

Abstract
Cellular senescence, first observed and defined through cell culture studies, is a cell fate associated with essentially permanent cell cycle arrest and that can be triggered by a variety of inducers. Emerging evidence suggests senescence is a dynamic process with diverse functional characteristics. Depending on the tissue, type of inducer, and time since induction, senescent cells can promote tissue repair and re-modeling, prevent tumor development, or contribute to age-related disorders and chronic diseases, including cancers. Senescent cell characteristics appear to depend on multiple factors and be influenced by the milieu and other senescent cells locally and at a distance. We review diverse phenotypes of senescent cells originating from different cell types, senescence inducers over time since induction of senescence, and across conditions and diseases. This background is essential to inform further understanding about senescent cell subtypes and will point towards rational senescence-modulating strategies for achieving therapeutic benefit.
Keywords: Senolytics, Cellular senescence, Deleterious senescent cell subtype, Helper senescent cell subtype
1. Introduction
Senescence is a cell fate that entails an essentially permanent cell cycle arrest in response to multiple factors, including repeated replication, potentially oncogenic mutations and activated oncogenes, DNA damage, metabolic insults, damage/danger signals, neutrophils, radiation, and cytotoxic and other drugs [1–4]. Senescent cells often undergo profound morphological and functional changes, including overall cellular, nuclear, and mitochondrial enlargement, chromosomal deconvolution and chromatin remodeling, gene expression changes, heightened metabolic activity, and sustained survival with resistance to apoptosis [5–9]. Some, but not all senescent cells acquire a senescence-associated secretary phenotype (SASP), with release of interleukins and other cytokines, chemokines that attract, activate, and anchor immune cells, pro-apoptotic factors, matrix metalloproteinases, growth factors, reactive metabolites, bioactive lipids including saturated fatty acids and ceramides, bradykines, and prostanoids, micro-RNAs, mitochondrial DNA, and other non-coding nucleotides, and extracellular vesicles, among other factors [10–12].
Senescence appears to be a programmed process that can contribute to tissue homeostasis and such physiological functions as embryonic and fetal development [13], parturition [14], tumor suppression [15], wound healing [16], and tissue repair [17]. Normally, senescent cells are cleared by the immune system soon after their appearance, but with chronic stress or immune dysfunction, senescent cells can persist and develop an increasingly pro-inflammatory, pro-apoptotic SASP [18–20]. Accumulation of senescent cells has emerged as a root cause contributor to stem cell, progenitor, tissue, and systemic dysfunction, predisposing to multiple disorders that account for much age- and chronic disease-related morbidity and mortality, including cancers, diabetes and its complications, neurodegenerative, cardiovascular, hepatic, and kidney diseases, osteoporosis, arthritis, and many more [10].
The molecular architecture of senescent cells determines their pathophysiological functions and secretory phenotype. Senescence is a dynamic multistep process that is impacted by intra- and extra-cellular signals leading to heterogeneity in functional outcomes [21]. The molecular characteristics of senescent cells depend on the stage of senescence progression, the nature of the inducer, and cell type of origin. The anti-apoptotic mechanisms of senescent cells and SASP profiles are shaped continuously by the microenvironment, immune cells, pathogens, and other neighboring senescent cells [20, 22].
2. Differences among senescent cells
2.1. Cell type of origin
Senescent cells in tissues and organs contribute to not only aging phenotypes but also to the genesis of multiple chronic diseases. Senescent cells can have SASP profiles that vary among cell types of origin. In vitro comparisons of senescent preadipocytes, endothelial cells, myoblasts, fibroblasts, and epithelial cells reveal differences in the extent of upregulation of SASP factors [23]. Senescent endothelial cells and preadipocytes tend to have greater SASP factor expression than other cell types, such as epithelial cells or myoblasts [23]. Higher SASP expression by such cells implies that some senescent cells may contribute more to chronic inflammation and organismal age-related dysfunction than others. Senescent cells in different tissues exert their effects through distinct mechanisms. For example, senescent preadipocytes release activin A, which impairs adipogenesis, leading to metabolic dysfunction [24, 25]. Senescent melanocytes induce telomere dysfunction in surrounding cells through the chemokine, CXCR3, contributing to impaired skin regenerative potential with aging [26]. Currently, it is unclear if all cell types can become senescent. Traditionally, only replicative or mitotically competent cells have been thought to be susceptible to senescence. However, it appears that terminally differentiated cells can also acquire features of senescence [27]. For example, cortical and hippocampal neurons from old mice can develop DNA damage, SASP production, heterochromatinization, and senescence-associated β-galactosidase (SA β-gal) activity, among other features of cellular senescence, many of which may contribute to neural aging phenotypes [28]. Similarly, several features of senescence have been identified in terminally differentiated, non-dividing hepatocytes [29] and cardiomyocytes [4] of aged mice, with increased expression of the DNA damage markers, p21Cip1 and p16Ink4a, along with pro-fibrotic SASP induction. Emerging evidence suggests that specific immune cell populations, such as macrophages [30] and T cells [31, 32], may display senescent-like features, which raises the question of whether these cells actually are or can become senescent. Additionally, the SASP comprises many molecules indicative of inflammation, adding to the complex nature of discerning whether immune cells are truly senescent or are reacting as non-senescent normal immune cells to age- or disease-related signals.
