

Abstract
Accumulating evidence suggests X‐linked dominant mutations in UBQLN2 cause amyotrophic lateral sclerosis (ALS) with frontotemporal dementia (FTD) through both loss‐ and gain‐of‐function mechanisms. However, the mechanisms by which the mutations cause disease are still unclear. The goal of the study was to uncover the possible pathomechanism(s) by which UBQLN2 mutations cause ALS/FTD. An analysis of proteomic changes in neuronal tissue was used to identify proteins with altered accumulation in the P497S UBQLN2 transgenic mouse model of ALS/FTD. We then used immunocytochemistry and biochemical techniques to confirm protein changes in the mutant P497S mice. Additionally, we used cell lines inactivated of UBQLN2 expression to determine whether its loss underlies the alteration in the proteins seen in P497S mice. The proteome screen identified a dramatic alteration of serine protease inhibitor (serpin) proteins in the mutant P497S animals. Double immunofluorescent staining of brain and spinal cord tissues of the mutant and control mice revealed an age‐dependent change in accumulation of Serpin A1, C1, and I1 in puncta whose staining colocalized with UBQLN2 puncta in the mutant P497S mice. Serpin A1 aggregation in P497S animals was confirmed by biochemical extraction and filter retardation assays. A similar phenomenon of serpin protein aggregation was found in HeLa and NSC34 motor neuron cells with inactivated UBQLN2 expression. We found aberrant aggregation of serpin proteins, particularly Serpin A1, in the brain and spinal cord of the P497S UBQLN2 mouse model of ALS/FTD. Similar aggregation of serpin proteins was found in UBQLN2 knockout cells suggesting that serpin aggregation in the mutant P497S animals may stem from loss of UBQLN2 function. Because serpin aggregation is known to cause disease through both loss‐ and gain‐of‐function mechanisms, we speculate that their accumulation in the P497S mouse model of ALS/FTD may contribute to disease pathogenesis through similar mechanism(s).
Keywords: amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), protein aggregation, serpin, Ubiquilin‐2 (UBQLN2)
We found aberrant accumulation and co‐aggregation of serpin proteins, particularly Serpin A1, with UBQLN2 inclusions in the brain and spinal cord of the P497S UBQLN2 mouse model of ALS/FTD. Similar aggregation of serpin proteins was found in UBQLN2 knockout cells suggesting serpin aggregation may stem from a loss of UBQLN2 function. Serpin aggregation is known to cause disease through both gain‐ and loss‐of‐function mechanisms, and we speculate that their accumulation in P497S mice may contribute to disease pathogenesis through similar mechanisms.

1. INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive fatal neurodegenerative disease characterized by loss of upper and lower motor neurons in the brain and spinal cord (SC) (1, 2). ALS symptoms typically involve progressive loss of muscle movement, frequently leading to paralysis and death (2). About 15–20% of people with ALS also have frontotemporal dementia (FTD) (3). Mutations in at least 30 different genes have now been linked to inheritance of either ALS or of ALS with FTD (ALS/FTD) (4, 5, 6). The largest category of the genes mutated in ALS are those that function in proteostasis, including Ubiquilin‐2 (UBQLN2), an intronless gene on the X‐chromosome (7). The UBQLN2 protein maintains proteostasis by clearing ubiquitinated and misfolded proteins through both the proteasome and autophagy degradation pathways as well as by assisting in protein refolding and chaperone activity (8, 9, 10). Most of the identified ALS/FTD mutations are missense mutations that map in or surround a PXX repeat motif, of unknown function, unique to UBQLN2 (8, 9, 10). Growing evidence suggests that the mutations severely impede both proteasomal degradation and autophagy, presumably resulting in a toxic buildup of protein aggregates within the cell (7, 11, 12, 13). The physiologic consequence of this disruption in proteostasis remains to be clarified. To address this, we compared the hippocampus and SC proteomes of the P497S UBQLN2 transgenic (Tg) mouse model of ALS/FTD (P497S line) (14) with a Tg line expressing wild‐type (WT) human UBQLN2 protein (WT356 line) and a Non‐Tg line. Pursuit of candidate proteins from the screen led to our discovery of major changes in serine protease inhibitor (serpin) protein localization in the P497S line, compared to the two control lines (WT356 and Non‐Tg).
Serpins are a large family of metastable proteins that primarily function to regulate proteolytic cascades through the inhibition of serine proteases (15, 16). The proteins have been classified into clades that are all secreted, except members of clade B, which are intracellular serpins (15, 17). Inhibitory serpins use an exposed reactive center loop (RCL) that baits their target proteases (15, 18, 19). Upon interaction with a target protease, the RCL is cleaved, and the serpin undergoes a large conformational change, inactivating the protease in the process (18, 20). This metastability makes serpins particularly prone to misfolding and polymerization. Indeed, a number of diseases, termed serpinopathies, have been linked to the polymerization and/or dysregulation of serpin expression (21, 22). Two primary examples are α1‐antitrypsin deficiency and familial encephalopathy with neuroserpin inclusion bodies (FENIB), which are caused by mutations in Serpin A1 and Serpin I1, respectively (21, 22). These serpinopathies arise chiefly because mutations in serpins cause them to misfold in the ER during synthesis. Misfolded serpins are removed from the ER and degraded by the proteasome by ER‐associated degradation (ERAD) (21, 23, 24, 25). Serpin depletion can lead to unrestricted activity of their protease targets and thus tissue damage (26, 27). The misfolded serpins proteins also exhibit increased tendency to polymerize in the ER causing ER stress, chronic induction of which causes cell death (28, 29, 30). The discovery that mutations in serpins can induce disease of both the tissues they are synthesized in and those the inhibitors act in provided the first clear example of how protein aggregation causes disease through both gain and loss‐of‐function mechanisms (31, 32, 33, 34). The proteomic alterations of serpins found in P497S mutant animals and their known connection to disease provided the foundation for our studies.
2. MATERIALS AND METHODS
2.1. UBQLN2 Tg mice and animal procedures
Three congenic C57BL6/J mouse lines were used for the studies: a Non‐Tg C57BL6/J line (Strain 000664, The Jackson Labs, Bar Harbor, ME) and two UBQLN2 Tg lines expressing equivalent levels of either WT human UBQLN2 (WT356 line) or the P497S ALS/FTD UBQLN2 mutant protein (P497S line) (14). The mutant P497S line was shown to model central features of human ALS/FTD including development of motor neuron disease, cognitive deficits, TDP‐43 pathology, and deposition of UBQLN2 inclusions in the brain and SC (14). By contrast, these phenotypes and pathology were not observed in the WT356 line (14). Both Tg lines used in the present study were backcrossed with C57BL6/J mice for at least 10 generations. During the backcrossing the P497S line developed a more attenuated phenotype, exhibiting milder disease symptoms. We recently described the behavioral deficits and pathology of this more attenuated phenotype, showing that at 52 weeks of age P497S mutant animals have diminished grip strength, reduced muscle fiber diameter, massive accumulation of UBQLN2 inclusions in the brain and SC, together with significant loss of neurons in both the dentate gyrus of the brain and of MNs in the SC compared to Non‐Tg animals (35). Husbandry, genotyping, and euthanasia of animals was as described previously (14, 35). All animal procedures were approved by University of Maryland Baltimore Animal Care and Use Committees and conducted in full accordance with the NIH Guide for the Care and Use of Laboratory Animals.
2.2. Preparation of protein lysates, sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE), and immunoblotting
Mouse tissue lysates were made from the hippocampus and lumbar region of the brain and SC isolated from three independent Non‐Tg, WT356, and P497S animals at 32 weeks of age. The hippocampus tissue was homogenized in 300 µl, and the SC tissue in 800 µl, of our standard protein lysis buffer (50 mM Tris pH 6.8, 150 mM NaCl, 20 mM EDTA, 1 mM EGTA, 0.5% SDS, 0.5% NP40, 0.5% N‐lauroylsarcosine (sarkosyl), 10 mM orthovanadate, 2.5 mM sodium fluoride) (36) using a Kontes‐type glass homogenizer with a Teflon pestle connected to a Yamato LSC LH21 homogenizer. Protein lysates from cell cultures were prepared by washing cultured cells twice with 1× PBS before collecting with cell scrapers in the same protein lysis solution followed by brief sonication (3 × 1 min periods) using a Branson Sonifier 450. Protein concentrations of both the tissue and cell lysates were measured by the BCA method (Pierce BCA Assay Kit, Thermo Fisher Scientific). Tissue lysates were then solubilized with five volumes of urea buffer (8 M urea, 50 mM EPPS pH 8.5, 0.5% sarkosyl, 20 mM EDTA, 1 mM EGTA, 2.5 mM sodium fluoride, and 1 mM Pefabloc (Sigma‐Aldrich, St. Louis, MO) and then mixed with one‐fourth of its volume with non‐SDS gel loading buffer (8 M urea, 40% glycerol, 0.25 M Tris‐HCl pH 6.8, 15 mM DTT, 1.43 M 2‐Mercaptoethanol, and 0.04% Bromophenol Blue) while cell lysates were diluted in the same manner with loading gel buffer containing 8% of SDS in place of urea. The urea mixtures were heated at 37ºC for 10 min, whereas the SDS mixtures were heated at 100ºC for 5 min before gel loading. Samples were resolved on 8.5–10% SDS‐PAGE polyacrylamide gels at 60–90 V and then transferred onto 0.45 µm PVDF membranes (Millipore, Billerica, MA, USA) using the semidry transfer Power Blotter XL System (Thermo Fisher Scientific). Membranes were probed with primary serpin antibodies overnight and washed five times before incubating with corresponding HRP‐conjugated secondary antibodies (all from Thermo Fisher Scientific, Waltham, MA) for 1.5 h. Chemiluminescence images were captured using a FluorChem E imager (Protein Simple, San Jose, CA) and the band intensities were quantified using AlphaView software (Protein Simple).
2.3. Antibodies
The following primary antibodies were used for immunoblotting (IB) and immunofluorescence (IF). Rat anti‐Serpin A1a (#MAB7690, IB, IF), rat anti‐Serpin C1 (#MAB1287, IB, IF, both from R&D Systems, Minneapolis, MN), mouse anti‐UBQLN2 #NBP2‐25164, IB, IF, Novus Biologicals, Littleton, CO), mouse anti‐neuroserpin (#66997‐1, IB, IF, Proteintech Group, Inc, Rosemont, IL), mouse anti‐ubiquitin (sc‐8017, Santa Cruz Biotechnology, IB, Santa Cruz, CA), Rabbit anti‐GFAP (#PA1‐10019 IF, Thermo Fisher Scientific), rabbit anti‐IBA1 (#019‐19741, IF, FUJIFILM Wako Pure Chemicals, Corp, Richmond, VA), rabbit anti‐Serpin B1 (#PA576875, IB, IF, Thermo Fisher Scientific), rabbit anti‐Serpin C1/antithrombin III (#NBP2‐76966, IB, Novus Biologicals), rabbit anti‐LC3A/B (#4108, IF, Cell Signaling Technology, Danvers, MA, USA), rabbit anti‐Myc (UMY81, IB, home‐made against a peptide with an amino acid sequence of MEQKLISEEDLN), rabbit anti‐Neuroserpin (#ab33077, IB, IF, abcam, Eugene, OR), rabbit anti‐p97/VCP (UMY475, IB, home‐made), rabbit anti‐UBQLN2 (#23449‐1‐AP, IF Proteintech Group, Inc.), goat anti‐ChAT (#AB144P, IF, Millipore‐Sigma, Burlington, MA), and goat anti‐LAMP1 (sc‐8098, IF, Santa Cruz Biotechnology (Dallas, TX, USA).
