

Abstract
The critical role of site-specific phosphorylation in eukaryotic transcription has motivated efforts to decipher the complex phosphorylation patterns exhibited by the carboxyl-terminal domain (CTD) of RNA polymerase II. Phosphorylation remains a challenging post-translational modification to characterize by mass spectrometry owing to the labile phosphate ester linkage and low stoichiometric prevalence, two features that complicate analysis by high-throughput MS/MS methods. Identifying phosphorylation sites represents one significant hurdle in decrypting the CTD phosphorylation, a problem exaggerated by a large number of potential phosphorylation sites. An even greater obstacle is decoding the dynamic phosphorylation pattern along the length of the periodic CTD sequence. Ultraviolet photodissociation (UVPD) is a high-energy ion activation method that provides ample backbone cleavages of peptides while preserving labile post-translational modifications that facilitate their confident localization. Herein, we report a quantitative parallel reaction monitoring (PRM) method developed to monitor spatiotemporal changes in site-specific Ser5 phosphorylation of the CTD by cyclin-dependent kinase 7 (CDK7) using UVPD for sequence identification, phosphosite localization, and differentiation of phosphopeptide isomers. We capitalize on the series of phospho-retaining fragment ions produced by UVPD to create unique transition lists that are pivotal for distinguishing the array of phosphopeptides generated from the CTD.
Graphical Abstract
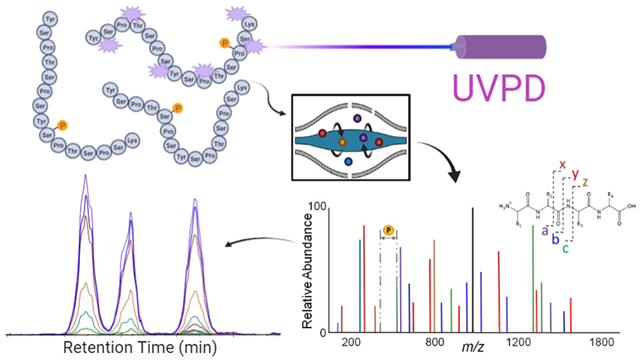
INTRODUCTION
The largest subunit of eukaryotic RNA polymerase II (RNAP II) consists of a unique C-terminal domain (CTD) containing multiple heptad repeats whose consensus sequence is Y1S2P3T4S5P6S7.1 Specific phosphorylation along the length of the CTD is vital for the recruitment of integral factors involved in transcription.2,3 Barring prolines, the remaining residues can undergo phosphorylation or dephosphorylation at various stages of transcription, all choreographed by an array of kinases and phosphatases. Of these residues, Ser2 and Ser5 are the most frequently modified residues found in almost every round of transcription.4,5 Hyper-phosphorylation of Ser5 is typically observed during the initiation of transcription, whereas Ser2 sites are predominantly phosphorylated during the transition from initiation to elongation.6 Precise phosphorylation on Ser2 and Ser5 is essential for successful eukaryotic transcription. Errors in transcription factor recruitment due to imprecise CTD phosphorylation can directly lead to severe cellular developmental defects or death.4,7,8 The critical role of site-specific phosphorylation in transcription has catalyzed extensive efforts to decipher the CTD phosphorylation pattern.9 However, it is unclear how CTD kinases distinguish the repeating heptad CTD motif once recruited to RNAP II. Identifying phosphorylation sites represents one significant challenge in decrypting the CTD phosphorylation; an even more daunting challenge is quantifying the dynamic phosphorylation pattern.
Although mass spectrometry is recognized as one of the best methods for qualitative and quantitative analysis in proteomics, tracking post-translational modifications (PTMs) adds an extra degree of complexity. In particular, phosphorylation may be difficult to localize and quantify owing to a labile phosphate ester linkage, low stoichiometric prevalence, and the acidic nature of the phosphoryl group, which suppresses ionization efficiencies of peptides in bottom-up proteomics workflows.10–13 Moreover, the challenges of identification and characterization of phosphopeptides are exacerbated by analysis of peptides that contain multiple potential phosphorylation sites, such as those containing multiple Ser, Thr, and Tyr residues. Site-specific quantification via bottom-up LC-MS/MS workflows has typically been accomplished by one of three isotope-centric techniques. The first entails derivatization of N-terminal amines or free side-chain amines with isobaric isotope-containing tags prior to peptide analysis.13–15 In isobaric labeling-based quantification, peptides in each sample set are derivatized with isotopic variants of an isobaric mass tag and pooled before analysis using mass spectrometry (MS). Because the tags are isobaric, peptides labeled with an isotopic variant of the tag appear as a single precursor m/z value, and fragmentation of these modified precursor ions generates two types of product ions: reporter ions with different isotopic masses and peptide fragment ions. Quantification of tagged peptides is achieved by directly comparing the relative abundances of reporter ions to that of the peptide selected for activation and fragmentation.14–16 A second prominent method utilizes metabolic incorporation of stable isotope-labeled amino acids during cell culture (SILAC).17–19 In SILAC methods, two identical cell populations are labeled with light and heavy isotope amino acids, respectively. Labeled cell lysates are combined and analyzed simultaneously by LC-MS/MS, in which each peptide appears as a doublet with distinct mass differences corresponding to the heavy or light amino acids used in culture. The quantitative difference between the two samples is calculated directly by comparing the difference in abundances of each pair of matching isotopic doublets.17–19 Alternatively, the use of stable isotopic peptide analogues as internal standards that mirror the target peptides allows a proficient means for absolute quantitation.20,21 Because the internal standard should have identical physicochemical characteristics as the peptide of interest, the ratio between their abundances is regarded as being proportional to their concentrations.20,21 However, using internal peptide standards requires amino acid synthesis and precise site modification of each target peptide of interest. Consequently, the cost associated with the stable isotope quantitative strategies may become cost-prohibitive with an extensive list of targets.
Label-free quantitation (LFQ) based on spectral counting or signal abundances of specific targeted ions offers a cost-effective alternative to stable isotope quantitative strategies and also minimizes the difficulty of sample preparation.22,23 In LFQ workflows, quantification is accomplished by integrating extracted ion chromatograms (XICs) generated for specific peptide ions derived from LC-MS data.24,25 The peptides of interest may be monitored based on MS1 or MS/MS features; however, the selection of ions based on MS1 analysis alone precludes the distinction of isomeric peptides or coeluting peptide targets. In contrast, MS/MS analysis yields more specific information about the peptide sequence and the potential for site-localization of PTMs, thus providing an advantage over ones based on MS1 methods. Data acquisition in MS-based quantitative assays uses one of two primary targeted methods: selected reaction monitoring (SRM) or parallel reaction monitoring (PRM). SRM acquisition is routinely performed using a triple quadrupole (QQQ) mass spectrometer, where predefined sets of transitions (precursor/product ion pairs) are monitored over time for precise quantification.26 Alternatively, PRM allows all fragment ions from a precursor ion to be monitored in parallel using an ion trap or Orbitrap instrument, thus generally increasing sensitivity and specificity.27 The attributes of high resolution and high mass accuracy with PRM have accelerated its widespread adoption for quantitative proteomics using various ion activation methods, including both collisional activation28–30 and electron-based techniques.31–33
Adaptation of PRM methods to other mass spectrometry platforms and alternative ion activation methods would further advance the scope of PRM strategies for quantitative proteomics. Alternate MS/MS methods offer new opportunities for enhancing the successful differentiation of isoform-specific peptides, particularly those having the same sequence but different sites of modification. Ultraviolet photodissociation (UVPD) is one of the newer ion activation methods that has emerged for proteomics applications.34,35 In addition to promoting extensive backbone cleavages of peptides that result in high sequence coverage, UVPD preserves labile post-translational modifications, allowing confident localization of PTMs.36–41 These features make UVPD a compelling option for implementation in quantitative proteomic workflows such as PRM. In this work, we report a quantitative method developed to monitor spatiotemporal changes in site-specific Ser5 phosphorylation of the periodic CTD sequence by the cyclin-dependent kinase 7 (CDK7) kinase module using UVPD for peptide identification and phosphosite localization. The capability of UVPD for pinpointing phosphorylation sites of peptides containing multiple Ser, Tyr, and Thr residues offers a unique asset that proves essential for quantitative mapping of phosphorylation of the CTD. We demonstrate targeted quantitation of stoichiometric changes of phosphorylation of the CTD based on XICs of fragment ions unique to each isomeric phosphopeptide via a targeted PRM approach on a high-resolution Orbitrap mass spectrometer. The yeast and human CTDs were analyzed to probe the spatiotemporal phosphorylation patterns of the entire CTD span in vitro to determine additional CTD control by CDK7.
RESULTS AND DISCUSSION
UVPD has been used for the qualitative characterization of phosphopeptides in several recent studies aimed at elucidating sites of CTD phosphorylation of RNAP II.37–39,42–44 Here, we extend the use of 193 nm UVPD to a quantitative PRM workflow (Figure 1). This study establishes the capacity of UVPD-based PRM analysis for determining with spatiotemporal resolution the dynamic site-specific pattern of phosphorylation of the CTD of RNA polymerases II by kinases. We used various substrates to interrogate the preference and specificity of CDK7 toward RNA polymerase II, including several 21-mer CTD constructs, a 26-repeat S. cerevisiae CTD variant, and human CTD in the distal region (sequences shown in Figure S1). Preliminary examination of phosphorylation of various CTD constructs and human CTD peptides using HCD as the MS/MS method resulted in several cases in which phosphorylation sites were ambiguously or incorrectly localized (see Figures S2–S4, Tables S1 and S2). The misassignments occurred owing to the absence of key fragment ions that retained the modifications or inadequate bracketing of the phosphorylation sites via lack of backbone cleavages at each side of the modified residues. The ability to bracket each residue is particularly crucial for pinpointing specific sites in CTD peptides which contain up to five potential phosphorylation sites per heptad. The identification of phosphorylation sites with single amino acid resolution based on the informative UVPD fragmentation patterns allows a better understanding of kinase activity along the periodic sequence composed of the heptad (Y1S2P3T4S5P6S7). Relative quantitation of the phosphorylation site-specific peptides is obtained by monitoring specific fragment ions (i.e., transitions from the precursor peptide to characteristic fragment ions upon UVPD) for each phosphopeptide isomer.
