Google Scholar]
- 6.Mori V, Amici A, Mazzola F, Di Stefano M, Conforti L, Magni G, Ruggieri S, Raffaelli N and Orsomando G. Metabolic profiling of alternative NAD biosynthetic routes in mouse tissues. PloS one. 2014;9:e113939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshino J, Mills KF, Yoon MJ and Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell metabolism. 2011;14:528–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Picciotto NE, Gano LB, Johnson LC, Martens CR, Sindler AL, Mills KF, Imai S and Seals DR. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell. 2016;15:522–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mills KF, Yoshida S, Stein LR, Grozio A, Kubota S, Sasaki Y, Redpath P, Migaud ME, Apte RS, Uchida K, Yoshino J and Imai SI. Long-Term Administration of Nicotinamide Mononucleotide Mitigates Age-Associated Physiological Decline in Mice. Cell metabolism. 2016;24:795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trammell SA, Weidemann BJ, Chadda A, Yorek MS, Holmes A, Coppey LJ, Obrosov A, Kardon RH, Yorek MA and Brenner C. Nicotinamide Riboside Opposes Type 2 Diabetes and Neuropathy in Mice. Sci Rep. 2016;6:26933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trammell SA, Schmidt MS, Weidemann BJ, Redpath P, Jaksch F, Dellinger RW, Li Z, Abel ED, Migaud ME and Brenner C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun. 2016;7:12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D’Amico D, Ropelle ER, Lutolf MP, Aebersold R, Schoonjans K, Menzies KJ and Auwerx J. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016;352:1436–43. [DOI] [PubMed] [Google Scholar]
- 13.Airhart SE, Shireman LM, Risler LJ, Anderson GD, Nagana Gowda GA, Raftery D, Tian R, Shen DD and O’Brien KD. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PloS one. 2017;12:e0186459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonkowski MS and Sinclair DA. Slowing ageing by design: the rise of NAD(+) and sirtuin-activating compounds. Nature reviews Molecular cell biology. 2016;17:679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karamanlidis G, Lee CF, Garcia-Menendez L, Kolwicz SC Jr., Suthammarak W, Gong G, Sedensky MM, Morgan PG, Wang W and Tian R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell metabolism. 2013;18:239–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee CF, Chavez JD, Garcia-Menendez L, Choi Y, Roe ND, Chiao YA, Edgar JS, Goo YA, Goodlett DR, Bruce JE and Tian R. Normalization of NAD+ Redox Balance as a Therapy for Heart Failure. Circulation. 2016;134:883–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ido Y, Kilo C and Williamson JR. Cytosolic NADH/NAD+, free radicals, and vascular dysfunction in early diabetes mellitus. Diabetologia. 1997;40 Suppl 2:S115–7. [DOI] [PubMed] [Google Scholar]
- 18.Williamson JR, Chang K, Frangos M, Hasan KS, Ido Y, Kawamura T, Nyengaard JR, van den Enden M, Kilo C and Tilton RG. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes. 1993;42:801–13. [DOI] [PubMed] [Google Scholar]
- 19.Wu J, Jin Z, Zheng H and Yan LJ. Sources and implications of NADH/NAD(+) redox imbalance in diabetes and its complications. Diabetes Metab Syndr Obes. 2016;9:145–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berthiaume JM, Hsiung CH, Austin AB, McBrayer SP, Depuydt MM, Chandler MP, Miyagi M and Rosca MG. Methylene blue decreases mitochondrial lysine acetylation in the diabetic heart. Mol Cell Biochem. 2017;432:7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vazquez EJ, Berthiaume JM, Kamath V, Achike O, Buchanan E, Montano MM, Chandler MP, Miyagi M and Rosca MG. Mitochondrial complex I defect and increased fatty acid oxidation enhance protein lysine acetylation in the diabetic heart. Cardiovasc Res. 2015;107:453–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsu CP, Oka S, Shao D, Hariharan N and Sadoshima J. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ Res. 2009;105:481–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carroll PA, Diolaiti D, McFerrin L, Gu H, Djukovic D, Du J, Cheng PF, Anderson S, Ulrich M, Hurley JB, Raftery D, Ayer DE and Eisenman RN. Deregulated Myc requires MondoA/Mlx for metabolic reprogramming and tumorigenesis. Cancer Cell. 2015;27:271–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiao YA, Kolwicz SC, Basisty N, Gagnidze A, Zhang J, Gu H, Djukovic D, Beyer RP, Raftery D, MacCoss M, Tian R and Rabinovitch PS. Rapamycin transiently induces mitochondrial remodeling to reprogram energy metabolism in old hearts. Aging (Albany NY). 2016;8:314–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gu H, Zhang P, Zhu J and Raftery D. Globally Optimized Targeted Mass Spectrometry: Reliable Metabolomics Analysis with Broad Coverage. Anal Chem. 2015;87:12355–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore A, Shindikar A, Fomison-Nurse I, Riu F, Munasinghe PE, Ram TP, Saxena P, Coffey S, Bunton RW, Galvin IF, Williams MJ, Emanueli C, Madeddu P and Katare R. Rapid onset of cardiomyopathy in STZ-induced female diabetic mice involves the downregulation of pro-survival Pim-1. Cardiovasc Diabetol. 2014;13:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wold LE and Ren J. Streptozotocin directly impairs cardiac contractile function in isolated ventricular myocytes via a p38 map kinase-dependent oxidative stress mechanism. Biochem Biophys Res Commun. 2004;318:1066–71. [DOI] [PubMed] [Google Scholar]