2.4. Serpin antibody validation
Complementary DNAs encoding the entire open reading frames of either full‐length human Serpin A1, human Serpin B1, a 258 amino acid spliced‐variant of human Serpin C1, and full‐length Serpin I1 (clones MHS6278‐202756262, MHS6278‐202755832, MHS6278‐202841233, and MHS6278‐202806491, respectively, Dharmacon, Inc, Chicago, IL) were cloned between the EcoRI and XhoI restrictions sites of the pCMV‐Myc‐C vector (Clontech) to express each serpin protein with a C‐terminal Myc‐tag. The recombinant plasmid DNAs were transfected into HeLa cell cultures using Lipofectamine LTX (Thermo Fisher Scientific, Waltham, MA). Lysates were collected 24 h post‐transfection using our standard lysis buffer, mixed with SDS gel loading buffer and, following separation by SDS‐PAGE, were immunoblotted with different serpin antibodies to ensure their specificity. The serpin antibodies we used for all of the immunostainings reacted only with their corresponding Myc‐tagged recombinant protein as well as with endogenous proteins of their expected size.
2.5. Tissue sectioning and immunofluorescence staining
Whole brains and spinal cords were dissected from perfused and non‐perfused P497S, WT356, and Non‐Tg animals. All tissues were fixed in 4% of paraformaldehyde in 1× PBS for 24 h at 4ºC prior to transferring to a 25% of sucrose solution in 1× PBS at 4ºC for another 24 h. Tissues were then embedded in OCT Compound (Sakura Finetek USA, Inc., Torrance CA). Sagittal sections (14–20 µm) of the brain and transverse sections of the lumbar region of the SC were cut on a Leica CM3050 cryostat and mounted onto glass slides that were pretreated with a 5% solution of 3‐Aminopropyltriethoxysilane. Brain and lumbar SC sections were fixed, permeabilized, and blocked by sequential incubation with 4% PFA, 0.1% Triton X‐100, and 0.8% BSA, before being incubated for 2 h in a 1:100 dilution of primary antibodies (listed above) at room temperature. Slides were then gently washed three times in 1× PBS prior to incubation for 2 h at room temperature in a 1:200 dilution of Alexa‐Fluor‐conjugated secondary antibodies of different wavelengths (488, 594, and 647 nm). Slides were again washed with 1× PBS and incubated with 4′,6‐diamidino‐2‐phenylindole (DAPI) (0.5 µg/ml) for 10 min. Coverslips were mounted with Aquamount and allowed to dry overnight. Slides were imaged using a Leica MZ 16 FA fluorescence stereo microscope to capture low magnification images of the hippocampus and with a Nikon A1R super resolution confocal microscope for high magnification images. For confocal imaging, a S Fluor 40× (1.30 oil) objective lens was used to capture immunohistochemical staining of brain (1× and a 5× zoom images) and SC (1×, 2×, and 5× zoom images) sections. Image channels were merged using iVision software (BioVision Technologies, Exton, PA).
2.6. Line scans of immunofluorescent images
Line scans were performed using Image J software. Regions of interest from zoom confocal images were analyzed using the Plot Profile function to analyze the pixel intensity along a defined region of interest. Plots were obtained from identical regions of interest drawn in each of the color channels and then overlaid to produce the final plots. Regions of interest were chosen so as to include representative puncta in order to analyze the degree of colocalization between the proteins.
2.7. Filter trap assay
Filter trap assays were performed with lysates made from the cortical region of the brain isolated from 8‐ to 32‐week‐old P497S, WT356, and Non‐Tg animals as well as with cell lysate and culture medium of HeLa and NSC34 mouse motor neuron cell cultures that were grown for 48 h. The cell culture conditions were described previously (12). The parental and two different UBQLN2 knockout (KO) lines for both the HeLa (KO8 and KO12) were analyzed by this assay. UBQLN2 KO NSC34 KO20 and KO69 cells were used for other aggregation assays. The generation and characterization of the UBQLN2 KO lines were described previously (12). All the UBQLN2 KO lines lack UBQLN2 expression, but some of them have subtle alterations in expression of other UBQLN isoforms or other proteins (12). All of the lysates were homogenized in our standard protein lysis buffer. Following homogenization, the cortical lysates were briefly centrifuged at 1000 g for 10 min to remove tissue debris. The protein concentration of the saved supernatant as well that of the lysates from the cell cultures were determined by the BCA method. Equal amounts of protein lysate for each sample was then adjusted with protein lysis buffer so that their volume was the same prior to their filtration. Equal volumes of the cell culture media were filtered. All the filter trap assays were conducted using 0.2 µm OE66 cellulose acetate membranes (Schleicher & Schuell, Dassel, Germany) using a Dot‐Blot Apparatus (Bio‐Rad, Hercules, CA) by first wetting the filters with wash buffer (0.1% SDS, 10 mM Tris pH 8.0, 150 mM NaCl) prior to gentle vacuum filtration of the samples through the membranes. The membranes were then washed twice with the wash buffer before being removed from the apparatus, blocked with 4% of BSA in TBS buffer (37) for 30 min, and probed by the method described in the immunoblotting section.
2.8. Differential extraction assay
Aggregated proteins in mouse brain tissue were characterized by their differential extraction properties using different biochemical buffers as described by Newmann et al. (38, 39). Equal weights of 8‐ and 32‐week brain cortical tissue from P497S, WT356, and Non‐Tg mice were homogenized in 5 ml/g of low salt (LS) buffer (10 mM Tris pH 7.5, 5 mM EDTA, 1 mM DTT, 10% sucrose, and protease inhibitor cocktail tablet). The homogenates were centrifuged at 25,000 g for 30 min at 4ºC and the supernatant was saved. The remaining pellet was re‐homogenized in Triton X‐100 (TX‐100) buffer (LS buffer plus 1.0% Triton X‐100, 0.5 M NaCl) and then centrifuged again at 180,000 g for 30 min at 4ºC and the supernatant was again saved. The resulting pellet was re‐homogenized a second time in the TX‐100 buffer with the addition of 30% sucrose to float and remove myelin debris. This supernatant was discarded. The remaining pellet was homogenized in sarkosyl (SARK) buffer (LS buffer, 1.0% sarkosyl, 0.5 M NaCl) and incubated on the shaker for 1 h at 22ºC. It was then centrifuged at 180,000 g for 30 min at 22ºC and supernatant collected. The final fraction was obtained by homogenizing the insoluble pellet in urea buffer (7 M urea, 2 M thiourea, 4% CHAPS, 30 mM Tris‐HCl pH 8.5), followed by centrifugation at 25,000 g for 30 min at 22ºC, and saving of the supernatant. SDS buffer (10 mmol/L Tris pH 6.8, 1 mmol/L EDTA, 40 mmol/L DTT, and 1.0% SDS) was added to each of the saved extractions (LS, TX, SARK, and urea) and adjusted with SDS gel loading buffer. All of the mixtures, except the urea mixtures, were heated at 100ºC for 5 min and equal portions of them were separated on 8–12% Bis‐Tris NuPage gels and immunoblotted with serpin or UBQLN2 antibodies.
2.9. Proteomics
The proteome of the hippocampus and lumbar SC regions from three independent 2‐month‐old Non‐Tg, WT356, and P497S were determined by isobaric labeling and mass spectroscopy quantification. Full details of this analysis, including links to access the raw data can be found in the publication (40). Comparison of the Log2 ratio of the average peptide expression between the genotypes indicated that many serpin proteins had higher ratios between P497S versus Non‐Tg and P497S versus WT356 compared WT356 versus Non‐Tg animals.
2.10. Statistical methods
Differences in protein expression were analyzed using either Student's t‐tests or one‐way ANOVA, or two‐way ANOVA in GraphPad Prism 9 software depending on the number of groups and factors being compared. p < 0.05 was considered statistically significant and *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
3. RESULTS
3.1. Alteration of serpin protein levels in P497S UBQLN2 mice by proteomic analysis
Proteome changes to the hippocampus and SC of congenic 8‐week‐old P497S Tg, WT356 Tg, and Non‐Tg mice were recently described (40). The P497S Tg line expresses human UBQLN2 under control of the neuron‐specific Thy1.2 promoter, whereas the WT356 line expresses an equivalent amount of WT UBQLN2 by the same promoter (12). Behavioral and pathologic studies have shown that the P497S line recapitulates hallmark features of the human disease, while the WT356 line does not (12, 35). Examination of the Log2 ratio of change in protein expression between the genotypes revealed an increase in several members of the serpin protein family (Table 1). In particular, expression of ten serpins was upregulated in either the hippocampus or SC in P497S animals but not in the WT356 or Non‐Tg controls. However, comparison of their alteration in the three animals for each genotype examined revealed wide fluctuation between individuals, likely resulting in many of the changes being nonsignificant. Nevertheless, because changes in serpin expression have been found in neurodegenerative diseases, including ALS (41, 42, 43, 44, 45, 46, 47), we conducted further investigations to determine if expression or localization of serpin members were altered in the mice. We focused our studies on Serpin A1 (alpha‐1‐antitrypsin), Serpin B1, and Serpin C1 (antithrombin) because they were the most highly expressed serpins in P497S animals compared to controls and represent well‐studied serpins from several different clades (15, 17). Additionally, we chose to investigate Serpin I1 (neuroserpin), because it is primarily expressed in the brain and involved in neurodegeneration (27). Furthermore, several of these serpins have been linked to different diseases (16, 22).
TABLE 1.