Figure 1.
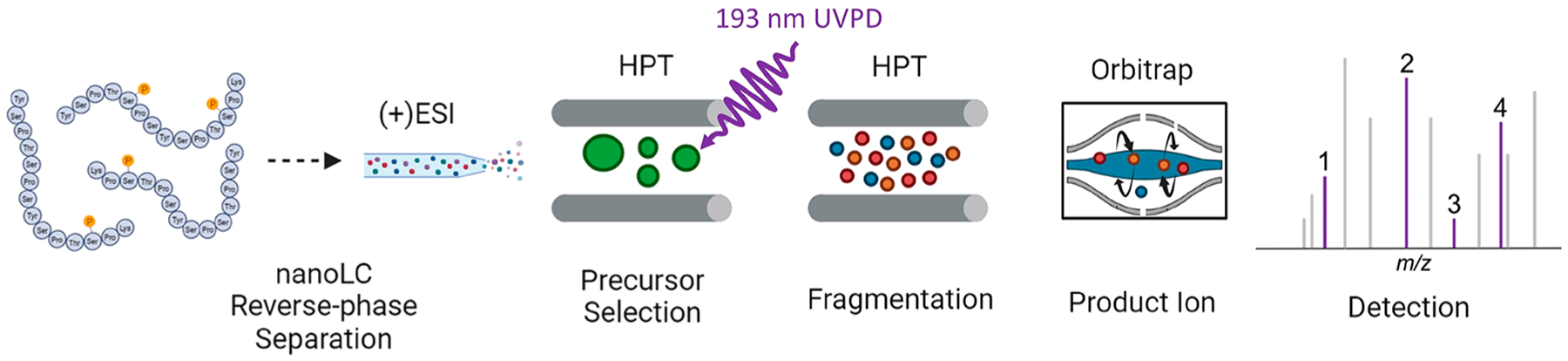
A 193 nm UVPD parallel-reaction monitoring (PRM) workflow schematic showing an isolated precursor fragmented in the Lumos high-pressure ion trap.
The earliest phosphorylation marks of the CTD during active transcription are Ser5 modifications;38 thus our particular interest in CDK7 activity is its reported role in these earliest phosphorylation marks.44,45 In addition, the kinase activity of CDK7 also activates other CTD kinases to promote transcription.46 As previously reported, CDK7 promoted preferential phosphorylation of Ser5 of the CTD, an outcome supported by modeling the active site of CDK7 and the favorable orientation of Ser7 at the active site and adjacent proline in a hydrophobic pocket.44 CDK7 is also believed to be responsible for the phosphorylation of Ser7 in vivo, but the activity seems to be very low in vitro.47 To understand the heptad activity and preference of CDK7, we used two heptad constructs as substrates for CDK7. We used a 21-mer S5E′ containing a surrogate Ser5 “phosphorylated” heptad bounded on either side of a wild-type heptad (YSPTSPS). The acidic nature of the glutamic acid at S5E in the middle heptad mimics the Ser5 phosphorylation and disallows modification at that site.48 A chromatographic trace of 3-S5E′ (GPGSGMEPSYSPTSPSYSPTEPSYSPT) is shown in Figure S5, and peptide assignments based on UVPD are provided in Table S3. Two distinct mono-phosphorylated sites emerge, one low-abundance species exhibiting Ser7 phosphorylation at s23 and another high-abundance Ser5 phosphorylation at s13 on the wild-type heptad. This result shows that CDK7 prefers the phosphorylation of Ser5 as long as it is available. We then tested the CDK7 activity in a 21-mer CTD sequence (3-S5E, GPGSGMEPSYSPTEPSYSPTEPSYSPT) in which every Ser5 is replaced by a phosphorylation surrogate glutamate in order to evaluate where CDK7 places the phosphorylation marks when all Ser5 sites are phosphorylated. Indeed, as shown in Figure S6 (along with MS/MS data in Table S4) two Ser7 modifications were observed at nearly equal abundance, suggesting that permissibility for Ser7 modification occurs after complete Ser5 modification. A minor mono-phosphorylated species is identified as phosphorylation at residue 25, corresponding to a Ser2 site. This modification suggests a degree of non-Ser5 specificity by CDK7, consistent with the previous observation of weak activity against Ser2.44 These LC-UVPD-MS results show that CDK7 can phosphorylate both Ser5 and Ser7 in vitro with a significant preference for Ser5. The results also reflect the importance of UVPD for confident localization of the phosphorylation sites even for low-abundance peptides.
The effect of the amino acid residue at the seventh position of the CTD heptad on CDK7 activity is of particular interest because this position represents the most divergent site during the evolution of the CTD. Human CTD contains a proximal region highly obeying the consensus sequence and a distal region with deviation from the consensus sequence in a few heptad repeats, predominantly in the seventh position with lysine point-mutations (S7K) as the most frequently occurring deviation from consensus in CTD spans. To explore the impact of Lys mutation at the seventh position in the heptads, we compared the rates of phosphorylation by CDK7 using three substrates of about the same length: a wild-type (WT) S. cerevisiae CTD (26 repeats, mainly containing a consensus sequence), an S. cerevisiae CTD variant where every Ser7 is mutated to lysine at every other heptad (26X S7K, 26 repeats with a total of 13 lysines at the seventh positions), and a WT human distal CTD sequence (26 repeats containing eight S7K variations) by using a kinase assay. In this assay, the kinase activity was monitored by measuring the accumulation of ADP (see Figure S7). The results suggest a CDK7 substrate preference for CTD containing a seventh position lysine mutation over the wild-type human distal CTD and even more so for the S. cerevisiae CTD with a serine residue at the same position.
To better understand the CDK7 preference of the substrate, we used a 21-mer S7K peptide (3-S7K, GPGSGMSPSYSPTSPKYSPTSPSYSPT) as the substrate for CDK7 kinase reaction and analyzed the products by LC-MS/MS. This construct contains two full heptad repeats with flanking residues from neighboring heptads to ensure sufficient binding containing 21 amino acids altogether. A typical chromatographic trace of the 3-S7K products is shown in Figure 2, illustrating the near baseline resolution of each mono- and bis-phosphorylated product. UVPD parameters were optimized to enhance the production of fragment ions with excellent signal-to-noise ratios that permit sequence assignments and localization of phosphorylation for peptides that differ only in their sites of phosphorylation. The UVPD mass spectra used to confidently identify the three separated mono-phosphorylated isomers are shown in Figure S8 (with m/z values and mass accuracies summarized in Table S5). CDK7 predominantly phosphorylates two Ser5 sites (residues 14 and 21). The Ser5 positional phosphorylation at residue number 7 was not detected, likely due to a lack of consensus sequence motif N-terminal to this residue. As observed on the 3-S5E peptide, a trace amount of phosphorylation occurred at residue 25, corresponding to a Ser2 site, once again showing some non-Ser5 specificity by CDK7 on such a small substrate. Although nominally detected and successfully characterized by UVPD (Figure 2), the bis-phosphorylated peptides were not targeted for quantitation due to inefficient modification of this short 27-residue peptide by CDK7.
Figure 2.
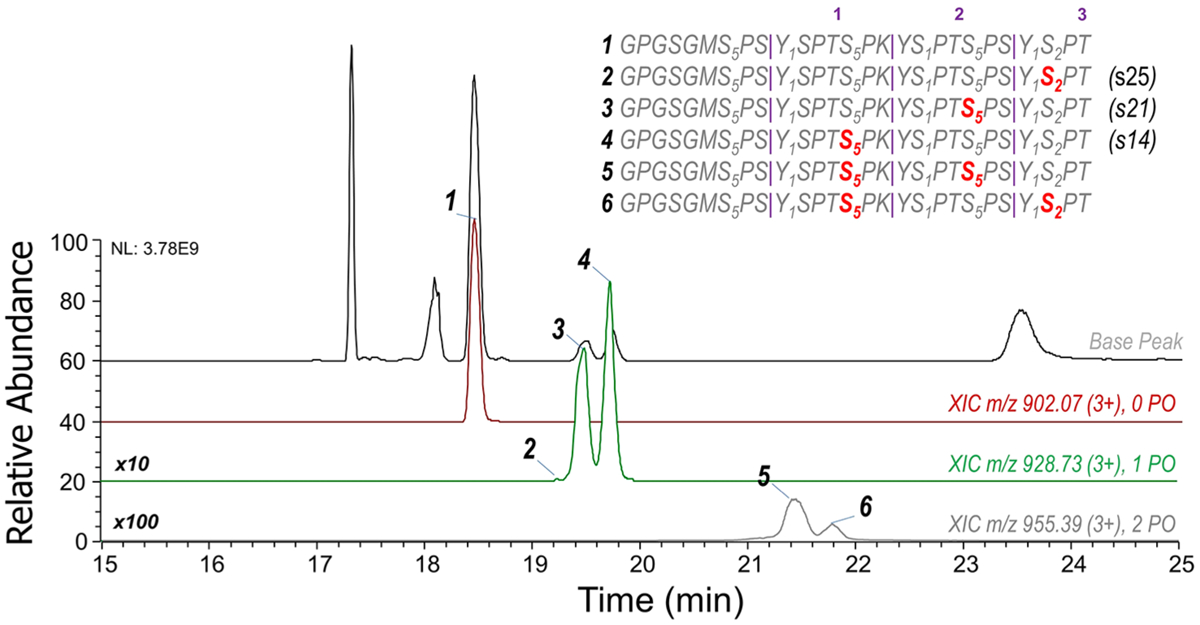
Chromatographic traces of each 3-S7K peptide. The top trace is the base peak chromatogram, followed by extracted-ion chromatograms of the unmodified, mono-phosphorylated, and bis-phosphorylated phosphopeptides. The numbers in purple above the sequence indicate the heptad repeat number, and subscript numbering indicates residue numbers in every heptad along the 3-S7K peptide.