2.2. Type of inducers
Many of the intra- and extra-cellular signals that contribute to cells’ entering the senescent cell fate include those related to tissue or cellular damage or cancer development. These include damaged DNA, telomeric uncapping or dysfunction, exposure to extra-cellular DNA, oncogene activation, replicative stress or inducers of proliferation (such as growth hormone/IGF-1), protein aggregates, misfolded proteins, failed protein removal through decreased autophagy, saturated lipids and other bioactive lipids (bradykines, ceramides, certain prostaglandins, etc.), reactive metabolites (e.g., reactive oxygen species [ROS], hypoxia, or hyperoxia), mechanical stress (e.g., bone-on-bone stress in osteoarthritis), inflammatory cytokines (e.g., TNFα)[33], damage-associated molecular patterns (DAMPs, e.g., released intracellular contents signaling breakage of neighboring cells), and pathogen-associated molecular patterns (PAMPs; e.g., bacterial endotoxins or components of viruses) [18, 22]. These inducers can activate one or more senescence-promoting transcription factor cascades, in some cases involving p16INK4A and retinoblastoma protein (Rb), in others, p53 and p21CIP1, both of these pathways, or other pathways [34]. These transcription factor cascades enforce replicative arrest and influence epigenetic changes, causing altered expression of hundreds of genes. The SASP may vary depending on the type of inducers. Variations in the SASP can occur both in the specific factors released and in the magnitude of their expression. For example, senescent fibroblasts can release factors such as platelet-derived growth factor AA (PDGF-AA) [16] and matricellular protein CCN1 [35], accelerating wound closure upon acute skin injury. Additionally, senescent fibroblasts can drive angiogenesis by secreting vascular endothelial growth factor (VEGF), promoting endothelial cell invasion and increasing vascularization [36]. Prolonged aberrant persistence of senescent cells can have detrimental effects in promoting cancer. For example, chemotherapy-induced senescent fibroblasts can promote tumor recurrence and metastasis by releasing SASP factors such as interleukins (IL)-6 and −8 [37].
2.3. Time since induction of senescence
Cellular senescence takes longer to become established than other cell fates: replication, differentiation, apoptosis, or necrosis. From initiation to the attainment of a complete state of cellular senescence takes from 10 days to 6 weeks, depending on the cell type and the inducers driving the cell into senescence, at least in vitro [38]. The growth arrest of senescent cells is related to increased p16INK4A or p21CIP1, which are inhibitors of the cyclin/cyclin-dependent kinase (CDK) complexes that mediate cell cycle progression, and this may or may not involve activation of p53 [39]. Activation of p53 and p21CIP1 in senescent cells can be transient; p53 and p21CIP1 protein levels generally decrease after growth arrest has become established [19]. p21CIP1 expression decreases after this peak, but p16INK4A can increase and contribute to maintaining growth arrest in some, but not all, senescent cells [19].
Although the initial senescence-inducing signals mediate cell cycle arrest, senescent cells can continue to remodel their chromatin and transcriptional landscape to develop other features of senescence, such as the SASP, in some, but not all, senescent cells. If these senescent cells persist, they can continue evolving toward a late phase of senescence. Activation of endogenous Line-1 (L1) transpositional elements can contribute to a pro-inflammatory SASP phenotype in late senescence [19]. L1 elements can lead to chromosomal rearrangements, loss of histones, and activation of cGAS/STING in response to transposon insertion-induced DNA damage, which exacerbates the inflammatory SASP through interferon 1 (IFN-1) [40]. More investigation is needed to identify molecular and functional characteristics of early vs. deep senescence across various cell types to understand potential beneficial vs. detrimental effects on health-span, and if targeting transposon-related reverse transcriptases impacts viability of pro-inflammatory late senescent cells of the D-subtype and their impact on other cells, tissues, and systemic function.
3. Differences in the SASP
3.1. The SASP is modifiable by the microenvironment
An intracellular IL-1α/ miR-146a/b/ IL-6/ C/EBP-β loop [41] and related p38/NF-κB [42], cGAS/STING [43], and mTOR-mediated pathways [44] appear to contribute to the changes in gene expression that result in the SASP [2]. Expression and characteristics of the SASP vary considerably with the location of the senescent cell, cell type of origin, cause of senescence, hormonal milieu, drugs, presence of pathogens, and the passage of time. Endogenous or pharmacological glucocorticoids can attenuate the release of a subset of SASP factors [45]. Further, the SASP can be downregulated or upregulated by circulating micro-RNAs as well as mt-DNA [12, 46, 47]. Inhibitors of pathways involved in SASP expression, such as metformin and rapamycin, attenuate the SASP, partly explaining their beneficial effects on age-related tissue dysfunction [48, 49]. JAK-STAT3 inhibition leads to reprogramming of the SASP, which decreases frailty and bone loss in old animals [50]. Even intermittent application of some SASP inhibitors appears to alleviate conditions related to aging [51]. However, drugs that are SASP inhibitors can act through other mechanisms, making it difficult to disentangle their effects on age-related phenotypes that are due to SASP modulation from other effects. These other mechanisms can also contribute to side effects that may not be related to SASP suppression. For example, rapamycin can induce insulin resistance by directly acting on TORC2 [52].