Proteomics analysis of 8‐week‐old P497S UBQLN2 mice and age‐matched controls (WT356 UBQLN2 and Non‐Tg animals) comparing the log2 ratios of P497S/WT356, P497/Non‐Tg, and WT356/Non‐Tg animals in the lumbar SC and hippocampus
| Protein Id | Gene symbol | Description | log2 Hippo (P497S/WT356) | p value | log2 Hippo (P497S/Non‐Tg) | p value | log2 Hippo (WT356/Non‐Tg) | p value | log2 Lumbar SC (P497S/WT356) | p value | log2 Lumbar SC (P497S/Non‐Tg) | p value | log2 Lumbar SC (Non‐Tg/WT356) | p value | log2 ratio |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sp|Q9CY58| | Serbp1 | PAIRB_MOUSE Plasminogen activator inhibitor 1 RNA‐bp | −0.066 | 0.176 | −0.064 | 0.558 | 0.002 | 0.986 | −0.117 | 0.330 | 0.0446745 | 0.4184232 | −0.083 | 0.244 | |
| sp|P07758| | Serpina1a* | A1AT1_MOUSE Alpha‐1‐antitrypsin 1‐1 | 1.347 | 0.191 | 1.035 | 0.243 | −0.312 | 0.640 | 0.416 | 0.252 | 0.0801778 | 0.1739446 | −0.036 | 0.366 | |
| sp|P22599| | Serpina1b | A1AT2_MOUSE Alpha‐1‐antitrypsin 1‐2 | 1.408 | 0.165 | 1.113 | 0.214 | −0.295 | 0.591 | 0.425 | 0.197 | 0.2422415 | 0.2390785 | −0.110 | 0.270 | |
| sp|Q00897| | Serpina1d | A1AT4_MOUSE Alpha‐1‐antitrypsin 1‐4 | 0.560 | 0.048 | 0.4144827 | 0.0665841 | 0.089 | 0.614 | |||||||
| sp|Q00898| | Serpina1e | A1AT5_MOUSE Alpha‐1‐antitrypsin 1‐5 | 1.486 | 0.190 | 0.796 | 0.322 | −0.690 | 0.386 | 0.652 | 0.313 | 0.1900601 | 0.5501875 | −0.235 | 0.589 | |
| sp|P07759| | Serpina3k | SPA3K_MOUSE Serine protease inhibitor A3K | 1.388 | 0.211 | 1.146 | 0.257 | −0.242 | 0.621 | 0.397 | 0.298 | 0.1034186 | 0.5541651 | −0.059 | 0.747 | |
| tr|G3X8T9| | Serpina3n | G3X8T9_MOUSE Serine (Or cys) peptidase inhibitor, 3N | 0.662 | 0.233 | 0.754 | 0.195 | 0.092 | 0.473 | 0.352 | 0.124 | 0.318729 | 0.353001 | −0.098 | 0.826 | |
| sp|Q9D154| | Serpinb1a* | ILEUA_MOUSE Leukocyte elastase inhibitor A | 0.570 | 0.169 | 0.761 | 0.096 | 0.191 | 0.332 | 0.044 | 0.524 | 0.182243792 | 0.729305304 | −0.192 | 0.700 | |
| sp|Q8VHP7| | Serpinb1b | ILEUB_MOUSE Leukocyte elastase inhibitor B | 1.254 | 0.241 | 1.270 | 0.237 | 0.016 | 0.832 | −0.008 | 0.905 | 0.3379195 | 0.3289914 | −0.059 | 0.887 | |
| sp|Q60854| | Serpinb6 | SPB6_MOUSE Serpin B6 | 0.085 | 0.483 | 0.039 | 0.731 | −0.046 | 0.182 | 0.116 | 0.099 | 0.0639798 | 0.7973305 | −0.588 | 0.395 | |
| tr|O08804| | Serpinb6b | O08804_MOUSE NK13 | 0.096 | 0.563 | 0.043 | 0.808 | −0.053 | 0.598 | 0.162 | 0.249 | −0.1165423 | 0.0305189 | −0.081 | 0.194 | −1.000 |
| sp|O08800| | Serpinb8 | SPB8_MOUSE Serpin B8 | −0.204 | 0.307 | −0.411 | 0.103 | −0.206 | 0.406 | −0.750 | ||||||
| tr|O08797| | Serpinb9 | O08797_MOUSE Protein Serpinb9 | 0.204 | 0.335 | 0.038 | 0.831 | −0.166 | 0.057 | 0.128 | 0.075 | −0.0034693 | 0.9759878 | −0.036 | 0.812 | −0.500 |
| sp|P32261| | Serpinc1* | ANT3_MOUSE Antithrombin‐III | 1.055 | 0.424 | 1.274 | 0.367 | 0.220 | 0.486 | 0.374 | 0.279 | 0.0343605 | 0.5043039 | 0.042 | 0.422 | −0.250 |
| sp|Q07235| | Serpine2 | GDN_MOUSE Glia‐derived nexin | 0.155 | 0.192 | 0.000 | 0.999 | −0.155 | 0.047 | −0.035 | 0.464 | −0.0394735 | 0.8658744 | −0.068 | 0.808 | 0.000 |
| sp|P97298| | Serpinf1 | PEDF_MOUSE Pigment epithelium‐derived factor | 0.208 | 0.073 | 0.025 | 0.823 | −0.183 | 0.203 | 0.029 | 0.919 | −0.0073329 | 0.9361391 | −0.012 | 0.755 | 0.250 |
| sp|P97290| | Serping1 | IC1_MOUSE Plasma protease C1 inhibitor | 0.325 | 0.125 | 0.667654 | 0.0549768 | 0.108 | 0.704 | 0.500 | ||||||
| sp|P19324| | Serpinh1 | SERPH_MOUSE Serpin H1 | 0.187 | 0.612 | 0.035 | 0.921 | −0.151 | 0.291 | 0.033 | 0.837 | 0.0278504 | 0.7998078 | 0.145 | 0.033 | 0.750 |
| sp|O35684| | Serpini1* | NEUS_MOUSE Neuroserpin | −0.277 | 0.203 | −0.298 | 0.166 | −0.021 | 0.696 | 0.005 | 0.958 | −0.012424 | 0.7553671 | −0.007 | 0.936 | 1.000 |
Several members of the serpin family are specifically upregulated in P497S animals. The color shades correspond to the Log2 ratios not significance.
Denotes the serpin proteins that were studied.
3.2. Serpin proteins accumulate and colocalize with UBQLN2 inclusions in the brain of P497S animals in an age‐dependent manner
We first determined whether localization of serpin proteins was altered in P497S mice. Accordingly, we performed double immunofluorescent staining of sagittal brain sections of 8‐ and 52‐week‐old P497S, WT356, and Non‐Tg animals for UBQLN2 and either Serpin A1, Serpin B1, Serpin C1, or Serpin I1. All the serpin antibodies were validated by correct recognition of only their corresponding recombinant protein or endogenous protein (Figure S1). Consistent with previous findings (12, 14), UBQLN2 staining of P497S animals showed the distinctive pattern of concentrated UBQLN2 inclusions in the dentate gyrus and CA1 to CA3 regions of the hippocampus, which were particularly prominent in the 52‐week‐old animals compared to the 8‐week‐old animals (Figure 1 and Figure S2). By contrast, age‐matched WT356 and Non‐Tg animals contained few, if any, such inclusions (Figure 1 and Figure S2). We did occasionally observe fibrillar‐like staining in control animals which we attributed to antibody cross‐reaction with blood vessels in non‐perfused animals as this staining was not observed in perfused animals of the same genotypes. Double staining of the sections for serpin proteins revealed dramatic alteration in localization of all four serpins in the 52‐week‐old P497S animals compared to the age‐matched WT 356 and Non‐Tg animals, which was most apparent in the hippocampal region (Figure 1). WT356 animals had similar serpin staining to the Non‐Tg animals, indicating that the alteration in P497S animals is a result of expression of the ALS/FTD P497S mutant UBQLN2 protein rather than simple overexpression of UBQLN2 protein. In the P497S animals, staining of Serpin A1, C1, and to a lesser extent Serpin I1, was coincident and closely aligned with the UBQLN2 inclusion pattern in the dentate gyrus, although subtle differences in the staining patterns of the serpins were noticeable (Figure 1 and Figure S2). Meanwhile, the staining of B1 was denser within the granule layer of the dentate gyrus and CA1 to CA3 regions of the hippocampus, in a pattern that was adjacent but did not exactly overlap the UBQLN2 inclusions.
FIGURE 1.
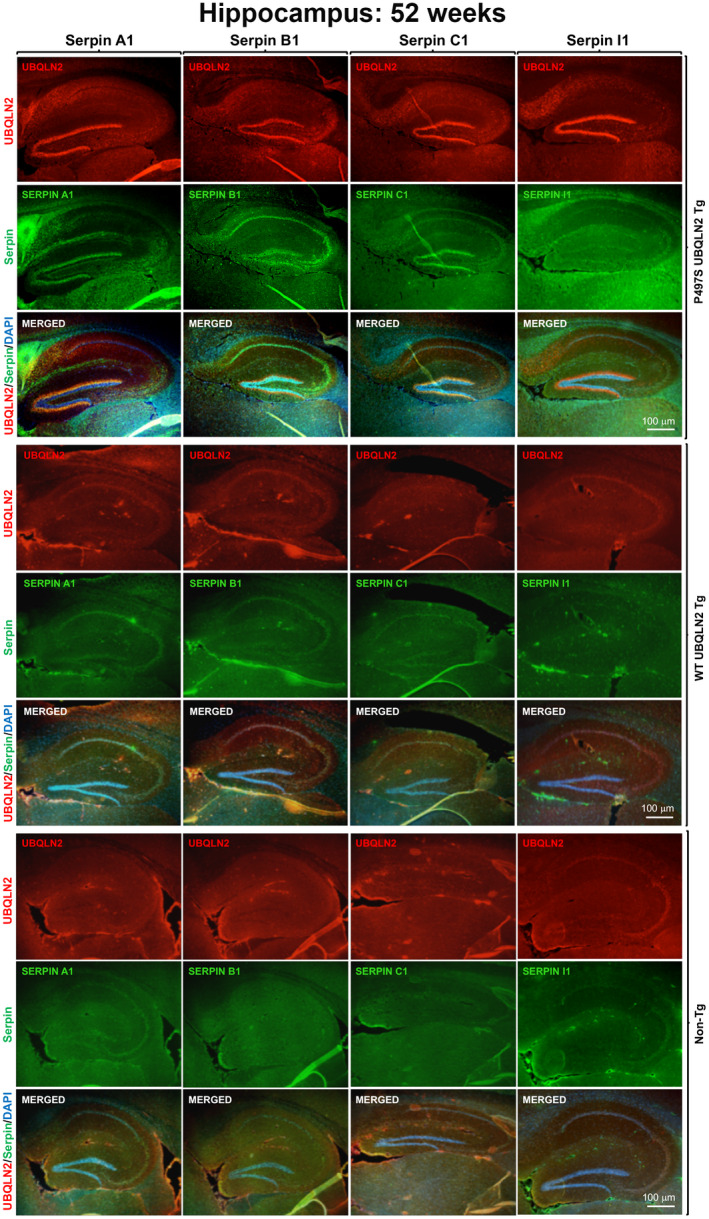