The monoisotopic m/z values of the nonphosphorylated precursor (m/z 902.41) and the mono-phosphorylated isomer (m/z 929.07) were used to create a scheduled UVPD MS/MS method. The 3-S7K peptide was incubated with CDK7 for varying reaction periods to determine its spatiotemporal phosphorylation (Figure 3). To correct for systematic run-to-run variations, a phosphopeptide reference standard was used to normalize all phosphorylated peptide signals in different runs. The peptide (TRDIYETDY[+80]YRK) phosphopeptide reference standard (Figure S9, Table S6) was spiked at a static concentration in all samples to allow normalization among different runs. This peptide displayed a retention time earlier than the scheduled CTD phosphopeptides, thus minimizing any overlap or interference in the data acquisition. For the UVPD-PRM quantitative strategy, identifying fragment ions were selected for each CTD phosphopeptide isomer (as listed in the insets of Figure 3), and the sums of integrated areas of the extracted ion chromatograms were normalized against the peptide standard included in each run. In line with our 21-mer work43 and the preferred heptad phosphorylation observed on human CTD by CDK7, the phosphorylation of the 3-S7K peptide shows a clear preference for Ser5 modification. Specifically, serines at positions 14 and 21 are predominantly modified, both of which correspond to Ser5 heptad positions. A minor amount of Ser2 phosphorylation is observed on s25 owing to the structural permissibility of CDK7 for Ser2 phosphorylation.44
Figure 3.
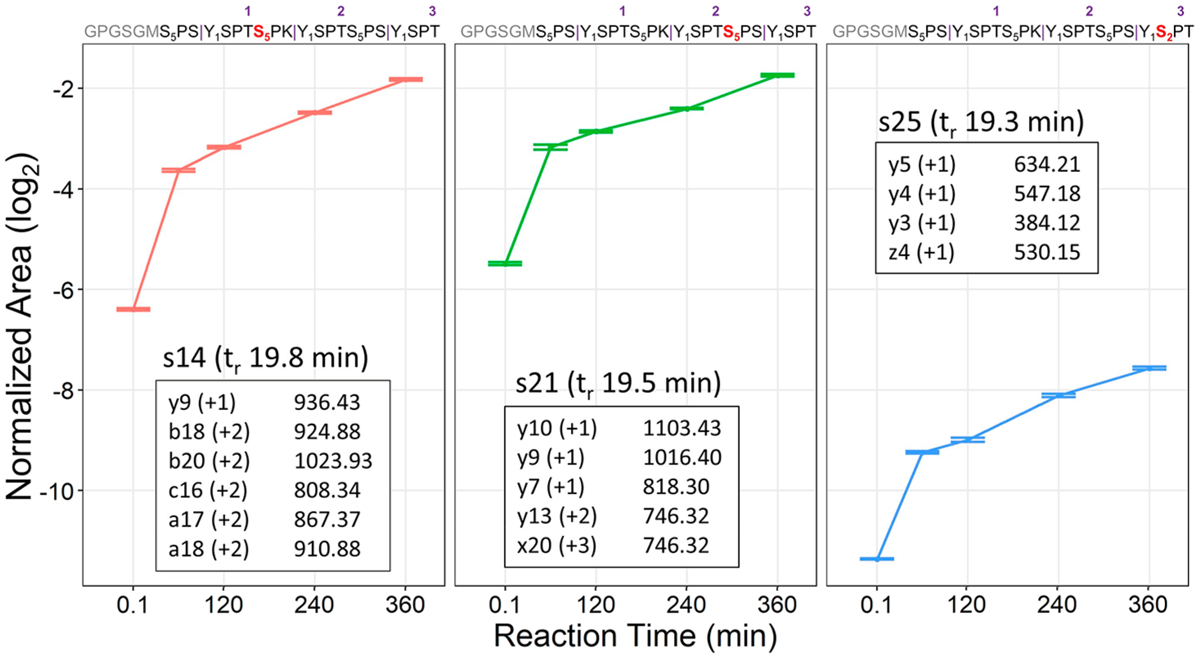
Normalized areas for each mono-phosphorylated 3-S7K peptide (corresponding to phosphorylation of s14, s21, or s25) generated from reaction with CDK7 and reported with 95% confidence intervals. The fragment ions generated by UVPD that are used to track each phosphopeptide for the PRM strategy are shown as insets based on isolation and activation of the 3+ charge state (m/z 928.73). The numbers in purple above the sequence indicate the heptad repeat number, and subscript numbering indicates residue numbers in every heptad along the 3-S7K peptide.
Successful demonstration of the UVPD-based PRM strategy for profiling phosphorylation of 3-S7K motivated the extension of the method to examine the phosphorylation of physiological length CTDs, a significantly greater challenge due to the added complexity of differential phosphorylation. The results for these more elaborate CTDs are showcased in numerous figures (Figures 4–7, S7–S64) as described in the following sections. S. cerevisiae CTD is known to faithfully conform to the consensus sequence in 19 of its 26 heptads. This proves a significant hindrance for MS analysis given the lack of basic/tryptic residues for digestion. On the basis of insight from previous studies, we engineered a CTD construct with lysine in every other heptad repeat to allow for time-dependent quantification of phosphorylation using LC-MS/MS analysis. The 26-repeat S7K CTD (26xS7K) construct is a 206-residue-long polypeptide with 26 CTD repeats of varying composition, as shown in Figure S1. We aimed to determine modifications across the entire 26xS7K construct and use it as a stepping stone to study the complex human distal CTD and delineate the specific motifs that modulate site-specific phosphorylation. Nine identical diheptads are composed of one consensus CTD heptad plus a second heptad containing a lysine residue to facilitate tryptic digestion. Three additional diheptad variants contain point-mutations in the heptad at the seventh position, originating from natural variance on the S. cerevisiae CTD sequence, plus a second heptad containing a lysine residue. This construct offered a glimpse into another layer of yeast transcriptional regulation with a simple CTD model containing a large stretch of known consensus region. Eight mono-phosphorylated (four pairs of isomers) and three bis-phosphorylated tryptic peptides containing different Ser5 phosphorylation sites were monitored, as illustrated in the chromatographic trace shown in Figure S10. The UVPD mass spectra for these 11 peptides are shown in Figures S18–S32, along with a summary of the unique fragment ions used to implement UVPD-PRM quantitative analysis (Supplementary Transition List). Figure 4 shows the phosphorylation level of the eight Ser5 phosphopeptides located in the four different diheptads after a 360 min reaction period. The preferential phosphorylation of the consensus heptad is an expected outcome. Among the three nonconsensus diheptads, CDK7 shows greater preferential phosphorylation of the Ala-containing peptide YSPTSPAYSPTSPK, followed by YSPTSPGYSPGSPK, and, lastly, YSPTSPNYSPTSPK.
Figure 4.
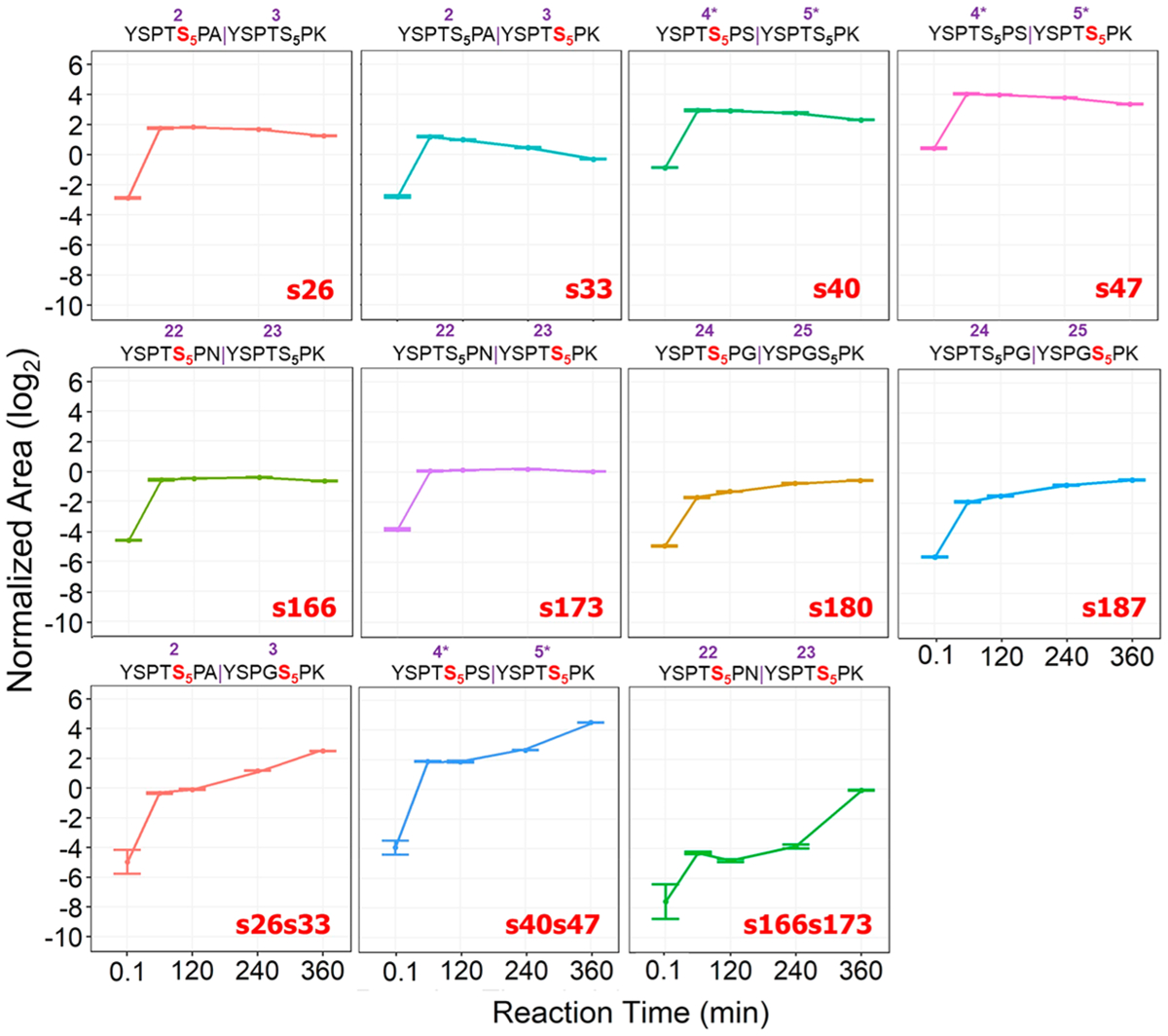
Summed and normalized area for each mono-phosphorylated and bis-phosphorylated 26xS7K yeast peptide, showing more enhanced CDK7 phosphorylation of the two consensus isomers represented by s40 and s47. Mono-phosphorylated peptides are phosphorylated a second time by CDK7, with an uptick in bis-phosphorylation observed at 240 and 360-minute marks aligning with the decrease in abundances of the consensus mono-phosphorylated peptides. Monitored transitions for the UVPD-PRM method are listed in the Supplementary Transition List. The numbers in purple above the sequence indicate the heptad repeat number along the 26xS7K yeast construct, and subscript numbering indicates residue numbers in every heptad along each tryptic 26xS7K yeast peptide.