3.2. Endocrine and paracrine impact of the SASP
Accumulation of senescent cells can cause local and systemic inflammation, tissue destruction, immune system inhibition, and stem and progenitor cell dysfunction due in part to the SASP. SASP factors can spread senescence to normal, non-senescent cells both locally and systemically: intraperitoneal transplantation of small numbers of senescent ear fibroblasts or preadipocytes in young mice was sufficient to cause early onset of physical dysfunction [53]. The transplanted cells remained within the peritonaeum, yet the recipient mice developed senescent cells in distant organs including the spleen, intestine, and fat depots that originated from the recipients’ own, previously normal cells, illustrating the autocrine and paracrine nature of the SASP [53]. This was linked to development of premature aging-like phenotypes, including decreased activity, reduced grip strength, and premature death from a range of conditions similar to those causing death in naturally-aged mice.
The SASP is regulated both at the transcriptional and post-translational levels and can intensify over time. Positive feedback loops that reinforce the SASP and other complex regulatory mechanisms may contribute to enhanced SASP factor secretion. An example of such a self-amplifying positive feedback loop involves regulation of IL-1α secretion [54]. IL-1α activates the transcription factors, C/EBPβ and NF-κB, leading to enhanced production of IL-6 and IL-8 and IL-1α itself. Similarly, the chemokine receptor CXCR2 and its ligand, GRO-α, can be upregulated in senescent cells, contributing to the formation of a network that reinforces growth arrest in a p53-dependent manner [55]. The SASP can also be regulated at an epigenetic level. For example, SASP induction involves recruitment of BRD4 to senescence-activated super-enhancers adjacent to SASP factor genes [56]. Among other pathways, mTOR and autophagy are also SASP regulators and can reinforce senescence [44, 57].
Through the SASP, senescent cells can signal and influence their surrounding environment and distant tissues, which can be beneficial or detrimental. For example, keratinocytes transiently exposed to SASP factors can have increased regenerative capacity. However, sustained exposure to the SASP can lead to the induction of senescence in these cells [58].
The SASP is the best-studied mechanism by which senescent cells influence their neighbors, but it is not the only one. For example, senescent cells can signal and influence adjacent cells through juxtacrine NOTCH/JAG1 signaling [59], ROS production [26], or by cargo transfer, which occurs through the formation of cytoplasmic bridges or release of exosomes [60]. Factors contributing to the spread of senescence vary among cell types and need to be explored further.
3.3. Anti-apoptotic defenses vary among senescent cells
Like cancer cells such those as in some types of B-lymphomas or lymphocytic leukemias, senescent cells can resist the pro-apoptotic effects of their own SASP through upregulating pro-survival/ anti-apoptotic defenses. The first identification of the responsible Senescent Cell Anti-apoptotic Pathways (SCAPs) was achieved by comparing the transcriptomic and proteomic profiles of senescent to non-senescent human cells (preadipocytes and Human Umbilical Vein Endothelial Cells [HUVECs]) [5]. This indicated higher expression of anti-apoptotic networks related to BCL-2/BCL-xL, PI3K/AKT, p53/p21CIP1/serpines, dependence receptors/Src and tyrosine kinases, and HIF-1α in senescent than non-senescent cells [5, 33]. Subsequently, it was discovered that senescent cells can also have upregulated chaperones, such as HSP-90 [61], which interact with AKT or ERK to promote apoptosis resistance. Up-regulation of these SCAPs might be related to senescence-associated mitochondrial dysfunction: mitochondria in senescent cells can have decreased membrane potential and increased resistance to fragmentation [62]. However, not all senescent cells are the same. Different types of senescent cells express different SASP factors, senescence markers, and, importantly, SCAP pathways to resist apoptosis. For example, senescent human preadipocytes do not depend on BCL- 2 family pro-survival proteins. On the other hand, senescent HUVECs display upregulation of BCL- 2 family members, particularly BCL- xL, as compared to non-senescent HUVECs [5]. Other SCAPs in senescent HUVECs include components of the PI3 kinase and HIF- 1α pathways. Senescent cells can have also increased FOXO4, which prevents cell death by sequestering p53 in the nucleus [63].
Only some senescent are pro-apoptotic and pro-inflammatory. Here, we term these cells Deleterious (D) senescent cells, which may comprise about 30 to 70 % of the senescent cell population (unpublished observations). Other senescent cells, termed here Helper- (H-) senescent cells, do not appear to release a substantial amount of inflammatory and pro-apoptotic factors (unpublished observations). H-senescent cells might promote stem cell function, wound healing, and tissue and remodelling through secretion of growth factors, such as PDGF-AA and TGF-β [13, 16, 64]. However, if H-senescent cells persist, they may promote cancer relapse (if they harbor oncogenic mutations and escape senescence-associated replicative arrest) and possibly other disorders. We speculate there could be mechanisms through which D- and H-senescent cells interconvert, but this has to be fully explored.