Alteration of serpin localization in the brain of P497S mutant mice. Staining of UBQLN2 and Serpin A1, B1, C1, and I1 proteins and their corresponding merged images, including of the DAPI panel (not shown), in the hippocampal region of sagittal brain sections from 52‐week‐old P497S (top panel), WT356 (middle panel), and Non‐Tg (bottom panel) animals. Scale bars shown = 100 μm for all images
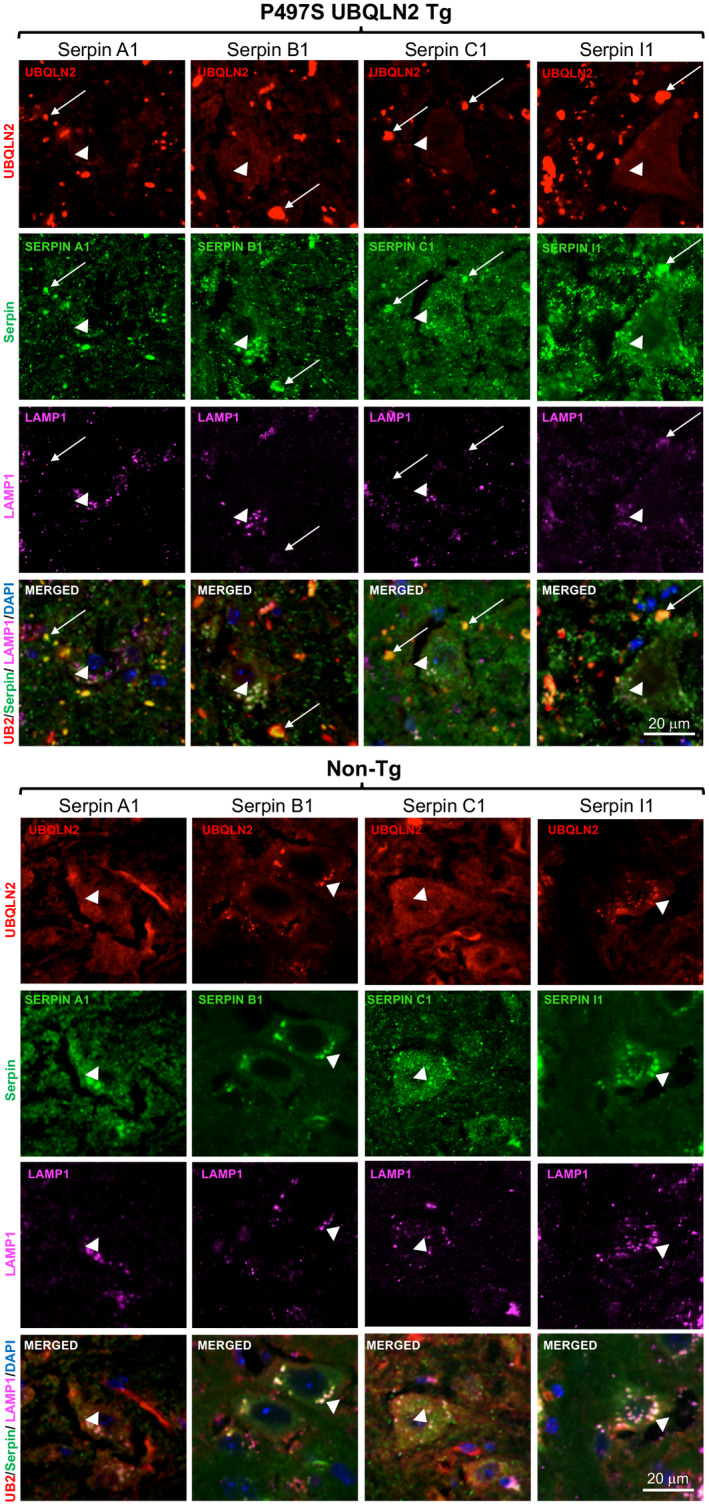
To determine the extent of colocalization of the serpin proteins with UBQLN2 inclusions, we conducted higher magnification confocal microscopy (Figure 2). The examination revealed that, compared to Non‐Tg and WT356 animals, P497S animals had highly concentrated staining of Serpin A1, C1, and I1 in numerous irregular shaped puncta, many of which colocalized with the UBQLN2 inclusions in the dentate gyrus, CA1, CA3, and cortex regions of the brain (Figure 2 and Figure S3). In the dentate gyrus region, the colocalization was mainly apparent in the molecular layer and less so in the granule layer where smaller serpin puncta were visible and UBQLN2 inclusions were less obvious. Analysis of line scans through representative areas of the staining confirmed the high degree of overlap between Serpin A1, C1, and I1 with UBQLN2 staining of puncta (Figure S4). The degree of overlap was more striking for Serpin A1 and Serpin C1 compared to Serpin I1 (Figure 2 and Figure S4). Interestingly, many Serpin A1 and Serpin I1 puncta did not show UBQLN2 staining. Serpin B1 showed increased perinuclear staining in granule cells of the dentate gyrus in P497S animals compared to the WT and Non‐Tg animals, but the staining did not overlap with that of the UBQLN2 inclusions (Figure 2 and Figure S4).
FIGURE 2.
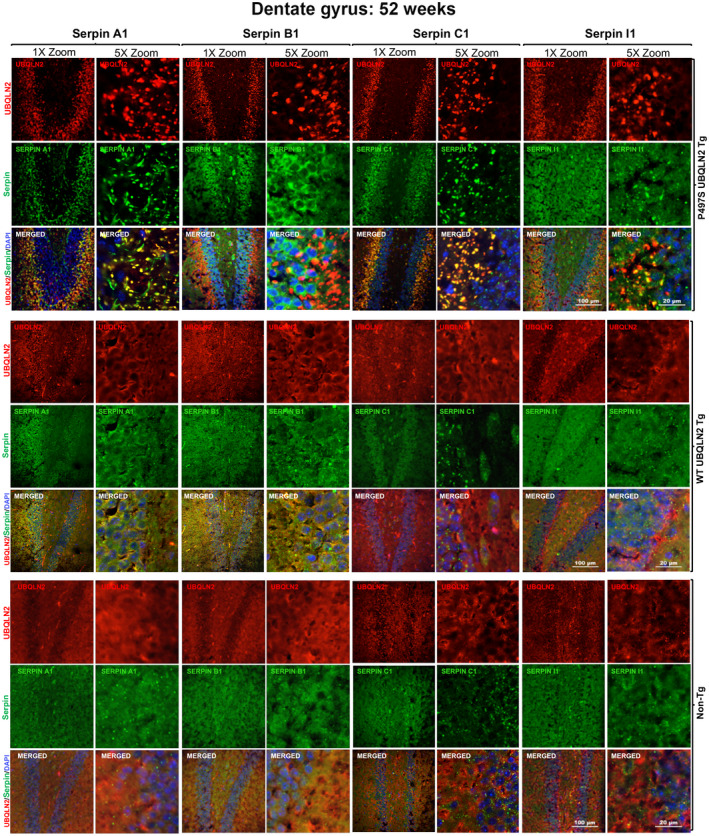
Colocalization of UBQLN2 and serpin proteins in puncta in the dentate gyrus region of the brain in P497S animals. Confocal microscopy images of the staining of UBQLN2 and Serpin A1, B1, C1, and I1 proteins and their corresponding merged images, including the DAPI panel, of the dentate gyrus of 52‐week‐old P497S (top panel), WT356 (middle panel), and Non‐Tg (bottom panel) animals. Scale bars shown = 100 μm and 20 μm for 1× and 5× zoom images, respectively
3.3. Colocalization of Serpin proteins with UBQLN2 inclusions in the SC of P497S animals
Because P497S animals develop MN disease, we also examined lumbar SC sections from all three genotypes for alteration in serpin staining (Figure 3). For this comparison, we used tissue from 32‐week‐old animals because MNs are still present in P497S animals at this age unlike at 52 weeks when most are lost (14, 35). The sections were triple stained for UBQLN2, one of the four serpin proteins, and choline acetyltransferase (ChAT), a MN marker (Figure 3 and Figure S5). As expected, P497S animals contained numerous UBQLN2 inclusions that were spread throughout the SC, with the majority located in structures outside the MNs. This contrasted with UBQLN2 staining in the WT356 and Non‐Tg animals, where it was more diffuse. Examination of serpin staining of the same sections revealed that all four serpins also had more punctate distribution in P497S animals compared to WT356 and Non‐Tg animals (Figure 3). Merging of the images and analysis of the overlap of the fluorescent signals by line scan revealed excellent colocalization of all four serpin proteins with the UBQLN2‐positive puncta (Figure S4). The staining of the four serpin proteins in WT356 and Non‐Tg animals was generally more diffuse, although more concentrated staining was evident in small puncta within MNs. The diffuse nature of the UBQLN2 and serpin staining patterns in control animals made it difficult to establish whether the proteins were truly colocalized with one another. Closer examination of magnified images of the MNs revealed clear evidence of UBQLN2 colocalization with several of the serpin proteins in round intracellular puncta in MN of the mice (Figure S5).
FIGURE 3.
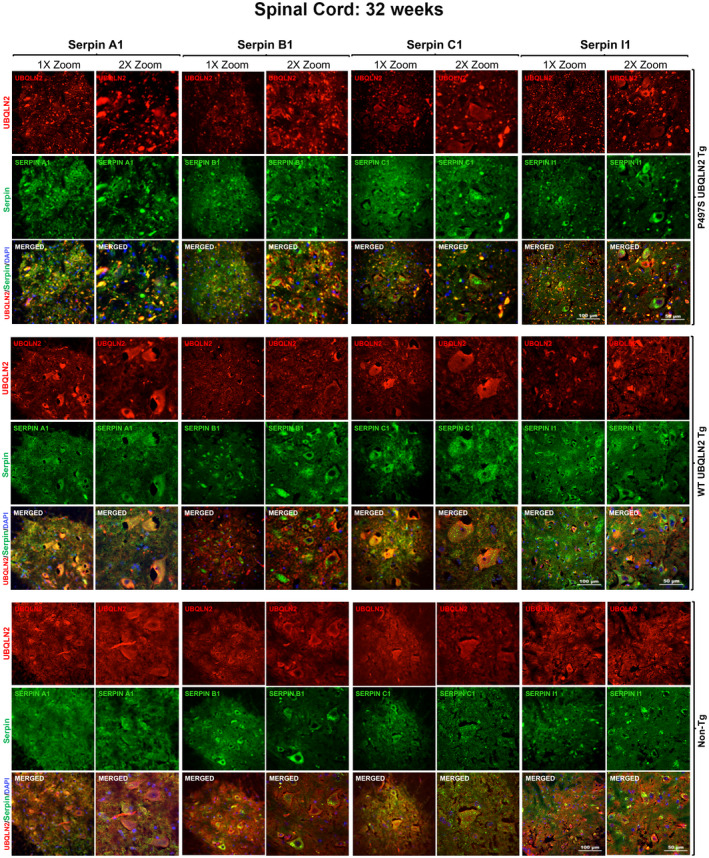
Colocalization of UBQLN2 and serpin proteins in puncta in the SC of P497S animals. Confocal microscopy images of the staining of UBQLN2 and Serpin A1, B1, C1, and I1 proteins and their corresponding merged image, including the DAPI panel, of the ventral horn region of the SC of 32‐week‐old P497S (top panel), WT356 (middle panel), and Non‐Tg (bottom panel) animals. Scale bars shown = 100 μm and 50 μm for 1× and 2× zoom images, respectively
3.4. UBQLN2 and serpins colocalize in autolysosomes in MN cells of the SC
Because UBQLN2 functions in autophagy, we postulated that the puncta double positive for UBQLN2 and serpins in MNs of Non‐Tg mice may correspond to autophagosomes or autolysosomes. To address this possibility, we co‐stained SC tissue from 32‐week‐old Non‐Tg, WT356, and P497S animals for each of the four serpin proteins, UBQLN2, and either LC3 or LAMP1, the latter two being autophagosome and lysosome markers, respectively (48) (Figures 4 and 5). The triple staining revealed excellent colocalization of both LAMP1 and LC3 markers with puncta double positive for UBQLN2 and serpins in MNs of the animals, suggesting the puncta are autolysosomes. Interestingly, line scans of these images showed a decrease in UBQLN2 colocalization with these puncta in P497S animals, particularly those positive for LAMP1, consistent with previous studies suggesting the P497S UBQLN2 mutation interferes with autophagy (Figures S6 and S7) (12). The larger, irregularly shaped puncta located outside of the MNs in P497S animals that stained double positive for UBQLN2 and the serpins were negative for either LC3 or LAMP1, suggesting they are unlikely to be autophagosomes. In order to further investigate the localization of the puncta outside of MNs, we conducted triple staining, for UBQLN2, serpins and either IBA1 or GFAP, markers of microglia and astrocytes, respectively (Figure 6 and Figures S8–S10). We predicted that these puncta were unlikely to be in microglia or astrocytes because expression of the P497S UBQLN2 transgene is only driven in neurons. Staining of 32‐week SC and 52‐week brain sections from P497S animals showed that the UBQLN2 and serpin double‐positive puncta were frequently juxtaposed to microglia decorated with IBA1 staining, while GFAP‐decorated astrocytes were rarely associated with the double‐positive puncta (Figure 6 and Figures S8–S10). These findings suggest the puncta may be either intracellular neuronal aggregates or extracellular remnants of them.