Figure 7.
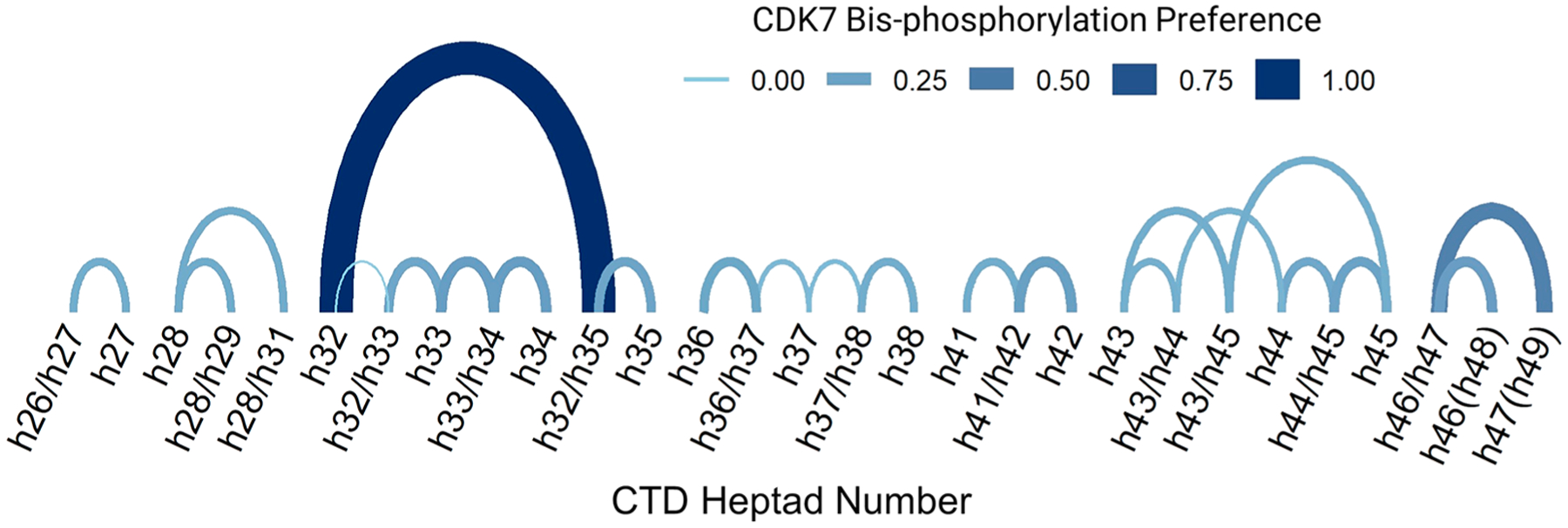
This arc diagram shows the interconnected and preferential CDK7 nonprocessive action after predominant mono-phosphorylation of heptad Ser5 positions between t = 0.1 min and t = 60 min. Each arc represents an interconnection between coupled heptads, e.g., single phosphorylation followed by bis-phosphorylation of a coupled heptad on tryptic peptides. The absolute difference (preference) between reaction times for each connected pair is shown by the arc’s thickness and color. Arc heights are proportional to the distance between each modified site, and coupled bis-phosphorylated heptads are shown with sites separated by a slash mark.
UVPD-PRM analysis allows the levels of site-specific phosphorylation to be monitored as a function of time for each category of phosphorylation site along with the 26-repeat S7K construct. Overall, mono-phosphorylation increases for all Ser5 positions after 60 min of reaction time (Figure 4). Virtually all Ser5 sites show a marked 2-fold increase from baseline phosphorylation levels after 60 min; however, all but two sites plateau and diminish beyond 1 h due to further phosphorylation of each diheptad span, as bis-phosphorylation becomes prevalent at longer reaction times. In other words, the apparent fold change of singly phosphorylated peptides appears to decrease owing to their additional interaction with CDK7 to become bis-phosphorylated peptides over time. Interestingly, except for the glycine-containing segment containing s180 and s187, all other portions of the construct show a ~10-fold increase in conversion to the bis-phosphorylated diheptad species after 360 min of incubation with CDK7 with a clear preference for consensus and the aliphatic alanine-containing portions of the construct (s26/s33). As expected, the consensus region between residue 26 and residue 161 displayed preferential phosphorylation consistent with CTD motif recognition by CDK7. It is speculated that after consensus Ser5 sites are phosphorylated; then CDK7 opts for nonconsensus regions of the S. cerevisiae CTD, choosing stretches with sterically similar alanine mutations in the seventh position preferentially over ones containing bulkier asparagine or a conformational change-inducing glycine mutation. This observed preference is consistent with our previous molecular modeling (Figure S11).44 Further evidence for the consensus region’s complete modification is the level of bis-phosphorylation observed after the 60 min time point. Bis-phosphorylation of the consensus peptide (YSPTSPSYSPTSPK, s40/s47) increases steadily throughout each reaction period, followed closely by complete phosphorylation of nonconsensus diheptads (YSPTSPAYSPTSPK, s26s33, and YSPTSPNYSPTSPK, s166s173). Phosphorylation of the nonconsensus diheptad containing two glycine point-mutations (YSPTSPGYSPGSPK) deviates from this behavior, as it does not exhibit bis-phosphorylation or phosphorylation on the C-terminus nonconsensus region of 26x-S7K.
The challenge of using MS/MS analysis to monitor phosphorylation of the CTD originates from the inability to differentiate the heptad number along this periodic sequence as well as the great potential for isomers having the same sequence but different phosphorylation sites. This ambiguity can be alleviated in part by studying the distal region of the human CTD sequence. The first 26 repeats of the human CTD (proximal CTD) exhibit considerable sequence conservation relative to the consensus CTD sequence, whereas the latter 26 repeats (distal CTD) significantly deviate from the consensus sequence (Figure S12). S7K is the most frequently occurring variation in the distal CTD, presenting a natural cleavage site for tryptic digestion and thus generating specific peptides that allow each to be matched to their original positions along the human distal CTD. There are two nonunique regions of the distal region: repeats of peptide YSPTSPK (repeats 39 and 40) and YSPTSPTYSPTSPK (repeats 46/47 and 48/49) (Figure 5A), respectively. These redundant spans mean that phosphorylation of sites Ser101 and Ser108 is represented by the same heptad, and the same duplicity arises for phosphorylation of sites Ser150 and Ser164 as well as sites Ser157 and Ser171 that occur in the repeated diheptad.
Figure 5.