4. Distinct effects of senolytics on different types of senescent cells
The identification of molecular pathways that permit viability of senescent cells provided the route to developing agents that selectively induce apoptosis in senescent cells vs. non-senescent cells, senolytic drugs. The Src/tyrosine kinase inhibitor, dasatinib, and the flavonoid, quercetin, were shown to induce apoptosis in senescent, but not non-senescent, primary human preadipocytes and HUVECs, respectively. Dasatinib targets, among others, dependence receptor/Src kinase SCAPs, while quercetin targets the BCL-2/BCL-xL, PI3K/AKT, and p53/p21CIP1/serpine SCAPs [5]. Based on this discovery, inhibitors of pro-survival BCL-2 family members, including navitoclax (ABT263), which inhibits BCL-xL/BCL-w/BCL-2, were later shown to induce apoptosis in senescent human fibroblasts and endothelial cells, but not senescent preadipocytes [6, 65]. The selective BCL-xL inhibitors, A1331852, and A1155463, were also shown to be senolytic against the same types of senescent cells [66]. Fisetin, which is related to quercetin, was discovered to be senolytic [66]. Several additional senolytic interventions have been identified based on the strategy of targeting SCAPs. For example, a FOXO-4-related peptide that inhibits the PI3K/AKT/p21CIP1/serpine SCAP by blocking interaction between p53 and the transcription factor FOXO4, leads to release of p53 from the nucleus and induction of cell-intrinsic apoptosis in some types of senescent cells [63].
In addition to pathway-based senolytic development, high-throughput approaches were also used to identify new agents [61]. Such an approach was used to identify HSP-90 inhibitors as a class of senolytics. HSP-90, a family of ubiquitously expressed molecular chaperones, can promote cell survival through stabilization of the AKT or ERK signaling pathways, that may become upregulated during senescence. Disruption of the HSP-90-AKT interaction inhibited the PI3K/AKT pathway, resulting in selective killing of senescent cells. The HSP-90 inhibitors, geldanamycin and tanespimycin (17-AAG), were senolytic in fibroblasts, endothelial cells, and mesenchymal stem cells [61]. In drug screens using models of oncogene-induced senescence in normal cells vs. therapy-induced senescent cells, cardiac glycosides (CGs) were identified as a class of senolytics [67, 68]. Cardiac glycosides, such as digoxin and ouabain, inhibit Na+, K+-ATPase, possibly contributing to their senolytic activity. Ouabain or digoxin also elevate several pro-apoptotic BCL-2 family proteins that can trigger apoptosis in senescent fibroblasts [68].
Elevated lysosomal SA β-gal activity in senescent cells has been exploited to develop drugs with selectivity for senescent cells. One example is a drug delivery system that involves encapsulating cytotoxic drugs with galacto-oligosaccharides. These capsules can be loaded with various cytotoxic compounds and take advantage of increased SA β-gal activity to release their cargo near senescent cells. A recent study used a similar strategy involving a galactose-modified prodrug [69]. Here, preferential processing of galactose-modified duocarmycin, a cytotoxic conjugate, was used to eliminate several senescent cell types. Potential problems with this approach are that not all senescent cells have increased SA β-gal activity and not every cell with high SA β-gal activity is senescent. For example, activated macrophages, which may not be truly senescent, can have increased SA β-gal activity[70]. This raises concerns about the senolytic specificity of approaches based on targeting SA β-gal activity.
Several senolytics, including dasatinib, quercetin, fisetin, navitoclax, the HSP-90 inhibitor, alvespimycin, and a peptide that targets the BCL-2- and p53-related SCAPs, have been demonstrated to be effective in reducing senescent cell burden in mice, with decreases in SA-βgal activity, p16Ink4a, p21Cip1, and SASP factor mRNAs, telomere-associated foci, and other senescent cell indicators [53, 71, 72]. Among the effects of senolytics in mice so far are: 1) restoration of metabolic function in obese mice [24]; 2) decreased hepatic steatosis in old mice [29]; 3) decreased frailty, osteoporosis, loss of intervertebral disc glycosaminoglycans, and spondylosis in progeroid Ercc1-/Δ mice [73]; 4) decreased gait disturbance in mice following radiation damage to a leg and hematological dysfunction caused by whole-body radiation [5, 74]; 5) improved cognitive and behavioral functions [75, 76]; 6) improved pulmonary function and reduced pulmonary fibrosis in mice with bleomycin-induced lung damage, a model of idiopathic pulmonary fibrosis [71]; 7) improved physical function and increased median lifespan in old mice [53, 73]; 8) improved cardiac ejection fraction and fractional shortening in old mice, enhanced vascular reactivity in old mice, and decreased vascular calcification and increased vascular reactivity in hypercholesterolemic, high fat-fed ApoE−/− mice [5, 72]; and 9) reduced senescent cell-like, intimal foam cell/macrophages in vascular plaques in high fat-fed LdlR−/− mice [30];
The target of senolytic drugs is senescent cells rather than a single receptor or an enzyme per se. Developing senolytic drugs arguably has more in common with developing antibiotics than, for example, an antihypertensive. In the cases of senolytics and antibiotics, the strategy is to transiently target cell-specific defense networks with the goal of eliminating a damaging cell type that depends on those networks, sometimes using agents or combinations of agents to hit multiple nodes on such networks. This can increase effectiveness. It also can reduce adverse effects by more specifically targeting pathogens or senescent cells that depend on a network than using approaches against a single, narrow target that necessitate sustained drug presence, such as a receptor or enzyme. The latter frequently can be shared across other cells types that are not the main target. Indeed, drugs with single or limited targets, such as navitoclax, have off- target apoptotic effects on multiple nonsenescent cell types, limiting clinical translation or requiring local application rather than systemic delivery, such as oral administration [77]. To most effectively target multiple senescent cell types in different diseases, a combination of senolytics or individual agents that have multiple pro-survival SCAP network targets, administered transiently in a hit-and-run fashion, may turn out to be the most effective approach, permitting oral/systemic use and minimizing adverse events from off target effects or the injections or other approaches needed for local administration.