FIGURE 4.
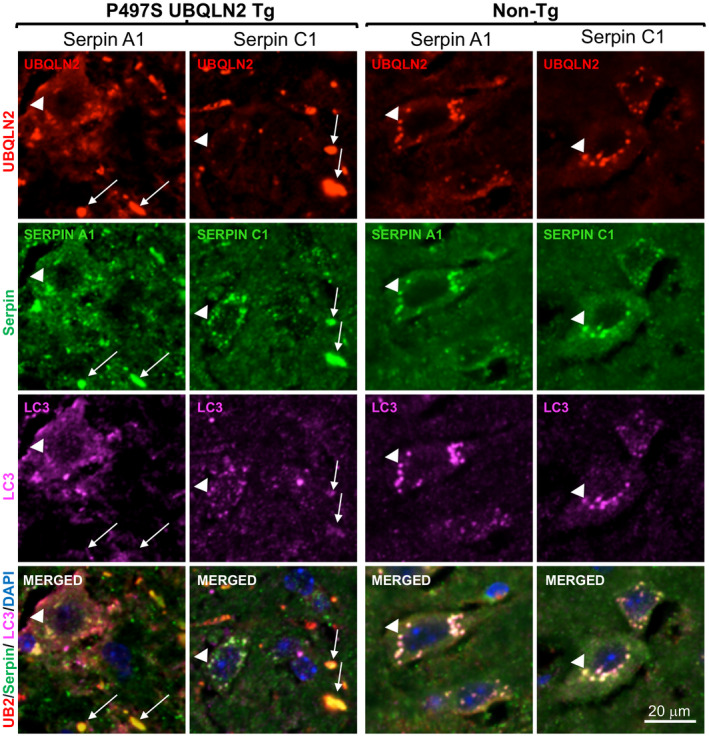
Colocalization of UBQLN2 and serpin proteins in autophagosomes. Confocal microscopy images of the staining of UBQLN2, Serpin A1 or C1 proteins, and LC3 in the SC of 32‐week‐old age‐matched P497S (left panel) and Non‐Tg (right panel) animals. UBQLN2 puncta outside MN in P497S animals are positive for serpins, but not for LC3 (arrows). UBQLN2 puncta within MN stain positive for all three proteins (arrow heads). Scale bars shown = 20 μm for all images
FIGURE 5.
Colocalization of UBQLN2 with serpin proteins in LAMP1‐positive puncta. Similar to Figure 4, but showing the staining of UBQLN2, Serpin A1 or C1 proteins, and LAMP1 in the SC of 32‐week‐old age‐matched P497S (top panel) and Non‐Tg (bottom panel) animals. Arrows indicate UBQLN2 puncta outside MN that stain positive for serpins, but negative for LAMP1. Puncta within MN stained positive for all three proteins (arrow heads), but UBQLN2 staining of the LAMP1 puncta was stronger in the Non‐Tg animals. Scale bars shown = 20 μm for all images
FIGURE 6.
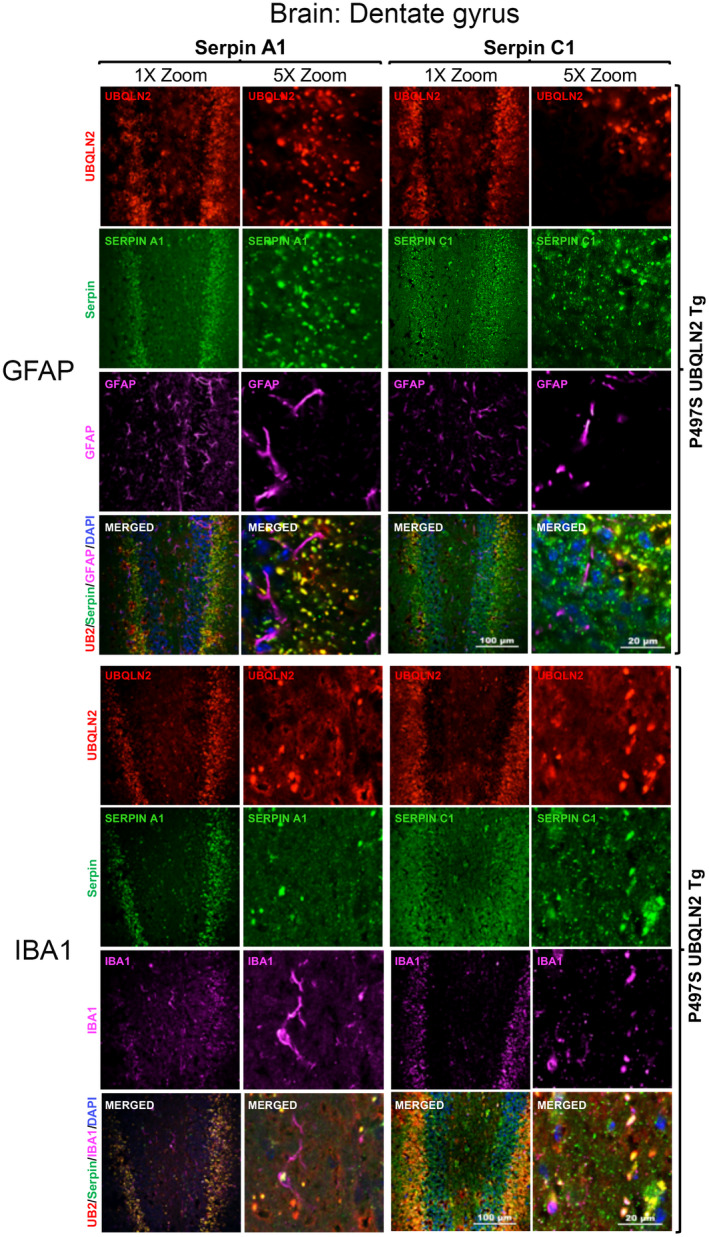
The UBQLN2 and serpin double‐positive puncta in the brain are more closely juxtaposed with microglia than with astrocytes. Confocal microscopy images of the staining of UBQLN2, Serpin A1 or C1 proteins with either GFAP (top panels) or IBA1 (bottom panels) in the dentate gyrus of 52‐week‐old P497S animals. Scale bars shown = 100 μm and 20 μm for 1× and 5× zoom images, respectively
3.5. Evidence of increased aggregation of serpin and UBQLN2 protein during aging in P497S UBQLN2 mutant mice
The mechanism by which serpins inactivate proteases is through a large change in their protein conformation, switching from a metastable native active state to a cleaved inactivated state (18, 31). However, this metastability makes the proteins highly prone to misfolding. Such misfolding chiefly occurs during biosynthesis, which by nature is error prone, and is further induced by mutations (18, 49, 50, 51, 52). We explored the possibility that the serpin puncta observed by immunofluorescent staining in the brain and SC of P497S animals are a result of aggregation of serpin proteins because of disturbances in proteostasis caused by the P497S UBQLN2 mutation. Accordingly, we conducted filter trap assays to determine whether the amount of UBQLN2 and serpin aggregates differed in the three mouse genotypes. In this assay, protein aggregates that are too large to pass through the pores of a 0.2 µm cellulose acetate membrane under vacuum filtration are retained and their amount can be quantified by antibody reactivity (35, 53). Filter trap assays were conducted with cortical and hippocampal lysates from three independent P497S, WT356, and Non‐Tg animals of 8 and 32 weeks of age. Immunoreactivity of the filters for UBQLN2 revealed little difference between the three genotypes in 8‐week‐old animals. However, the amount of UBQLN2 aggregates quantified by this assay was sixfold higher in 32‐week‐old P497S animals, compared to the age‐matched WT356 and Non‐Tg animals (Figure 7A,B). The increase in UBQLN2 aggregation in P497S animals is consistent with the age‐dependent increase in UBQLN2 puncta seen in this line (12, 14). Quantification of the four serpin proteins retained on the filters by this same assay revealed significant differences in Serpin A1 and Serpin I1 between the different genotypes, but not for Serpin B1 or C1 proteins, whose accumulation was more variable between samples of the same genotypes at both ages (Figure 7A,B). The high variability of Serpin B1 and C1 made quantification of their change unreliable. The amount of Serpin A1 and Serpin I1 retained on the filters was not significantly different for the three genotypes in 8‐week‐old animals (Figure 7A). However, higher amounts of both proteins were retained in samples of 32‐week‐old P497S animals compared to age‐matched WT356 and Non‐Tg animals (Figure 7A,B). This finding is also consistent with the age‐dependent increase observed for both serpins by immunofluorescence microscopy.
FIGURE 7.