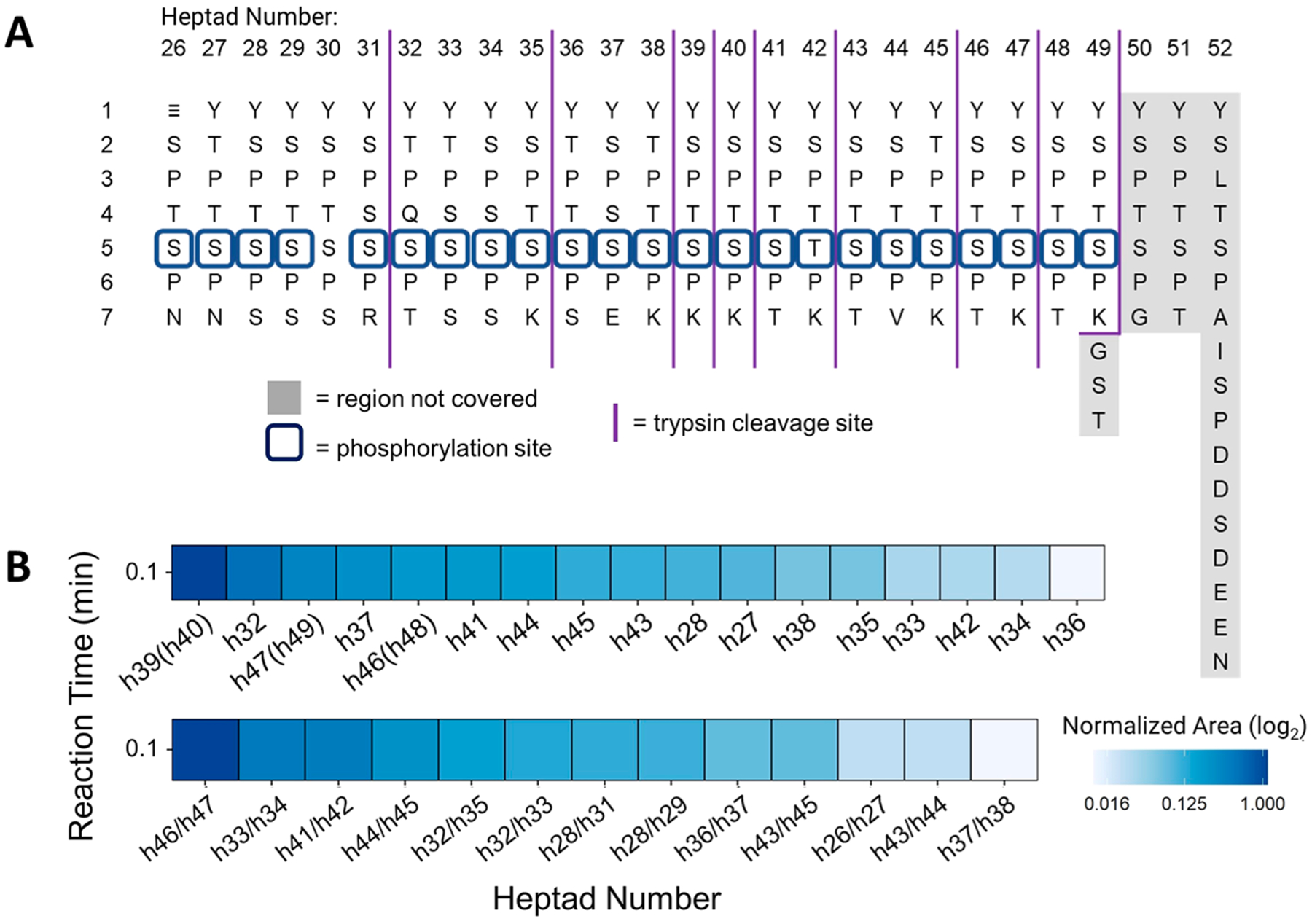
(A) Human distal CTD sequence arranged vertically with numbers along the horizontal axis indicating the repeat number and the vertical axis indicating residue numbers in every heptad along the CTD. Dark blue squares show the phosphorylation Ser5 species identified, and the light gray shade shows the region not covered in mapping. Purple vertical solid lines mark the boundaries of peptide fragments generated by trypsin digestion. (B) Normalized areas of each phosphorylated tryptic peptides generated from the 26-repeat human distal CTD after phosphorylation by CDK7, showing the distribution of Ser5 phosphorylation on each heptad repeat (each heptad number denoted as hN). CDK7 preference for heptad phosphorylation is shown as area at time 0.1 min. h39(h40), h46(h48), and h47(h49) refer to the YSPTSPTYSPTSPK peptides which are repeated twice and thus correspond to phosphorylation of sites on heptads 39 or 40, 46 or 48, and 47 or 49. The lower panel shows phosphorylated heptads with bis-phosphorylated sites separated by a slash mark. Monitored transitions are listed in the Supplementary Transition List. ≡ corresponds to the GPGSGM tag remaining from the GST-tag after 3C digestion.
Figure 5 summarizes the overall site-specific Ser5 phosphorylation observed along the length of the 26-repeat human distal construct and the preference for heptad phosphorylation at t = 0.1 min. Note that the human distal CTD is unphosphorylated prior to the addition of kinase, and thus a t = 0.1 min baseline immediately at the start of the reaction was necessary to capture the rapid “burst” phase of pre-steady-state preferential phosphorylation by CDK7, which occurs in the first few seconds. The UVPD mass spectra acquired for the 37 peptides are shown in Figures S33–S69, along with a summary of the unique fragment ions used to implement UVPD-PRM quantitative analysis provided in the Supplementary Transition List. As expected, the consensus sequence heptad YSPTSPK shows the highest phosphorylation levels [h39(h40)]. Despite the duplicate occurrence of YSPTSPTYSPTSPK (e.g., heptads h46(h48) and h47(h49)), the abundance of these phosphorylated peptides in the tryptic digest was lower compared to the heptads h32 and h37. Interestingly, the heptads with increased acidic character, such as those containing Ser or Thr residues adjacent to the phosphorylation site, also exhibited high phosphorylation levels (including h(32), h(37), h46/h47, and h33/h34). Figure S13A details phosphorylation levels over a 360 min period, showing site-specific phosphorylation along the 26-repeat human distal construct length. Similar to the 3-S7K S. cerevisiae construct, a significant 5-fold increase is observed along the consensus repeat sequence [h39(h40)] as well as the acidic regions of the human distal CTD (h(32), h(37), and h(41)). The phosphorylation curves flatten or decrease at longer times, corresponding to the increase in bis-phosphorylation on adjacent heptads as summarized in Figure S13B. Moreover, a 4-fold increase in bis-phosphorylation of the acidic regions of the human distal sequence (h32/h33, h33/h34, h32/h35) is observed. Unlike the trend in bis-phosphorylation observed for the 3-S7K S. cerevisiae construct, bis-phosphorylation or thorough heptad modification along the length of the 26-repeat human distal construct was significantly less prominent for the human distal construct.
Compiling the phosphorylation trends of each Ser5 site in the 26-repeat human distal construct at each reaction time allows the construction of a spatiotemporal heat map of CDK7 phosphorylation (Figure 6). This map highlights fast phosphorylation at t = 0.1 (first few seconds) and stochastic modification of Ser5 toward the C-terminal end of the human distal CTD construct. The increased phosphorylation levels observed at h(32) and h39(h40) correspond to a particularly serine-rich portion of the N-terminus. The N-terminus itself shows lower phosphorylation levels, likely due to the substantial frequency of deviation from the consensus heptad sequence. In using a bottom-up approach, information is lost about the directionality of CDK7 along the length of the human distal construct; however, in analyzing progressive phosphorylation of each tryptic peptide over time, an interconnected pattern emerges. The arc diagram in Figure 7 reveals the nonprocessive action of CDK7 after predominant mono-phosphorylation of Ser5 positions between t = 0.1 min and t = 60 min using min–max normalization. Here mono-phosphorylated heptads on each tryptic peptide set up the potential for a coupled bis-phosphorylation relationship. In network analysis of coupled sites, we are able to observe CDK7’s preferential phosphorylation of heptads over time. For example, we observe that after CDK7 phosphorylates h32 there is a very high probability that adjacent h35 will be phosphorylated to yield the h32/h35 bis-phosphorylated peptide. Moreover, h32 mono-phosphorylation is preferred over h35, allowing a significant probability that it will solicit further CDK7 modification to create the h32/h35 bis-phosphorylated species.
Figure 6.
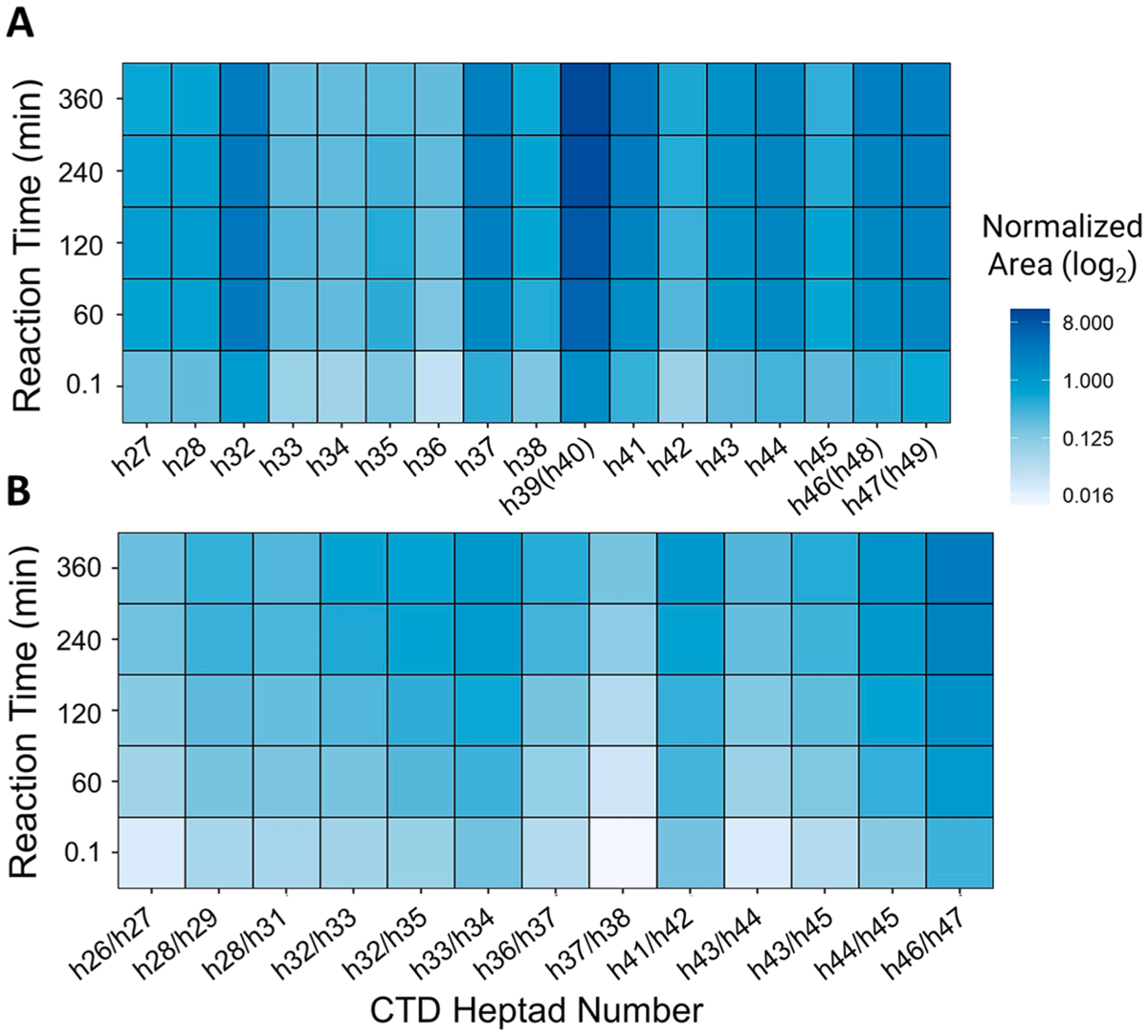
Heat maps illustrating the time-dependent variations in Ser5 phosphorylation by CDK7 of human distal CTD. (A) Mono-phosphorylated heptads. (B) Bis-phosphorylated heptads with pairs of Ser5 positions separated by a slash mark. h39(h40), h46(h48), and h47(h49) refer to the YSPTSPTYSPTSPK peptides, which are repeated twice and thus correspond to phosphorylation of sites on heptads 39 or 40, 46 or 48, and 47 or 49. The y-axis is normalized using log2 area and shows phosphorylation levels at each Ser5 site localization stacked for each time point.