- 28.Karunanayake EH, Hearse DJ and Mellows G. Streptozotocin: its excretion and metabolism in the rat. Diabetologia. 1976;12:483–8. [DOI] [PubMed] [Google Scholar]
- 29.Russo I and Frangogiannis NG. Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and therapeutic opportunities. Journal of molecular and cellular cardiology. 2016;90:84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dai DF, Chen T, Szeto H, Nieves-Cintron M, Kutyavin V, Santana LF and Rabinovitch PS. Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol. 2011;58:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nixon BR, Walton SD, Zhang B, Brundage EA, Little SC, Ziolo MT, Davis JP and Biesiadecki BJ. Combined troponin I Ser-150 and Ser-23/24 phosphorylation sustains thin filament Ca(2+) sensitivity and accelerates deactivation in an acidic environment. Journal of molecular and cellular cardiology. 2014;72:177–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Copeland O, Sadayappan S, Messer AE, Steinen GJ, van der Velden J and Marston SB. Analysis of cardiac myosin binding protein-C phosphorylation in human heart muscle. Journal of molecular and cellular cardiology. 2010;49:1003–11. [DOI] [PubMed] [Google Scholar]
- 33.Oliveira SM, Zhang YH, Solis RS, Isackson H, Bellahcene M, Yavari A, Pinter K, Davies JK, Ge Y, Ashrafian H, Walker JW, Carling D, Watkins H, Casadei B and Redwood C. AMP-activated protein kinase phosphorylates cardiac troponin I and alters contractility of murine ventricular myocytes. Circ Res. 2012;110:1192–201. [DOI] [PubMed] [Google Scholar]
- 34.Nixon BR, Thawornkaiwong A, Jin J, Brundage EA, Little SC, Davis JP, Solaro RJ and Biesiadecki BJ. AMP-activated protein kinase phosphorylates cardiac troponin I at Ser-150 to increase myofilament calcium sensitivity and blunt PKA-dependent function. The Journal of biological chemistry. 2012;287:19136–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia D and Shaw RJ. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Molecular cell. 2017;66:789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jeon SM. Regulation and function of AMPK in physiology and diseases. Exp Mol Med. 2016;48:e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gowans GJ, Hawley SA, Ross FA and Hardie DG. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell metabolism. 2013;18:556–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berthiaume JM, Kurdys JG, Muntean DM and Rosca MG. Mitochondrial NAD(+)/NADH Redox State and Diabetic Cardiomyopathy. Antioxid Redox Signal. 2019;30:375–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goodman RP, Markhard AL, Shah H, Sharma R, Skinner OS, Clish CB, Deik A, Patgiri A, Hsu YH, Masia R, Noh HL, Suk S, Goldberger O, Hirschhorn JN, Yellen G, Kim JK and Mootha VK. Hepatic NADH reductive stress underlies common variation in metabolic traits. Nature. 2020;583:122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, Stancakova A, Goetzman E, Lam MM, Schwer B, Stevens RD, Muehlbauer MJ, Kakar S, Bass NM, Kuusisto J, Laakso M, Alt FW, Newgard CB, Farese RV Jr., Kahn CR and Verdin E. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Molecular cell. 2011;44:177–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eto K, Tsubamoto Y, Terauchi Y, Sugiyama T, Kishimoto T, Takahashi N, Yamauchi N, Kubota N, Murayama S, Aizawa T, Akanuma Y, Aizawa S, Kasai H, Yazaki Y and Kadowaki T. Role of NADH shuttle system in glucose-induced activation of mitochondrial metabolism and insulin secretion. Science. 1999;283:981–5. [DOI] [PubMed] [Google Scholar]
- 42.Tan C, Tuch BE, Tu J and Brown SA. Role of NADH shuttles in glucose-induced insulin secretion from fetal beta-cells. Diabetes. 2002;51:2989–96. [DOI] [PubMed] [Google Scholar]
- 43.Lewandowski ED, O’Donnell JM, Scholz TD, Sorokina N and Buttrick PM. Recruitment of NADH shuttling in pressure-overloaded and hypertrophic rat hearts. Am J Physiol Cell Physiol. 2007;292:C1880–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Donnell JM, Kudej RK, LaNoue KF, Vatner SF and Lewandowski ED. Limited transfer of cytosolic NADH into mitochondria at high cardiac workload. Am J Physiol Heart Circ Physiol. 2004;286:H2237–42. [DOI] [PubMed] [Google Scholar]
- 45.Gowans GJ and Hardie DG. AMPK: a cellular energy sensor primarily regulated by AMP. Biochem Soc Trans. 2014;42:71–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Auciello FR, Ross FA, Ikematsu N and Hardie DG. Oxidative stress activates AMPK in cultured cells primarily by increasing cellular AMP and/or ADP. FEBS Lett. 2014;588:3361–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pillai VB, Sundaresan NR, Kim G, Samant S, Moreno-Vinasco L, Garcia JG and Gupta MP. Nampt secreted from cardiomyocytes promotes development of cardiac hypertrophy and adverse ventricular remodeling. Am J Physiol Heart Circ Physiol. 2013;304:H415–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mericskay M Nicotinamide adenine dinucleotide homeostasis and signalling in heart disease: Pathophysiological implications and therapeutic potential. Arch Cardiovasc Dis. 2016;109:207–15. [DOI] [PubMed] [Google Scholar]
- 49.Diguet N, Trammell SAJ, Tannous C, Deloux R, Piquereau J, Mougenot N, Gouge A, Gressette M, Manoury B, Blanc J, Breton M, Decaux JF, Lavery GG, Baczko I, Zoll J, Garnier A, Li Z, Brenner C and Mericskay M. Nicotinamide Riboside Preserves Cardiac Function in a Mouse Model of Dilated Cardiomyopathy. Circulation. 2018;137:2256–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.