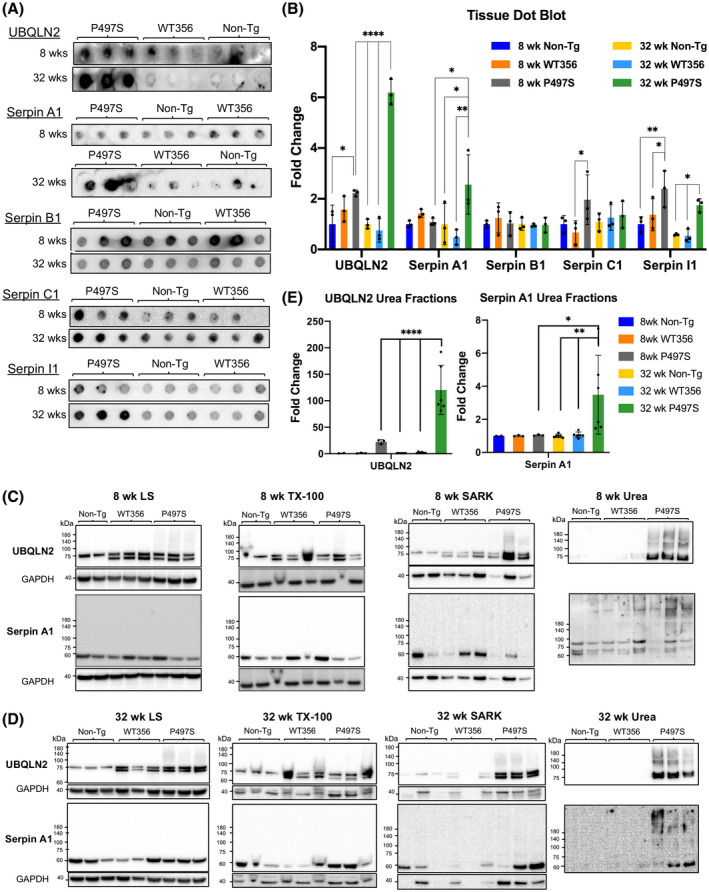
UBQLN2 and serpin proteins form aggregates in P497S animals in an age‐dependent manner. (A) Representative images of filter trap assays of cortical tissue from 8‐ to 32‐week‐old P497S, WT356, and Non‐Tg animals indicates a significant increase in aggregation of UBQLN2, Serpin A1, and Serpin I1 in 32‐week‐old, but not 8‐week‐old, animals. (B) Quantification of filter trap assays: *p < 0.05, **p < 0.01, ****p < 0.0001. n = 3‐6 animals/age and genotype. (C and D) Immunoblots of equal portion of fractions following sequential extraction of cortical tissues from 8‐ (C) to 32‐week‐old (D) Non‐Tg, WT356, and P497S mice with increasingly harsh solubilizing buffers (low salt buffer, Triton X‐100 (TX‐100), sarkosyl (SARK), and urea). The GAPDH signal was virtually undetectable in the urea samples, and therefore, is not shown. (E) Quantification of the UBQLN2 and Serpin A1 signals in the urea fractions for 8‐ and 32‐week‐old animals. *p < 0.05, **p < 0.01, ****p < 0.0001
To further verify that serpin proteins formed aggregates in the P497S mice, we compared the properties of serpins proteins in tissues of the different animals to biochemical extraction with increasingly harsh buffers, reasoning that the aggregated proteins should be more resilient to extraction with milder buffers. Equal amounts of cortical tissue from 32‐week‐old Non‐Tg, WT356, and P497S animals were homogenized beginning with a low salt buffer followed by Triton X‐100, sarkosyl, and urea buffers. The same procedure was successfully used to characterize TDP‐43 aggregates (38, 39). Equal volumes of the extracted material from each extraction were then immunoblotted for UBQLN2 and the four serpins. As expected, the amount of UBQLN2 present in the ultimate urea‐extracted fraction was higher in the P497S sample compared to the WT356 and Non‐Tg samples (Figure S11E). The P497S sample was also enriched for proteins that were strongly reactive for ubiquitin, a signature feature of ubiquitin‐tagged misfolded proteins (Figure S11D,E). Evaluation of Serpin A1, B1, C1, and I1 in the same fractions revealed the presence of multiple bands for several of the serpin proteins, consistent with aggregation and/or modification of the protein. Quantification of all bands for the three genotypes in the penultimate sarkosyl and ultimate urea extractions revealed a dramatic increase in the levels of Serpin A1 in P497S animals compared to the WT356 and Non‐Tg animals (Figure S11C). The reason why an increase in the other serpins was not detected in P497S animals is not clear, but we speculate it is most likely related to their resistance to solubility and/or detection by this methodology.
To confirm these findings, we repeated the analysis, comparing the extraction properties of Serpin A1 and UBQLN2 in cortical tissue from 8‐ to 32‐week‐old animals for different animals for the three genotypes. We only focused on these two proteins because material needed for the analysis was limited. The blots for these extractions revealed that, as expected, the major difference in UBQLN2 extraction was in P497S mutant animals, where the protein exhibited more resistance to extraction with the milder buffers, as evidenced by an increase in its amounts after extraction with the urea buffer, compared to the Non‐Tg and WT356 animals (Figure 7C,D). Moreover, the UBQLN2 protein in the urea‐extracted fractions ran as a smear in the polyacrylamide gels, a characteristic of misfolded ubiquitinated proteins. This pattern was evident in both 8‐ and 32‐week‐old animals, demonstrating the presence of the misfolded UBQLN2 protein at both ages. The blots for Serpin A1 in the same fractions, likewise, revealed increased presence of the protein, particularly of high molecular forms of it, in the urea fractions of the P497S animals compared to the Non‐Tg and WT 356 animals (Figure 7C,D). Again, increased amounts of the high molecular weight Serpin A1 species were observed in the urea‐extracted material of 8‐week‐old P497S animals, and more clearly apparent in P497S animals at 32 weeks of age. Quantification of the UBQLN2 and Serpin A1 signals in the urea fractions revealed P497S animals, compared to the WT356 and Non‐Tg animals, had significantly higher amounts of both proteins at the 32 week time point (Figure 7E).
Taken together with our immunological findings that the serpin proteins form puncta and have increased retention in filter trap assays, these biochemical fractionation findings provide strong support for the age‐dependent increase of serpin protein aggregation, particularly Serpin A1, in P497S animals.
3.6. Inconsistency in detection of serpin protein accumulation in P497S animals by immunoblotting
We also attempted to quantify changes of the four serpin proteins in brain and SC using SDS‐PAGE and immunoblotting. However, we were mindful of known problems in accurately quantifying changes in serpin proteins by this method based on prior studies that had found serpin proteins, once aggregated, are resistant to entry in SDS‐polyacrylamide gels (51, 54). Nevertheless, we conducted immunoblots probing equal amounts of protein lysate from both the hippocampus and lumbar SC tissue of three independent 32‐week‐old WT356, Non‐Tg, and P497S animals (n = 3/genotype) for all four serpin proteins. Interestingly, the quantifications revealed a significant decrease in Serpin A1 and I1 levels in the hippocampus of P497S animals as well as a significant decrease of Serpin C1 in the SC of P497S mice compared to the WT356 or Non‐Tg animals (Figure S12A,B). Other serpin proteins in these tissues followed this trend but did not decrease significantly compared to protein levels in control animals. Based on the increased aggregation of several of the serpin proteins that we found in P497S animals, we postulate that the decreases noted for some of the serpins in P497S mutant animals by immunoblotting is probably an artifact arising from the known insolubility and/or resistance of serpin aggregates to separation by SDS‐PAGE.
3.7. Inactivation of UBQLN2 expression in cell lines induces serpin aggregation
Our studies of the P497S mouse model of ALS/FTD suggests the P497S UBQLN2 mutation induces disease through both loss‐ and gain‐of‐function mechanisms (12, 14). To determine if loss of UBQLN2 affects serpin expression and/or aggregation, we used UBQLN2 knockout HeLa human epithelial and NSC34 mouse motor neuron cells. The properties of these knockout lines were described previously (12). Two independent clones from each cell line (HeLa KO8 and KO12 and NSC34 KO20 and KO69) were tested for Serpin A1, Serpin B1, Serpin C1, and Serpin I1 expression by immunoblots and compared to serpin expression in the parental HeLa and NSC34 cell lines (Figure 8A). Few changes in the protein bands were visible in the resolving part of the gels. However, major changes were seen in the upper portion of the gels. For example, the level of immunoreactivity of Serpin A1 and Serpin B1 in the high molecular weight region of the gels containing HeLa UBQLN2 KO8 and KO12 lysates was notably higher than in the wild‐type HeLa cells (Figure 8A,B). Similar findings were noted for Serpin B1 in NSC34 UBQLN2 KO20 cells, compared to the parental WT UBQLN2‐expressing line (Figure 8A,B). We speculated that the increased accumulation of serpins proteins in the stacking gel in the UBQLN2 KO cells was because of increased aggregation of the proteins.
FIGURE 8.
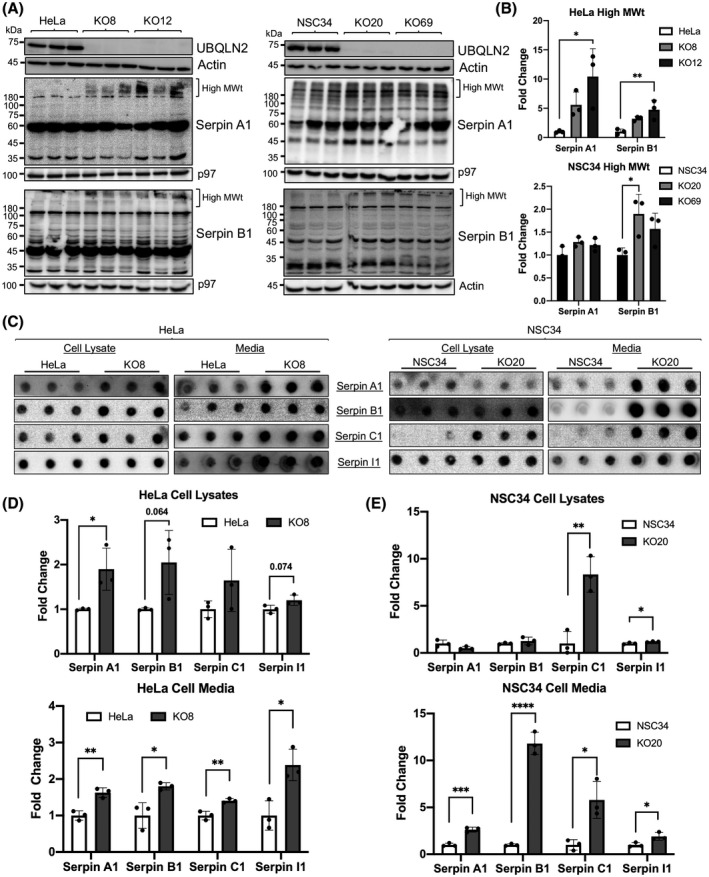
Inactivation of UBQLN2 in cells induces serpin aggregation. (A) Immunoblots comparing Serpin A1 and B1 levels in parental HeLa and NSC34 lines to two independent UBQLN2 knockout lines (KO8/KO12 and KO20/KO69, respectively) for each line. (B) Both HeLa and NSC34 UBQLN2 knockout cell lines show increased accumulation of high molecular weight (High MWt) serpin bands, suggesting protein aggregation. *p < 0.05, **p < 0.01. (C) Filter trap assays using cell lysate and media aliquots from the parental HeLa and UBQLN2 KO8 cells as well as the parental NSC34 and UBQLN2 KO20 cells. UBQLN2 knockout in either cell type results in increased serpin aggregation in cell lysates and the media. (D and E) Quantification of the signals for the filter trap assays shown in B. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. n = 3–6 replicates/cell line and results were reproduced in at least two independent experiments
To determine whether UBQLN2 inactivation alters serpin aggregation, we conducted filter trap assays comparing retention of the four serpin proteins between the HeLa KO8 and NSC34 KO20 lines and their parental lines. As the majority of serpins are secreted proteins, we also performed filter trap assays of the media in equal portions collected after a 48 h culturing of the lines in equal cell densities to determine if the proteins accumulate extracellularly. Quantification of the serpin proteins retained after filtration of equal amounts of the HeLa cell lysates (of three independent cultures) indicated a significant increase in the amount of Serpin A1 that was trapped on the filters for the KO8 line compared to WT HeLa control line (Figure 8C,D). Increases were also detected for Serpin B1, Serpin C1, and Serpin I1, although the changes were just outside significance. Quantification of the serpins retained after filtration of the media from these cultures revealed a significant increase for all four serpin proteins in the HeLa KO8 compared to the WT control line. Similar comparison for the NSC34 cell lines revealed significant increases in the amount of Serpin C1, and Serpin I1 in the NSC34 KO20 cell lysates compared to the parental WT line (Figure 8C,E). Paradoxically, we did not detect increases in Serpin A1 and a small but nonsignificant increase in Serpin B1 levels in the KO20 lysates. By contrast, the amount of serpin proteins trapped after filtration of the media was significantly higher for all of the serpins in the KO20 compared to the WT NSC34 line (Figure 8C,E). Taken together, these results suggest that the aggregation of serpin proteins observed in the brain and SC of P497S animals may arise from loss of UBQLN2 function.