RNA polymerase II-mediated transcription is a highly dynamic process. Although it has been well acknowledged that different PTMs dominate at different stages of transcription, it is almost impossible to trap the polymerase in a particular stage and assess the phosphorylation marks on its CTD. Our method follows the dynamic phosphorylation changes to the level of single amino acid accuracy and quantitatively analyzes phosphorylation marks of the CTD in a time-dependent manner. This method will be beneficial for the field of transcription to correlate PTM patterns of RNA polymerase II to transcriptional functions. Since PTM patterns recruit different transcriptional functions regulators to RNA polymerase II, this method can significantly advance our understanding of how PTMs of RNA polymerase II regulate eukaryotic transcription.
CONCLUSION
The use of targeted UVPD-MS in a PRM strategy establishes a powerful method to identify and precisely map isomeric phosphorylation sites with quantitative insight about the spatiotemporal phosphorylation of the C-terminal domain of RNA polymerase II. The rich array of phospho-retaining fragment ions produced by UVPD are leveraged to create site-specific transition lists critical for differentiating phosphopeptide isomers generated from the CTD. Furthermore, network analyses (as done in Figure 7) could be easily interpreted as probabilistic measures of CDK7’s preference for particular heptads at a specific time. Although reaction time points have minimalistic meaning in vitro, this study repeated in vivo would directly correspond to transcriptional progress, linking preferences of a kinase for CTD heptads with different transcriptional stages. We anticipate combining this method with in-depth eukaryotic transcriptional studies will provide additional information into how the CTD code orchestrates the transcriptional cycle.
EXPERIMENTAL SECTION
The three-repeat CTD constructs were ordered as primers, amplified, and cloned using sequence- and ligation-independent cloning (SLIC).49 The 26-repeat S7K construct with Lys in every other heptad repeat was ordered a synthetic gene (Genescript). Human distal CTD was cloned from a full-length human CTD construct from pYFP-RPB1-αAmr.45 All constructs were cloned into a PET28a vector (Novagene) containing a 6X-histidine tag and a glutathione-S-transferase (GST) tag with a 3C protease cleavage site added after the two tags. T4 DNA polymerase (NEB) was used to create 5′ overhangs for cloning into the vectors using SLIC.
Protein expression and purification were done as described previously.38,39,43,44,50 E. coli BL21 (DE3) cells were used as the protein expression system. The transformation was carried out by thawing the cells on ice for 10 min, adding the DNA, incubating on ice for 30 min, heat shocking at 42 °C for 1 min, and cooling on ice for 5 min. The cells were recovered in Super Optimal Broth medium for 1 h at 37 °C and were plated on Luria–Bertani agar plates with 50 μg/mL of kanamycin for selection using the spread plate technique. Individual colonies were grown in Luria–Bertani medium at 37 °C containing 50 μg/mL of kanamycin in 50 mL flasks. Ten milliliters of inoculum was used for inoculating 1 L of terrific broth (Thermo Fisher), and the culture was grown to an OD of 0.4–0.6. Isopropyl β-d-1-thiogalactopyranoside was used to induce the expression at a final concentration of 0.5 mM. The cultures were pelleted by centrifugation after overnight growth (16 h at 16 °C), and the cells were lysed through sonication in a lysis buffer (50 mM Tris-Cl pH 8.0, 500 mM NaCl, 10% glycerol, 0.1% Triton-X 100, 15 mM imidazole, and 10 mM β-mercaptoethanol (BME)). Sonication of the samples was carried out on ice, at 90 amplitude for 3 min per cycle (1 s on and 5 s off) for five cycles with a 3 min break between each cycle. The lysate was cleared by centrifugation at 27000g for 45 min at 4 °C. The proteins were purified through affinity column chromatography using Ni2+/NTA beads (Qiagen). Briefly, the column was equilibrated with lysis buffer; then, the cleared lysate supernatant was run through the column. A wash (50 mM Tris-Cl pH 8.0, 500 mM NaCl, and 10 mM BME) was done before eluting with an elution buffer (50 mM Tris-Cl pH 8.0, 500 mM NaCl, 200 mM imidazole, and 10 mM BME). Proteins were dialyzed in a gel filtration buffer (50 mM NaCl, 20 mM Tris-Cl pH 8.0, and 10 mM BME) at 4 °C overnight with a suitable dialysis membrane. Proteins were concentrated using centrifugal filtration and cleaned up by size exclusion chromatography using a Superdex 200 column (GE Life Sciences). The integrity of the proteins was assessed by polyacrylamide gel electrophoresis (Coomassie Brilliant Blue Staining) (Figure S14).
Kinase Reaction Assays.
All kinase reaction assays were performed in triplicate with 2 mM ATP, 50 mM Tris pH 8, and 10 mM MgCl2. A 0.2 μM concentration of CDK7/Cyclin-H/Ménage à trois 1 (Millipore-Sigma) was used to treat 1 mg/mL of CTD substrate for 360 min (buffer containing 50 mM Tris-Cl pH 8.0 and 10 mM MgCl2). The reaction time of CDK7 was optimized for substrates such as full-length yeast and human distal CTD until phosphorylation saturation in vitro, as monitored by MALDI and an electrophoretic mobility shift assay.51 The reactions were stopped at specific time points through ion quenching using 10 mM ethyl-enediaminetetraacetic acid and were flash-frozen in liquid nitrogen within 6 s for storage before further analysis.
Sample Preparation.
All CTD samples were digested using 3C-protease at a molar ratio of 100:1 protein/protease in a reaction volume of 100 μL. All GST-26-repeat CTD constructs were prepared for bottom-up analysis by overnight digestion with trypsin at 37 °C using a 1:50 enzyme-to-substrate ratio and spiked with 1 pmol/μL of phosphopeptide standard (AnaSpec, Inc.). CTD digests were desalted on C18 spin columns and resuspended to 0.5 μg/μL with 2% acetonitrile in 0.1% formic acid in HPLC-grade water for LC-MS analysis.
UVPD-MS and Data Analysis.
Five technical replicates of each of the three biological replicates were separated using a Dionex Ultimate 3000 nano liquid chromatography (LC) system (Thermo Scientific) plumbed for direct injection onto a 20 cm C18 (1.8 μm, 300 Å pore size, 75 um ID, packed in-house) Picofrit analytical column (New Objective, Woburn, MA, USA). Mobile phases A and B were composed of HPLC-grade water and acetonitrile, respectively, each containing 0.1% formic acid. Separations were carried out using gradients that were optimized for various samples as follows: a linear gradient of 2% to 24% B in 30 min was used for 26-repeat S7K and 3-repeat CTD (S7K, S5E, and S5E′). A linear gradient of 2% to 28% B for 40 min was used for the human distal CTD samples. The flow rate was maintained at 0.300 μL/min for all samples. Eluted peptides were analyzed in positive polarity mode using an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) using a NanoFlex electrospray source. The mass spectrometer was equipped with an excimer laser operated at 193 nm (Coherent, ExciStar, Santa Clara, CA, USA) and modified to enable UVPD in the dual linear ion trap as described previously.52 All spectra were acquired in the Orbitrap mass analyzer using resolution settings of 60 and 30 K (at m/z 200) for MS1 and MS/MS events, respectively. Targeted peptides were activated using two laser pulses of 1.5 mJ for UVPD or an NCE of 28 for HCD, and scans were triggered by time-scheduled targeted CTD ions assessed with a ±1 min elution window (Supporting Information).
MS/MS spectra were deconvoluted in the XCalibur QualBrowser software using the Xtract algorithm with a signal-to-noise threshold of 3. Fragments were matched to the nine ion types observed from UVPD of peptides (a, a+1, b, c, x, x+1, y, y–1, z) using ProSight Lite. As reported before, phosphosite confirmation was achieved by adding the mass of a phospho group (+79.97 Da) at each of the possible Tyr or Ser residues to identify fragment ions that were phosphorylated.37,38,44,53 All PRM-MS raw files were processed in Skyline v20.1 to generate XICs and allow targeted peak integration.54 The summed peak areas of the 3 to 5 most abundant fragment ions that allowed confident phosphosite differentiation of peptides were used to quantify CTD phosphopeptides. Following ProSight and Skyline data processing, visualization and sample mix-modeling statistics were achieved using scripts written in R-programming.55 To determine the relative abundance of the target peptides, the summed peak area of unique UVPD-generated product ions was first normalized to the surrogate reference peptide (TRDIYETDpYYRK). Ion fragment XICs unique to each phosphopeptide precursor ion were integrated, totaled, and normalized to that of the surrogate reference peptide to overcome differences in the heterogeneous nature of peptide composition and phosphorylation level, thus accounting for the potential differences in peptide ionization efficiency. Specifically, the relative site-specific phosphorylation level of each CTD peptide was calculated as the ratio of the summed signal intensities of individual CTD phosphopeptide XICs and the summed signal intensity of reference peptide XICs.
Mixed-Effects Statistical Analysis of CTD Phosphopeptide Quantitation.
A mixed-effects model was fit to the set of peptides originating from each CTD construct for which peptides containing different phosphorylation sites were expected to contribute different abundances over time. Mixed-effects modeling is an established statistical methodology for the analysis of time-course data or when data are organized in different levels or groups, each categorized based on distinctive conditions.23,56,57 Consequently, this modeling is particularly well-suited for tracking significant changes in specific peptide abundance from an overall population, outside of random variation. The methodology includes terms for both fixed effects, such as the factors of interest that are directly manipulated in the study, and random effects such as random error, which we wish to generalize into our model, but not empirically measure. Specifically, replicate LC-MS runs were used to monitor at five time points the different peptides originating from one protein possessing multiple phosphorylation sites. Mixed modeling has a couple of key advantages: first, unlike routine analysis of variance, mixed modeling is not affected by unbalanced data or scenarios where one peptide may contain more phosphorylation sites than another. Second, mixed modeling can be useful in handling outliers or scenarios where statistically errant values can be dropped without any impact on global analysis by missing observations.