5. Threshold Theory of Cellular Senescence
The Threshold Theory of Cellular Senescence holds that once senescent cell burden increases beyond the point that clearance by the immune system can keep pace with feed-forward generation of new senescent cells caused by the SASP, senescent cell burden becomes self-amplifying, leading to progressive tissue dysfunction and occurrence of co-morbidities [18, 78]. Several findings support this theory. 1) Senescent cell turnover slows with age in mice, and this correlates with mortality [79]. 2) Patients who receive chemotherapy for cancer have a higher burden of senescent cells and develop age-related diseases faster than age-matched individuals [80, 81]; 3) Total body clearance of senescent cells alleviates age-related brain inflammation, cognitive impairment, and psychiatric deficits in mice [75]. 4) Fewer senescent cells had to be transplanted into obese than age-matched lean mice to cause dysfunction and early death[53]; obesity is associated with increased senescent cell burden[25, 82]; 5) Transplanting 106 senescent preadipocytes intraperitoneally into younger mice caused decreased treadmill endurance, grip strength, daily activity, food intake, and lifespan, while transplanting 2x105 senescent cells did not [53]. Hence, reducing the burden of D senescent cells to below a certain threshold may prove to have a positive impact on overall health and alleviate multiple disease conditions, a possibility that remains to be tested. Permanently reducing the ability to generate H senescent cells might impair such physiological functions as tissue repair and wound healing. Data about the abundance and characteristics of D and H senescent cells across tissues are necessary for devising interventions to selectively lower the burden of persistent, pro-inflammatory, pro-apoptotic D senescent cells, while not eliminating potentially beneficial H senescent cells, or at least preserving ability for H cells to be generated when needed.
6. Conclusions
Cellular senescence is a dynamic process that can be modified by the local microenvironment, host milieu, passage of time, drugs, and disease states. Senescent cells may be of helper or deleterious subtypes, depending on context and type of inducer of cellular senescence. However, little is currently known about regulation and progression of the SASP, windows of vulnerability/resistance of senescent cells to different types of senolytics, and impact of disease states and the microenvironment on different populations of senescent cells. It is critically important to understand the diverse populations of senescent cells and their functional relevance across tissues and diseases. It will be important to compare variation in senescence markers between the D and H senescent cell subtypes in vivo. Such knowledge could contribute to developing strategies for selectively removing D senescent cells at the right time points, while leaving H senescent cells available for physiological functions such as tissue repair. Also, we anticipate that H senescent cells might be interconvertible with the D subtype through as yet unknown mechanisms that need to be explored further. Efforts must be taken to understand which tissues preferentially harbor D and H senescent cells to determine if local or systemic clearance of senescent cells may be the better option in aged or diseased patients.
Figure 1.