4. DISCUSSION
Missense mutations in UBQLN2 cause X‐linked dominant ALS/FTD (7). The mutations are thought to disrupt proteostasis causing disease through both loss‐ and gain‐of‐function mechanisms (12). However, the pathophysiology of how the disturbances cause disease is still largely unknown. Here, we provide evidence that serpin proteins form insoluble protein aggregates within the brain and SC in the P497S Tg mouse model of ALS/FTD. We further show that knockout of UBQLN2 expression in cell lines leads to similar aggregation of serpin proteins, suggesting that the aberrant accumulation and aggregation of serpin proteins in this ALS/FTD disease model may stem from loss of UBQLN2 function. We discuss the potential implications of this abnormal accumulation of serpin proteins, suggesting it may contribute to disease pathogenesis through both loss‐ and gain‐of‐function mechanisms.
We obtained clear evidence that serpin proteins, in particular Serpin A1, become mislocalized and aggregate during disease progression in the P497S model of ALS/FTD. The mislocalization was evident by a dramatic alteration in the immunofluorescent staining patterns of several serpins, including Serpin A1, C1, and I1 in the brain and SC tissues of P497S Tg animals. Serpin proteins accumulated in an age‐dependent manner in puncta very similar to the UBQLN2 inclusions that develop in the P497S animals, differing from the more diffuse localization in age‐matched WT356 and Non‐Tg animals. Subtle differences were found in the colocalization of the three serpin proteins with UBQLN2 puncta in both the brain and SC suggesting region‐ or cell‐specific differences in serpin or UBQLN2 expression may influence their colocalization.
Further insight into the identity of the serpin puncta was obtained by the results of the filter trap and biochemical fractionation assays, both of which strongly suggest they are most likely to be inclusions composed of aggregated misfolded serpin proteins. The strongest evidence was seen for Serpin A1, both by the significant increase in the amounts of the protein retained after filtration of P497S brain lysates through 0.2 µm cellulose acetate filters and by the high molecular weight smear of the protein in gels in the urea‐containing fraction of mouse tissue extracted with increasingly harsh buffers. The same urea fractions from aged P497S animals were also highly enriched for UBQLN2 and ubiquitin reactivity, consistent with the expected properties of misfolded and aggregated proteins. Similar evidence for aggregation of Serpin C1 and I1 in P497S animals by the same criteria was more variable, with stronger evidence seen by the dot‐blot analysis than by the tissue extractions. It is not clear why an increase in the latter two proteins was not as well detected by these methods considering their strong colocalization with UBQLN2 in puncta in P497S animals. It is possible that epitope masking or the insoluble nature of the proteins has precluded their detection in the assays.
A key question arising from these observations is how expression of the P497S mutant UBQLN2 protein causes serpin proteins to aggregate in tissues of the P497S animals. We considered two chief, but non‐mutually exclusive possibilities by which this could occur. One possibility is that the P497S mutation inactivates UBQLN2 function in ERAD, which is required for clearing serpin proteins that misfold during synthesis in the ER (24, 55), leading to accumulation and aggregation of the serpin proteins. This could occur either by dominant‐negative loss of UBQLN2 function or by the mutation crippling the UBQLN2 protein such that it can no longer function in ERAD. Another possibility is that the P497S mutation causes UBQLN2 proteins to misfold, which in turn induces aggregation or coaggregation with the misfolded serpin proteins.
Our findings that serpin proteins aggregate following knockout of UBQLN2 in cells is consistent with the loss‐of‐function possibility. This loss of function could be particularly problematic for serpins because of their high propensity to misfold. Serpins are typically synthesized in high amounts in the ER (56), and, because protein folding is inherently error prone, the high rate of synthesis generates a corresponding high amount of terminally misfolded serpin proteins that must be efficiently removed from cells that otherwise can wreak havoc (52, 57, 58). Further, serpin polypeptides need to be folded into a metastable conformation for their activity. Deviation in their correct folding can cause collapse of the metastable structure into an inactive misfolded state, placing high demand on an efficient ERAD system for their removal (18, 59, 60, 61, 62, 63). UBQLN proteins are known to function in ERAD, including in removal of serpin proteins (64). Additionally, ALS/FTD mutations in UBQLN2 stall clearance of serpin proteins (13, 65). Accordingly, we propose that loss of UBQLN2 function in ERAD, caused by mutation or knockout of UBQLN2, could lead to serpin aggregation. Consistent with this idea, ER stress, which is induced by a buildup of misfolded proteins in the ER, is increased in P497S animals (35). Thus, the colocalization of UBQLN2 and serpins could represent the proteins that are entrapped because of failure to be cleared by ERAD. The absence of any noticeable colocalization of Serpin B1 with UBQLN2 inclusions in the P497S animals supports our idea that defects in ERAD could underlie the accumulation of serpins seen in our study. Serpin B1 differs from the secreted Serpin A1, C1, and I1 proteins by being synthesized in the cytoplasm (19). Thus, deficiencies in ERAD are not expected to affect its degradation. Unexpectedly, we found Serpin B1 expression was increased in the cell bodies of a number of neuronal cells in the brain and SC. Although the reason for its upregulation is not known, we speculate that dysregulation of proteostasis caused by the UBQLN2 mutation may underlie its upregulation (66).
Another mechanism by which serpin proteins could aggregate is through loss of UBQLN2 function in autophagy. We cannot discount this possibility because serpins, including the extracellularly secreted proteins and their inhibitory complexes can be reinternalized and cleared by autophagy (61, 67, 68, 69). Furthermore, ALS/FTD mutations in UBQLN2 have been found to impede autophagy because of a failure in autophagosome acidification from loss of UBQLN2 function (12). The importance of this clearance route for serpins is further highlighted by our findings that serpin proteins colocalize with UBQLN2, LAMP1, and LC3 proteins in puncta of spinal MNs in the Non‐Tg and WT356 animals, consistent with normal disposal of the proteins via autophagosomes/autolysosomes.
Further studies are needed to identify the mechanism underlying the increase of serpin aggregates in the culture media of UBQLN2 knockout cells. However, it is similar to the phenomenon seen for misfolded mutant Serpin A1 and neuroserpin proteins, where aggregated forms of both mutant serpin proteins were found in the culture media expressing cells despite evidence of increased retention in the ER (24, 70).
The pathologic consequence of the accumulation of serpin aggregates in the P497S animals may contribute to disease pathogenicity from gain‐of‐function toxicity based on other serpinopathies (22, 31, 50). For example, mutations in Serpin A1 and I1 are both thought to drive disease through aggregation (22, 31, 50, 71). Specifically, cognitive deficits and dementia symptoms seen in FENIB patients correlate with the rate of Serpin I1 polymerization and inclusion formation, suggesting formation of Serpin I1 inclusions drive neurodegeneration (27). Additionally, abnormal aggregation of Serpin A1, Serpin A3, and Serpin I1, have been identified in ALS/FTD and other neurodegenerative diseases (41, 42, 44, 45, 47, 72). Thus, it is conceivable that the serpin inclusions seen in the P497S mice may play a direct or indirect role in neurodegeneration in the P497S mouse model.
In addition to the potential gain of toxic function because of serpin aggregation, a loss of normal serpin function may also have important consequences in ALS/FTD pathology. Based on current evidence that serpins play critical roles in the regulation of proteolytic cascades and inflammatory pathways, we suggest that loss of normal serpin function may contribute to the uncontrolled inflammation and increased neuronal death which occur in ALS/FTD (73, 74). Several of the proteases that serpins regulate can induce an inflammatory response. Serpin A1 and B1 are both inhibitors of neutrophil elastase, which is released by neutrophils and responds to acute inflammation. Both ALS patient tissue and rodent models reveal an upregulation of neutrophils along peripheral motor pathways and dystrophic muscle, correlated with progressive degeneration of MN axons and muscular dystrophy (75, 76). Other studies of Serpin B1 provide evidence that the protein inhibits pro‐inflammatory cytokine production and is upregulated when in complex with neutrophil elastase in diseased lungs (77, 78). Loss of Serpin C1 function also affects the body's ability to properly mediate inflammation as inhibition of thrombin by Serpin C1 is imperative in the protection of motor axons (79). Moreover, thrombin has been shown to activate the complement cascade, which promotes inflammatory responses and is activated in the skeletal muscles of the hSOD1G93A ALS mouse model (80, 81). Taken together, these studies suggest that a loss of serpin inhibitory function and unregulated levels of proteases are linked to neuroinflammation and motor deficits, common features of ALS/FTD.
Our observation that UBQLN2‐serpin puncta in the brain and SC of P497S mice partially colocalize with the microglia marker IBA1 suggest that an immune response is related to the aberrant accumulation of serpin proteins in our model. It is important to note that the mouse models used in this study do not express the UBQLN2 transgene in glial cells. Thus, we propose two possibilities for these observations. First, as serpin colocalization with UBQLN2 puncta occurs in an age‐dependent manner, it is possible that serpin proteins are being released by microglia in response to the formation of UBQLN2 aggregates. In contrast, formation of serpin‐UBQLN2 inclusions could occur upstream of microglial association. In this case, colocalization with IBA1 could indicate that microglia are phagocytosing these inclusions in an attempt to clear the aggregates, as occurs in FTD, multiple sclerosis, Alzheimer's, and Parkinson's disease (82, 83, 84, 85).
In summary, our study has identified a novel pathological feature of serpin misaggregation in the P497S mouse model of ALS/FTD. Further insight into the mechanism of serpin aggregation and its relationship to neuroinflammation and toxicity is needed to fully understand the consequences that they could have in driving pathogenesis in ALS/FTD. Understanding the mechanistic connection between UBQLN2 and serpin proteins may provide important information concerning therapeutic targets for the treatment and prevention of ALS/FTD.
CONFLICT OF INTEREST
The authors declare no conflict of interests.
AUTHOR CONTRIBUTIONS
NRH, JEG, and MJM conceived and designed the experiments. NRH, JEG, JJW, and MJM performed the experiments. EM and GG provided reagents and advice. All authors read, revised, and approved the manuscript.
ETHICS AND INTEGRITY
All authors read and approved the manuscript. The work and figures were done with the highest ethical standards.