After normalization against the surrogate reference peptide, differences in abundances of peptides between reaction conditions are inferred from the log2-transformed ratio of each peptide’s summed fragment ion abundance and that of the surrogate reference peptide. This ratio significantly diminishes the systematic errors of LC-MS/MS analysis and subsequent data processing such as sample handling, matrix effects, and ESI ionization effects. A mixed-effects model was used to correlate the abundances of the observed peptides produced from each protein and calculate the significance in changes of phosphorylation across varying reaction times (Tables S7–S9). As described, this model includes terms for both fixed effects for which all levels of experimentation are of interest and directly manipulated in the study, such as the CDK7 kinase reaction time and random effects. Random effects are effects for which measurements are not included in the experiment and out of the control of experimentation, such as biological variations and measurement errors. Their impact can be considered to be drawn at random from a normal population, address issues of random error, and enable a valid inference on the fixed effects of interest whose normality assumption was verified through residual plots (Figures S15–S17). R programming provides a reliable and efficient suite of mixed-effect modeling packages used in a variety of standard quantitative proteomic tools.55
The equation above shows the common notation used in mixed-effect modeling with designated inputs for fixed and random effects particular to this study. Similar to the linear regression model Y = a + bX, area is the dependent variable with all independent variables listed after the “~” in the equation above, including reaction time and each specific phosphopeptide sequence. The mixed-effect model estimates a fixed intercept at reaction time t = 0 and is noted as a fixed effect as we directly controlled reaction time for each CTD construct. Random effects are noted within the parentheses of the model and delineate any contributions to the slope. The variable to the left of the vertical bar indicates random effects originating from replicate measurements such as differing kinase activity at each reaction time. The variable to the right of the bar indicates random effects for each phosphopeptide sequence stemming from kinase substrate specificity for different amino acid regions along each CTD construct after tryptic digestion. In essence, this model fits different intercepts for each reaction time and varies the slopes in response to random error observed after replicate measurements. Changes in phosphorylation are communicated using a log2 transform to easily communicate +1-fold or −1-fold proportional changes. Analysis of each CDK7 modification site along a CTD sequence shows significant changes in phosphorylation over each reaction experiment, disproving the null hypothesis.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported by grants from the National Institutes of Health (R01GM104896 and R01GM125882 to Y.J.Z. and National Institute of General Medical Sciences of the National Institutes of Health under awards R01GM121714 and R35GM139658 to J.S.B.) and the Welch Foundation (F-1778 to Y.Z. and F-1155 to J.S.B.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the Robert A. Welch Foundation or the National Institutes of Health. Funding from the UT System for support of the UT System Proteomics Core Facility Network is gratefully acknowledged.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c03321.
Parallel-reaction monitoring transition mass list for each CTD model organized by tab (XLSX)
Supplementary ADP accumulation assay, chromatographic traces, MS/MS spectra and their associated mass lists, and normalized area plots with statistical analysis (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c03321
The authors declare no competing financial interest.
Contributor Information
Edwin E. Escobar, Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, United States.
Mukesh Kumar Venkat Ramani, Department of Molecular Biosciences, University of Texas at Austin, Austin, Texas 78712, United States.
Yan Zhang, Department of Molecular Biosciences and Institute for Cellular and Molecular Biology, University of Texas at Austin, Austin, Texas 78712, United States;.
Jennifer S. Brodbelt, Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, United States;.
REFERENCES
- (1).Jeronimo C; Collin P; Robert F The RNA Polymerase II CTD: The Increasing Complexity of a Low-Complexity Protein Domain. J. Mol. Biol 2016, 428 (12), 2607–2622. [DOI] [PubMed] [Google Scholar]
- (2).Jeronimo C; Bataille AR; Robert F The Writers, Readers, and Functions of the RNA Polymerase II C-Terminal Domain Code. Chem. Rev 2013, 113 (11), 8491–8522. [DOI] [PubMed] [Google Scholar]
- (3).Buratowski S Progression through the RNA Polymerase II CTD Cycle. Mol. Cell 2009, 36 (4), 541–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Buratowski S The CTD Code. Nat. Struct. Mol. Biol 2003, 10 (9), 679–680. [DOI] [PubMed] [Google Scholar]
- (5).Komarnitsky P; Cho E-J; Buratowski S Different Phosphorylated Forms of RNA Polymerase II and Associated MRNA Processing Factors during Transcription. Genes Dev. 2000, 14 (19), 2452–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Eick D; Geyer M The RNA Polymerase II Carboxy-Terminal Domain (CTD) Code. Chem. Rev 2013, 113 (11), 8456–8490. [DOI] [PubMed] [Google Scholar]
- (7).Bataille AR; Jeronimo C; Jacques P-É; Laramée L; Fortin M-È; Forest A; Bergeron M; Hanes SD; Robert F A Universal RNA Polymerase II CTD Cycle Is Orchestrated by Complex Interplays between Kinase, Phosphatase, and Isomerase Enzymes along Genes. Mol. Cell 2012, 45 (2), 158–170. [DOI] [PubMed] [Google Scholar]
- (8).Jeronimo C; Bataille AR; Robert F The Writers, Readers, and Functions of the RNA Polymerase II C-Terminal Domain Code. Chem. Rev 2013, 113 (11), 8491–8522. [DOI] [PubMed] [Google Scholar]
- (9).Venkat Ramani MK; Yang W; Irani S; Zhang Y Simplicity Is the Ultimate Sophistication—Crosstalk of Post-Translational Modifications on the RNA Polymerase II. J. Mol. Biol 2021, 166912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Solari FA; Dell’Aica M; Sickmann A; Zahedi RP Why Phosphoproteomics Is Still a Challenge. Mol. BioSyst 2015, 11 (6), 1487–1493. [DOI] [PubMed] [Google Scholar]
- (11).Hunter T Why Nature Chose Phosphate to Modify Proteins. Philos. Trans. R. Soc., B 2012, 367 (1602), 2513–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Potel CM; Lemeer S; Heck AJR Phosphopeptide Fragmentation and Site Localization by Mass Spectrometry: An Update. Anal. Chem 2019, 91 (1), 126–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Osinalde N; Aloria K; Omaetxebarria MJ; Kratchmarova I Targeted Mass Spectrometry: An Emerging Powerful Approach to Unblock the Bottleneck in Phosphoproteomics. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci 2017, 1055–1056, 29–38. [DOI] [PubMed] [Google Scholar]
- (14).Hughes CS; Spicer V; Krokhin OV; Morin GB Investigating Acquisition Performance on the Orbitrap Fusion When Using Tandem MS/MS/MS Scanning with Isobaric Tags. J. Proteome Res 2017, 16 (5), 1839–1846. [DOI] [PubMed] [Google Scholar]
- (15).Schweppe DK; Rusin SF; Gygi SP; Paulo JA Optimized Workflow for Multiplexed Phosphorylation Analysis of TMT-Labeled Peptides Using High-Field Asymmetric Waveform Ion Mobility Spectrometry. J. Proteome Res 2020, 19 (1), 554–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Rauniyar N; Yates JR Isobaric Labeling-Based Relative Quantification in Shotgun Proteomics. J. Proteome Res 2014, 13 (12), 5293–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Deng J; Erdjument-Bromage H; Neubert TA Quantitative Comparison of Proteomes Using SILAC. Curr. Protoc. Protein Sci 2019, 95 (1), e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Hoedt E; Zhang G; Neubert TA; Woods AG; Darie CC Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) for Quantitative Proteomics. Advancements of Mass Spectrometry in Biomedical Research 2019, 531–539. [DOI] [PubMed] [Google Scholar]
- (19).Wang X; He Y; Ye Y; Zhao X; Deng S; He G; Zhu H; Xu N; Liang S SILAC–Based Quantitative MS Approach for Real-Time Recording Protein-Mediated Cell-Cell Interactions. Sci. Rep 2018, 8 (1), 8441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lee N; Lee JW; Kang G-Y; Park S-H; Kim KP Quantification of the Dynamic Phosphorylation Process of ERK Using Stable Isotope Dilution Selective Reaction Monitoring Mass Spectrometry. Proteomics 2019, 19 (17), 1900086. [DOI] [PubMed] [Google Scholar]
- (21).Villanueva J; Carrascal M; Abian J Isotope Dilution Mass Spectrometry for Absolute Quantification in Proteomics: Concepts and Strategies. J. Proteomics 2014, 96, 184–199. [DOI] [PubMed] [Google Scholar]
- (22).Neilson KA; Ali NA; Muralidharan S; Mirzaei M; Mariani M; Assadourian G; Lee A; van Sluyter SC; Haynes PA Less Label, More Free: Approaches in Label-Free Quantitative Mass Spectrometry. Proteomics 2011, 11 (4), 535–553. [DOI] [PubMed] [Google Scholar]
- (23).Clough T; Key M; Ott I; Ragg S; Schadow G; Vitek O Protein Quantification in Label-Free LC-MS Experiments. J. Proteome Res 2009, 8 (11), 5275–5284. [DOI] [PubMed] [Google Scholar]