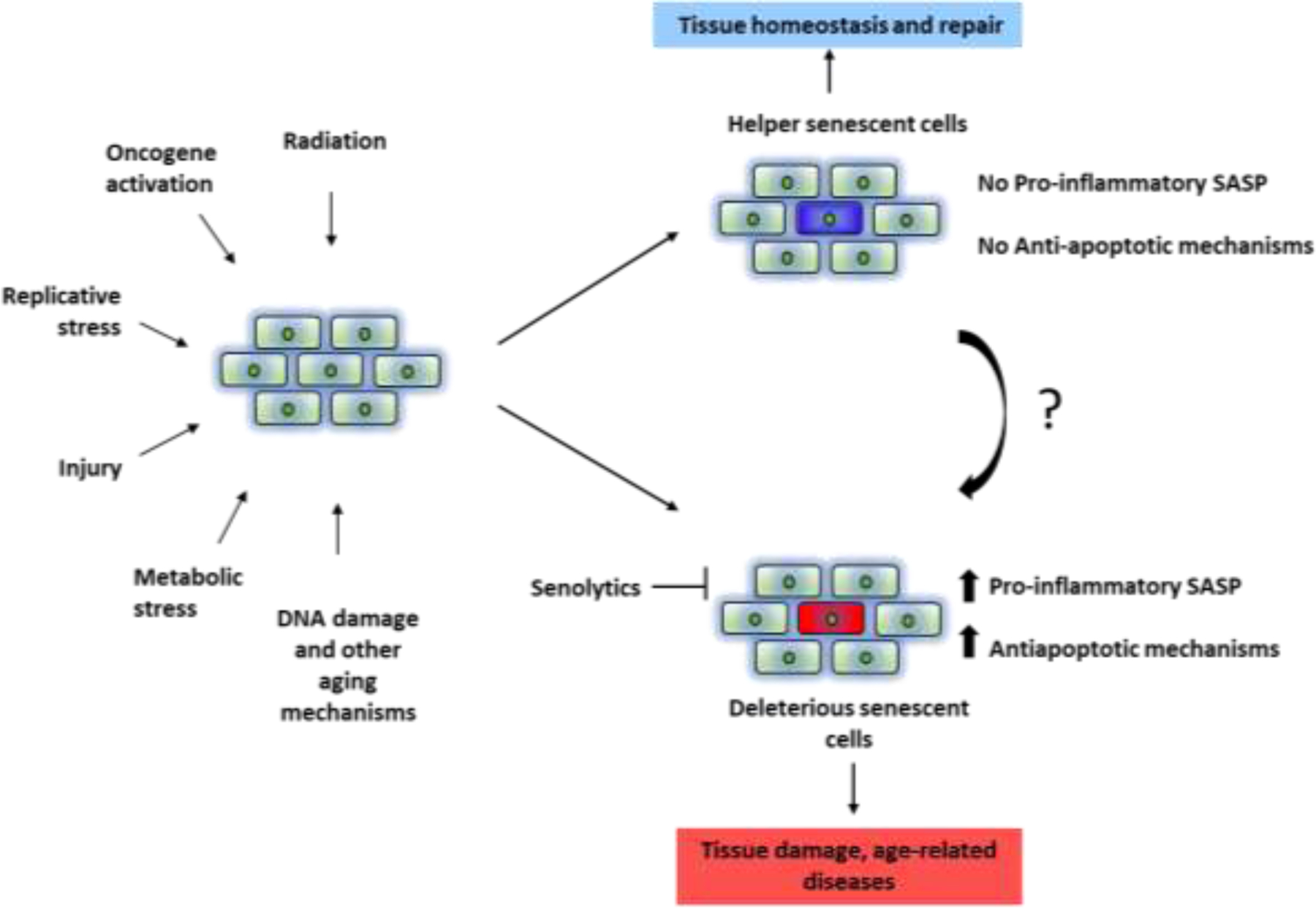
Theoretical Characteristics of Helper (H) and Deleterious (D) Senescent Cells. D-senescent cells may have a pro-inflammatory, pro-apoptotic, tissue-damaging SASP and be susceptible to senolytics. H-senescent cells may not have a SASP, may not have upregulated SCAPS, and might not be cleared by senolytics. H senescent cells may be convertible into D senescent cells through as yet unknown mechanisms.
HIGHLIGHTS.
Cellular senescence is a dynamic process and appears to be a heterogenous cell fate.
Senescent cells can be of helper (H) or deleterious (D) subtypes.
Lowering the burden of the D-subtype below a threshold may alleviate co-morbidities and enhance health-span, while reducing adverse effects from targeting the H-subtype.
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health (grants R37 AG13925, R33 AG61456, R01 AG072301, R01 AG61414, P01 AG62413, and UH3 AG56933), Robert and Arlene Kogod, the Connor Fund, Robert J. and Theresa W. Ryan, and the Noaber Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Hayflick L and Moorhead PS, The serial cultivation of human diploid cell strains. Experimental Cell Research, 1961. 25(3): p. 585–621. [DOI] [PubMed] [Google Scholar]
- 2.Tchkonia T, et al. , Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. Journal of Clinical Investigation, 2013. 123(3): p. 966–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lagnado A, et al. , Neutrophils induce paracrine telomere dysfunction and senescence in ROS-dependent manner. The EMBO journal, 2021: p. e106048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson R, et al. , Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. The EMBO journal, 2019. 38(5): p. e100492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu Y, et al. , The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell, 2015. 14(4): p. 644–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang E, Senescent Human Fibroblasts Resist Programmed Cell Death, and Failure to Suppress bcl2 Is Involved. Cancer Research, 1995. 55(11): p. 2284–2292. [PubMed] [Google Scholar]
- 7.Pathak RU, Soujanya M, and Mishra RK, Deterioration of nuclear morphology and architecture: A hallmark of senescence and aging. Ageing Research Reviews, 2021. 67: p. 101264. [DOI] [PubMed] [Google Scholar]
- 8.Xiong Y and Zhou L, The Signaling of Cellular Senescence in Diabetic Nephropathy. Oxidative Medicine and Cellular Longevity, 2019. 2019: p. 7495629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumari R and Jat P, Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Frontiers in Cell and Developmental Biology, 2021. 9(485). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khosla S, et al. , The role of cellular senescence in ageing and endocrine disease. Nature Reviews Endocrinology, 2020. 16(5): p. 263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coppé J-P, et al. , The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annual Review of Pathology: Mechanisms of Disease, 2010. 5(1): p. 99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iske J, et al. , Senolytics prevent mt-DNA-induced inflammation and promote the survival of aged organs following transplantation. Nature Communications, 2020. 11(1): p. 4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muñoz-Espín D, et al. , Programmed Cell Senescence during Mammalian Embryonic Development. Cell, 2013. 155(5): p. 1104–1118. [DOI] [PubMed] [Google Scholar]
- 14.Cubro H, et al. , Mechanisms of vascular dysfunction in the interleukin-10–deficient murine model of preeclampsia indicate nitric oxide dysregulation. Kidney International, 2021. 99(3): p. 646–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dimri GP, et al. , Regulation of a senescence checkpoint response by the E2F1 transcription factor and p14(ARF) tumor suppressor. Molecular and cellular biology, 2000. 20(1): p. 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Demaria M, An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell, 2014. 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim K-H, et al. , Matricellular Protein CCN1 Promotes Regression of Liver Fibrosis through Induction of Cellular Senescence in Hepatic Myofibroblasts. Molecular and Cellular Biology, 2013. 33(10): p. 2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kirkland JL and Tchkonia T, Senolytic drugs: from discovery to translation. Journal of internal medicine, 2020. 288(5): p. 518–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Cecco M, et al. , L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature, 2019. 566(7742): p. 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prata LGPL, et al. , Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Seminars in immunology, 2018. 40: p. 101275–101275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herranz N and Gil J, Mechanisms and functions of cellular senescence. The Journal of clinical investigation, 2018. 128(4): p. 1238–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Camell CD, et al. , Senolytics reduce coronavirus-related mortality in old mice. Science, 2021: p. eabe4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schafer MJ, et al. , The senescence-associated secretome as an indicator of age and medical risk. JCI insight, 2020. 5(12): p. e133668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palmer AK, et al. , Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging cell, 2019. 18(3): p. e12950–e12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu M, et al. , Targeting senescent cells enhances adipogenesis and metabolic function in old age. eLife, 2015. 4: p. e12997–e12997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Victorelli S, et al. , Senescent human melanocytes drive skin ageing via paracrine telomere dysfunction. The EMBO journal, 2019. 38(23): p. e101982–e101982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.von Zglinicki T, Wan T, and Miwa S, Senescence in Post-Mitotic Cells: A Driver of Aging? Antioxidants & redox signaling, 2021. 34(4): p. 308–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jurk D, et al. , Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging cell, 2012. 11(6): p. 996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ogrodnik M, et al. , Cellular senescence drives age-dependent hepatic steatosis. Nature communications, 2017. 8: p. 15691–15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Childs BG, et al. , Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science, 2016. 354(6311): p. 472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lucas CL, et al. , Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nature Immunology, 2014. 15(1): p. 88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Covre LP, et al. , The role of senescent T cells in immunopathology. Aging Cell, 2020. 19(12): p. e13272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beyne-Rauzy O, et al. , Tumor necrosis factor alpha induces senescence and chromosomal instability in human leukemic cells. Oncogene, 2004. 23(45): p. 7507–7516. [DOI] [PubMed] [Google Scholar]
- 34.Alcorta DA, et al. , Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proceedings of the National Academy of Sciences, 1996. 93(24): p. 13742–13747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jun J-I and Lau LF, The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nature cell biology, 2010. 12(7): p. 676–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coppé J-P, et al. , Secretion of Vascular Endothelial Growth Factor by Primary Human Fibroblasts at Senescence*. Journal of Biological Chemistry, 2006. 281(40): p. 29568–29574. [DOI] [PubMed] [Google Scholar]
- 37.Davalos AR, et al. , Senescent cells as a source of inflammatory factors for tumor progression. Cancer metastasis reviews, 2010. 29(2): p. 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hernandez-Segura A, Brandenburg S, and Demaria M, Induction and Validation of Cellular Senescence in Primary Human Cells. Journal of visualized experiments : JoVE, 2018(136): p. 57782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Passos JF, et al. , Feedback between p21 and reactive oxygen production is necessary for cell senescence. Molecular Systems Biology, 2010. 6(1): p. 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zierhut C and Funabiki H, Regulation and Consequences of cGAS Activation by Self-DNA. Trends in Cell Biology, 2020. 30(8): p. 594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhaumik D, et al. , MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging, 2009. 1(4): p. 402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Freund A, Patil CK, and Campisi J, p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. The EMBO Journal, 2011. 30(8): p. 1536–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Glück S, et al. , Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nature Cell Biology, 2017. 19(9): p. 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laberge R-M, et al. , MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nature Cell Biology, 2015. 17(8): p. 1049–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laberge R-M, et al. , Glucocorticoids suppress selected components of the senescence-associated secretory phenotype. Aging cell, 2012. 11(4): p. 569–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baker JR, et al. , MicroRNA-570 is a novel regulator of cellular senescence and inflammaging. The FASEB Journal, 2019. 33(2): p. 1605–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Badi I, et al. , MicroRNA-34a Induces Vascular Smooth Muscle Cells Senescence by SIRT1 Downregulation and Promotes the Expression of Age-Associated Pro-inflammatory Secretory Factors. The Journals of Gerontology: Series A, 2015. 70(11): p. 1304–1311. [DOI] [PubMed] [Google Scholar]
- 48.Moiseeva O, et al. , Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell, 2013. 12(3): p. 489–498. [DOI] [PubMed] [Google Scholar]
- 49.Wang R, et al. , Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging cell, 2017. 16(3): p. 564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu M, et al. , JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proceedings of the National Academy of Sciences, 2015. 112(46): p. E6301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bitto A, et al. , Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. eLife, 2016. 5: p. e16351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lamming DW, et al. , Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science (New York, N.Y.), 2012. 335(6076): p. 1638–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu M, et al. , Senolytics improve physical function and increase lifespan in old age. Nature Medicine, 2018. 24(8): p. 1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Orjalo AV, et al. , Cell surface-bound IL-1α is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proceedings of the National Academy of Sciences, 2009. 106(40): p. 17031–17036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Acosta JC, et al. , Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell, 2008. 133(6): p. 1006–1018. [DOI] [PubMed] [Google Scholar]