- (24).Blein-Nicolas M; Zivy M Thousand and One Ways to Quantify and Compare Protein Abundances in Label-Free Bottom-up Proteomics. Biochim. Biophys. Acta, Proteins Proteomics 2016, 1864 (8), 883–895. [DOI] [PubMed] [Google Scholar]
- (25).Goeminne LJE; Gevaert K; Clement L Experimental Design and Data-Analysis in Label-Free Quantitative LC/MS Proteomics: A Tutorial with MSqRob. J. Proteomics 2018, 171, 23–36. [DOI] [PubMed] [Google Scholar]
- (26).Lange V; Picotti P; Domon B; Aebersold R Selected Reaction Monitoring for Quantitative Proteomics: A Tutorial. Mol. Syst. Biol 2008, 4, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ronsein GE; Pamir N; von Haller PD; Kim DS; Oda MN; Jarvik GP; Vaisar T; Heinecke JW Parallel Reaction Monitoring (PRM) and Selected Reaction Monitoring (SRM) Exhibit Comparable Linearity, Dynamic Range and Precision for Targeted Quantitative HDL Proteomics. J. Proteomics 2015, 113, 388–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Karayel O; Tonelli F; Winter SV; Geyer PE; Fan Y; Sammler EM; Alessi DR; Steger M; Mann M Accurate MS-Based Rab10 Phosphorylation Stoichiometry Determination as Readout for LRRK2 Activity in Parkinson’s Disease. Mol. Cell. Proteomics 2020, 19 (9), 1546–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Park J; Oh HJ; Han D; Wang JI; Park IA; Ryu HS; Kim Y Parallel Reaction Monitoring-Mass Spectrometry (PRM-MS)-Based Targeted Proteomic Surrogates for Intrinsic Subtypes in Breast Cancer: Comparative Analysis with Immunohistochemical Phenotypes. J. Proteome Res 2020, 19 (7), 2643–2653. [DOI] [PubMed] [Google Scholar]
- (30).Barthélemy NR; Mallipeddi N; Moiseyev P; Sato C; Bateman RJ Tau Phosphorylation Rates Measured by Mass Spectrometry Differ in the Intracellular Brain vs. Extracellular Cerebrospinal Fluid Compartments and Are Differentially Affected by Alzheimer’s Disease. Front. Aging Neurosci 2019, 11, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sweredoski MJ; Moradian A; Raedle M; Franco C; Hess S High Resolution Parallel Reaction Monitoring with Electron Transfer Dissociation for Middle-Down Proteomics. Anal. Chem 2015, 87 (16), 8360–8366. [DOI] [PubMed] [Google Scholar]
- (32).Andjelković U; Josić D Mass Spectrometry Based Proteomics as Foodomics Tool in Research and Assurance of Food Quality and Safety. Trends Food Sci. Technol 2018, 77, 100–119. [Google Scholar]
- (33).Jung HR; Sidoli S; Haldbo S; Sprenger RR; Schwämmle V; Pasini D; Helin K; Jensen ON Precision Mapping of Coexisting Modifications in Histone H3 Tails from Embryonic Stem Cells by ETD-MS/MS. Anal. Chem 2013, 85 (17), 8232–8239. [DOI] [PubMed] [Google Scholar]
- (34).Brodbelt JS; Morrison LJ; Santos I Ultraviolet Photodissociation Mass Spectrometry for Analysis of Biological Molecules. Chem. Rev 2020, 120 (7), 3328–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Macias LA; Santos IC; Brodbelt JS Ion Activation Methods for Peptides and Proteins. Anal. Chem 2020, 92 (1), 227–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Escobar EE; King DT; Serrano-Negrón JE; Alteen MG; Vocadlo DJ; Brodbelt JS Precision Mapping of O-Linked N-Acetylglucosamine Sites in Proteins Using Ultraviolet Photodissociation Mass Spectrometry. J. Am. Chem. Soc 2020, 142 (26), 11569–11577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Burkholder NT; Sipe SN; Escobar EE; Venkatramani M; Irani S; Yang W; Wu H; Matthews WM; Brodbelt JS; Zhang Y Mapping RNAPII CTD Phosphorylation Reveals That the Identity and Modification of Seventh Heptad Residues Direct Tyr1 Phosphorylation. ACS Chem. Biol 2019, 14 (10), 2264–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Mayfield JE; Irani S; Escobar EE; Zhang Z; Burkholder NT; Robinson MR; Mehaffey MR; Sipe SN; Yang W; Prescott NA; Kathuria KR; Liu Z; Brodbelt JS; Zhang Y Tyr1 Phosphorylation Promotes Phosphorylation of Ser2 on the C-Terminal Domain of Eukaryotic RNA Polymerase II by P-TEFb. eLife 2019, 8, e48725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Irani S; Sipe SN; Yang W; Burkholder NT; Lin B; Sim K; Matthews WL; Brodbelt JS; Zhang Y Structural Determinants for Accurate Dephosphorylation of RNA Polymerase II by Its Cognate C-Terminal Domain (CTD) Phosphatase during Eukaryotic Transcription. J. Biol. Chem 2019, 294 (21), 8592–8605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Robinson MR; Taliaferro JM; Dalby KN; Brodbelt JS 193 Nm Ultraviolet Photodissociation Mass Spectrometry for Phosphopeptide Characterization in the Positive and Negative Ion Modes. J. Proteome Res 2016, 15 (8), 2739–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Fort KL; Dyachenko A; Potel CM; Corradini E; Marino F; Barendregt A; Makarov AA; Scheltema RA; Heck AJR Implementation of Ultraviolet Photodissociation on a Benchtop Q Exactive Mass Spectrometer and Its Application to Phosphoproteomics. Anal. Chem 2016, 88, 2303–2310. [DOI] [PubMed] [Google Scholar]
- (42).Portz B; Lu F; Gibbs EB; Mayfield JE; Rachel Mehaffey M; Zhang YJ; Brodbelt JS; Showalter SA; Gilmour DS Structural Heterogeneity in the Intrinsically Disordered RNA Polymerase II C-Terminal Domain. Nat. Commun 2017, 8 (1), 15231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Mayfield JE; Robinson MR; Cotham VC; Irani S; Matthews WL; Ram A; Gilmour DS; Cannon JR; Zhang YJ; Brodbelt JS Mapping the Phosphorylation Pattern of Drosophila Melanogaster RNA Polymerase II Carboxyl-Terminal Domain Using Ultraviolet Photodissociation Mass Spectrometry. ACS Chem. Biol 2017, 12 (1), 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Ramani MKV; Escobar EE; Irani S; Mayfield JE; Moreno RY; Butalewicz JP; Cotham VC; Wu H; Tadros M; Brodbelt JS; Zhang YJ Structural Motifs for CTD Kinase Specificity on RNA Polymerase II during Eukaryotic Transcription. ACS Chem. Biol 2020, 15 (8), 2259–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Darzacq X; Shav-Tal Y; de Turris V; Brody Y; Shenoy SM; Phair RD; Singer RH In Vivo Dynamics of RNA Polymerase II Transcription. Nat. Struct. Mol. Biol 2007, 14 (9), 796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Ebmeier CC; Erickson B; Allen BL; Allen MA; Kim H; Fong N; Jacobsen JR; Liang K; Shilatifard A; Dowell RD; Old WM; Bentley DL; Taatjes DJ Human TFIIH Kinase CDK7 Regulates Transcription-Associated Chromatin Modifications. Cell Rep. 2017, 20 (5), 1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Glover-Cutter K; Larochelle S; Erickson B; Zhang C; Shokat K; Fisher RP; Bentley DL TFIIH-Associated Cdk7 Kinase Functions in Phosphorylation of C-Terminal Domain Ser7 Residues, Promoter-Proximal Pausing, and Termination by RNA Polymerase II. Mol. Cell. Biol 2009, 29 (20), 5455–5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Pérez-Mejías G; Velázquez-Cruz A; Guerra-Castellano A; Banos-Jaime B; Díaz-Quintana A; González-Arzola K;Ángel De la Rosa M; Díaz-Moreno I Exploring Protein Phosphorylation by Combining Computational Approaches and Biochemical Methods. Comput. Struct. Biotechnol. J 2020, 18, 1852–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Li MZ; Elledge SJ Harnessing Homologous Recombination in Vitro to Generate Recombinant DNA via SLIC. Nat. Methods 2007, 4 (3), 251–256. [DOI] [PubMed] [Google Scholar]
- (50).Burkholder NT; Sipe SN; Escobar EE; Venkatramani M; Irani S; Yang W; Wu H; Matthews WM; Brodbelt JS; Zhang Y Mapping RNAPII CTD Phosphorylation Reveals That the Identity and Modification of Seventh Heptad Residues Direct Tyr1 Phosphorylation. ACS Chem. Biol 2019, 14 (10), 2264–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Mayfield JE; Irani S; Escobar EE; Zhang Z; Burkholder NT; Robinson MR; Mehaffey MR; Sipe SN; Yang W; Prescott NA; Kathuria KR; Liu Z; Brodbelt JS; Zhang Y Tyr1 Phosphorylation Promotes Phosphorylation of Ser2 on the C-Terminal Domain of Eukaryotic RNA Polymerase II by P-TEFb. eLife 2019, 8, e48725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Klein DR; Holden DD; Brodbelt JS Shotgun Analysis of Rough-Type Lipopolysaccharides Using Ultraviolet Photodissociation Mass Spectrometry. Anal. Chem 2016, 88 (1), 1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Burkholder NT; Sipe S; Kumar M; Escobar E; Irani S; Matthews W; Sim K; Brodbelt J; Zhang YJ Modification of Ser7 of the RNA Polymerase II C-Terminal Domain (CTD) Regulates CTD Phosphorylation Patterns and Transcription. FASEB J. 2019, 33, 458–458. [Google Scholar]
- (54).Pino LK; Searle BC; Bollinger JG; Nunn B; MacLean B; MacCoss MJ The Skyline Ecosystem: Informatics for Quantitative Mass Spectrometry Proteomics. Mass Spectrom. Rev 2020, 39 (3), 229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).R Core Team. R: A Language and Environment for Statistical Computing; Vienna, Austria, 2013. [Google Scholar]
- (56).Daly DS; Anderson KK; Panisko EA; Purvine SO; Fang R; Monroe ME; Baker SE Mixed-Effects Statistical Model for Comparative LC–MS Proteomics Studies. J. Proteome Res 2008, 7 (3), 1209–1217. [DOI] [PubMed] [Google Scholar]
- (57).Webb-Robertson B-JM; McCue LA; Waters KM; Matzke MM; Jacobs JM; Metz TO; Varnum SM; Pounds JG Combined Statistical Analyses of Peptide Intensities and Peptide Occurrences Improves Identification of Significant Peptides from MS-Based Proteomics Data. J. Proteome Res 2010, 9 (11), 5748–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.