- 56.Tasdemir N, et al. , BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance. Cancer discovery, 2016. 6(6): p. 612–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kang C, et al. , The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science, 2015. 349(6255): p. aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ritschka B, et al. , The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes & development, 2017. 31(2): p. 172–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parry AJ, et al. , NOTCH-mediated non-cell autonomous regulation of chromatin structure during senescence. Nature Communications, 2018. 9(1): p. 1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alibhai FJ, et al. , Cellular senescence contributes to age-dependent changes in circulating extracellular vesicle cargo and function. Aging Cell, 2020. 19(3): p. e13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fuhrmann-Stroissnigg H, et al. , Identification of HSP90 inhibitors as a novel class of senolytics. Nature Communications, 2017. 8(1): p. 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dalle Pezze P, et al. , Dynamic Modelling of Pathways to Cellular Senescence Reveals Strategies for Targeted Interventions. PLOS Computational Biology, 2014. 10(8): p. e1003728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baar MP, et al. , Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell, 2017. 169(1): p. 132–147.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saito Y, et al. , Exercise enhances skeletal muscle regeneration by promoting senescence in fibro-adipogenic progenitors. Nature Communications, 2020. 11(1): p. 889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhu Y, et al. , Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell, 2016. 15(3): p. 428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhu Y, et al. , New agents that target senescent cells: the flavone, fisetin, and the BCL-X(L) inhibitors, A1331852 and A1155463. Aging, 2017. 9(3): p. 955–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guerrero A, et al. , Cardiac glycosides are broad-spectrum senolytics. Nature Metabolism, 2019. 1(11): p. 1074–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Triana-Martínez F, et al. , Identification and characterization of Cardiac Glycosides as senolytic compounds. Nature Communications, 2019. 10(1): p. 4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cai Y, et al. , Elimination of senescent cells by β-galactosidase-targeted prodrug attenuates inflammation and restores physical function in aged mice. Cell Research, 2020. 30(7): p. 574–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hall BM, et al. , p16(Ink4a) and senescence-associated β-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging, 2017. 9(8): p. 1867–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schafer MJ, et al. , Cellular senescence mediates fibrotic pulmonary disease. Nature communications, 2017. 8: p. 14532–14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roos CM, et al. , Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging cell, 2016. 15(5): p. 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yousefzadeh MJ, et al. , Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine, 2018. 36: p. 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chang J, et al. , Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nature medicine, 2016. 22(1): p. 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ogrodnik M, et al. , Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging cell, 2021. 20(2): p. e13296–e13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ogrodnik M, et al. , Obesity-Induced Cellular Senescence Drives Anxiety and Impairs Neurogenesis. Cell Metabolism, 2019. 29(5): p. 1061–1077.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sharma AK, et al. , The Senolytic Drug Navitoclax (ABT-263) Causes Trabecular Bone Loss and Impaired Osteoprogenitor Function in Aged Mice. Frontiers in Cell and Developmental Biology, 2020. 8(354). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Short S, et al. , Senolytics and senostatics as adjuvant tumour therapy. EBioMedicine, 2019. 41: p. 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Karin O, et al. , Senescent cell turnover slows with age providing an explanation for the Gompertz law. Nature Communications, 2019. 10(1): p. 5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ness KK, et al. , Premature Physiologic Aging as a Paradigm for Understanding Increased Risk of Adverse Health Across the Lifespan of Survivors of Childhood Cancer. Journal of Clinical Oncology, 2018. 36(21): p. 2206–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cupit-Link MC, et al. , Biology of premature ageing in survivors of cancer. ESMO Open, 2017. 2(5): p. e000250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Minamino T, et al. , A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nature Medicine, 2009. 15(9): p. 1082–1087. [DOI] [PubMed] [Google Scholar]