Supporting information
Fig S1‐S12
FIGURE S1 Immunoblots of HeLa cell lysates transfected with Myc‐tagged Serpin A1, B1, C1 and I1 expression cDNAs to validate the specificity of serpin antibodies used in this study. All antibodies detect only their cognate recombinant protein and the correct size endogenous protein
FIGURE S2 Age‐dependent alteration of UBQLN2 and serpin staining in P497S mutant mice. (A and B) Immunofluorescence staining of UBQLN2 and Serpin A1, B1, C1, or I1 in the hippocampal region of sagittal brain sections from 8‐week‐old (A) and 52‐weekold (B) P497S animals. Scale bars shown = 100 μm for all images
FIGURE S3 Colocalization of serpin and UBQLN2 proteins in different brain regions of P497S animals. Confocal microscopy images of 52‐week‐old P497S animals similar to Fig 2, but showing the UBQLN2 and Serpin A1, C1, and I1 protein staining in the CA1 (top panel), CA3 (middle panel), and cortex (bottom panel) of the brain. Scale bars shown = 20 μm for all images
FIGURE S4 Line scan analysis showing colocalization of serpins with UBQLN2 staining in the dentate gyrus and spinal cord sections of P497S animals. Line scans were performed through representative regions of the magnified images shown in Figs 2 and 3. Plots depict pixel intensity of the individual UBQLN2 (red) and serpin (green) image channels for the lines analyzed in the images shown to the right of each plot. Lines are 2 drawn through the same region of interest in each image to determine the extent of overlap between the UBQLN2 and serpin staining
FIGURE S5 Colocalization of UBQLN2 and serpins proteins in puncta within spinal MN. Confocal microscopy images of 32‐week‐old P497S animals similar to Fig 3, but showing higher magnification images of MN, identified by ChAT‐positive staining, in the SC of age‐matched P497S (top panel), WT356 (middle panel), and Non‐Tg (bottom panel) animals. Scale bars shown = 20 μm for all images
FIGURE S6 Line scans showing the degree of overlap between UBQLN2, serpin, and LC3 staining in P497S and Non‐Tg SC sections shown in Fig 4. Plots depict pixel intensity for UBQLN2 (red), serpin (green), and the autophagosome marker LC3 (magenta) for the lines shown in images below each plot. Lines are drawn through the same region of interest containing representative puncta for either the larger, extracellular puncta in P497S animals (left column) or the smaller puncta within motor neurons of P497S and Non‐Tg animals (middle and right columns, respectively)
FIGURE S7 Line scans showing the degree of overlap between UBQLN2, serpins and LAMP1 in P497S and Non‐Tg spinal cord sections shown in Fig 5. Similar to Fig S6, but measuring colocalization between UBQLN2 (red), serpins (green), the LAMP1 lysosomal marker (magenta)
FIGURE S8 The UBQLN2 and serpin double‐positive puncta in the SC are more closely juxtaposed with microglia than with astrocytes. Confocal microscopy images of the staining of UBQLN2, Serpin A1 or C1 proteins with either GFAP (top panels) or IBA1 (bottom panels) in the SC of 32‐week‐old P497S animals. Scale bars shown = 100 μm and 20 μm for 1× and 5× zoom images, respectively
FIGURE S9 Comparison of the UBQLN2, Serpin A1 and C1 staining patterns in the dentate gyrus of P497S, WT 356 and Non‐Tg animals. Similar to Fig 6, but showing staining for all three genotypes, as indicated. The P497S staining panels are the same as Fig 6. Scale bars shown = 100 μm and 20 μm for 1× and 5× zoom images, respectively
FIGURE S10 Comparison of the UBQLN2, Serpin A1 and C1 staining patterns in the SC of P497S, WT 356 and Non‐Tg animals. Similar to Fig S8, but showing staining in the SC for all three genotypes, as indicated. The P497S staining panels are the same as Fig 6. Scale bars shown = 100 μm and 20 μm for 1× and 5× zoom images, respectively. Scale bars shown = 100 μm and 20 μm for 1× and 5× zoom images, respectively
FIGURE S11 Decreased solubility of UBQLN2 and Serpin A1 in P497S mutant animals. (A) Immunoblots of equal portions of low salt (LS), Triton‐X100 (TX‐100) sarkosyl (SARK) and urea fractions following sequential biochemical extraction of equal weight of cortical brain tissue from 32‐week‐old Non‐Tg, WT356, and P497S mice. The fractions analyzed were obtained by progressive extraction of the tissues with low salt, Triton X4 100, sarkosyl, and urea buffers. The Triton X‐100 fraction was not included because of aberrant migration of proteins extracted with the buffer. (B) Quantification of penultimate SARK fractions for UBQLN2 and serpin proteins. (C) Quantification of ultimate urea fractions for each serpin protein. (D) Ubiquitin (UBQTN) immunoblots of urea fractions from 8‐ and 32‐week‐old animals. (E) Quantification of 32 week urea fractions for UBQLN2 and UBQTN
FIGURE S12 Quantification of serpin protein levels in mouse tissues by SDS‐PAGE analysis. (A) Immunoblots of hippocampus (left) and lumbar SC tissue (right) of 3 WT356, Non‐Tg, and P497S animals per genotype analyzed after separation by SDSPAGE. (B) Quantification of immunoblots in A shows a significant decrease in Serpin I1 in the hippocampus and a significant decrease in Serpin C1 in the spinal cord of 32‐ week‐old P497S animals. Other serpins trended toward a decrease in P497S animals though not to a level of significance. As expected, levels of the UBQLN2 transgene in both the hippocampus and spinal cord were significantly increased in P497S animals
ACKNOWLEDGMENTS
We thank Trong Phung for helpful comments on the manuscript.
Nicole R. Higgins and Jessie E. Greenslade contributed equally to this work.
Funding information
The work was funded by NIH grants R01NS098243 and R01NS100008 and in part by The Robert Packard Center for ALS research at Johns Hopkins to MJM
DATA AVAILABILITY STATEMENT
All of the data in contained in either the body of the manuscript or included in the Supplemental material provided. Any additional data used in the study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Brown RH, Al‐Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377:162–72. [DOI] [PubMed] [Google Scholar]
- 2.van Es MA, Hardiman O, Chio A, Al‐Chalabi A, Pasterkamp RJ, Veldink JH, et al. Amyotrophic lateral sclerosis. Lancet. 2017;390:2084–98. [DOI] [PubMed] [Google Scholar]
- 3.Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet. 2015;386:1672–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mejzini R, Flynn LL, Pitout IL, Fletcher S, Wilton SD, Akkari PA. ALS genetics, mechanisms, and therapeutics: where are we now? Front Neurosci. 2019;13:1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nguyen HP, Van Broeckhoven C, van der Zee J. ALS genes in the genomic era and their implications for FTD. Trends Genet. 2018;34:404–23. [DOI] [PubMed] [Google Scholar]
- 6.Taylor JP, Brown RH Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, et al. Mutations in UBQLN2 cause dominant X‐linked juvenile and adult‐onset ALS and ALS/dementia. Nature. 2011;477:211–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Higgins N, Lin B, Monteiro MJ. Lou Gehrig's disease (ALS): UBQLN2 mutations strike out of phase. Structure. 2019;27:879–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Renaud L, Picher‐Martel V, Codron P, Julien JP. Key role of UBQLN2 in pathogenesis of amyotrophic lateral sclerosis and frontotemporal dementia. Acta Neuropathol Commun. 2019;7:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng T, Yang Y, Castaneda CA. Structure, dynamics and functions of UBQLNs: at the crossroads of protein quality control machinery. Biochem J. 2020;477:3471–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang L, Monteiro MJ. Defective proteasome delivery of polyubiquitinated proteins by Ubiquilin‐2 proteins containing ALS mutations. PLoS One. 2015;10:e0130162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu JJ, Cai A, Greenslade JE, Higgins NR, Fan C, Le NTT, et al. ALS/FTD mutations in UBQLN2 impede autophagy by reducing autophagosome acidification through loss of function. Proc Natl Acad Sci U S A. 2020;117:15230–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xia Y, Yan LH, Huang B, Liu M, Liu X, Huang C. Pathogenic mutation of UBQLN2 impairs its interaction with UBXD8 and disrupts endoplasmic reticulum‐associated protein degradation. J Neurochem. 2014;129:99–106. [DOI] [PubMed] [Google Scholar]
- 14.Le NT, Chang L, Kovlyagina I, Georgiou P, Safren N, Braunstein KE, et al. Motor neuron disease, TDP‐43 pathology, and memory deficits in mice expressing ALS‐FTD‐linked UBQLN2 mutations. Proc Natl Acad Sci U S A. 2016;113:E7580–E7589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heit C, Jackson BC, McAndrews M, Wright MW, Thompson DC, Silverman GA, et al. Update of the human and mouse SERPIN gene superfamily. Hum Genomics. 2013;7:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huntington JA. Serpin structure, function and dysfunction. J Thromb Haemost. 2011;9(Suppl 1):26–34. [DOI] [PubMed] [Google Scholar]
- 17.Gettins PG. Serpin structure, mechanism, and function. Chem Rev. 2002;102:4751–804. [DOI] [PubMed] [Google Scholar]
- 18.Gettins PG, Olson ST. Inhibitory serpins. New insights into their folding, polymerization, regulation and clearance. Biochem J. 2016;473:2273–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Law RH, Zhang Q, McGowan S, Buckle AM, Silverman GA, Wong W, et al. An overview of the serpin superfamily. Genome Biol. 2006;7:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wright HT, Scarsdale JN. Structural basis for serpin inhibitor activity. Proteins. 1995;22:210–25. [DOI] [PubMed] [Google Scholar]
- 21.Davies MJ, Lomas DA. The molecular aetiology of the serpinopathies. Int J Biochem Cell Biol. 2008;40(6–7):1273–86. [DOI] [PubMed] [Google Scholar]
- 22.Lomas DA, Carrell RW. Serpinopathies and the conformational dementias. Nat Rev Genet. 2002;3:759–68. [DOI] [PubMed] [Google Scholar]
- 23.Le A, Ferrell GA, Dishon DS, Le QQ, Sifers RN. Soluble aggregates of the human PiZ alpha 1‐antitrypsin variant are degraded within the endoplasmic reticulum by a mechanism sensitive to inhibitors of protein synthesis. J Biol Chem. 1992;267:1072–80. [PubMed] [Google Scholar]
- 24.Miranda E, Romisch K, Lomas DA. Mutants of neuroserpin that cause dementia accumulate as polymers within the endoplasmic reticulum. J Biol Chem. 2004;279:28283–91. [DOI] [PubMed] [Google Scholar]
- 25.Ying Z, Wang H, Fan H, Wang G. The endoplasmic reticulum (ER)‐associated degradation system regulates aggregation and degradation of mutant neuroserpin. J Biol Chem. 2011;286:20835–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brantly M, Nukiwa T, Crystal RG. Molecular basis of alpha‐1‐antitrypsin deficiency. Am J Med. 1988;84(6A):13–31. [DOI] [PubMed] [Google Scholar]
- 27.Davis RL, Shrimpton AE, Holohan PD, Bradshaw C, Feiglin D, Collins GH, et al. Familial dementia caused by polymerization of mutant neuroserpin. Nature. 1999;401:376–9. [DOI] [PubMed] [Google Scholar]
- 28.Costa‐Mattioli M, Walter P. The integrated stress response: From mechanism to disease. Science. 2020;368(6489):eaat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sano R. Reed JC (2013) ER stress‐induced cell death mechanisms. Biochim Biophys Acta. 1833;12:3460–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belorgey D, Hagglof P, Karlsson‐Li S, Lomas DA. Protein misfolding and the serpinopathies. Prion. 2007;1:15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carrell RW, Jeppsson JO, Laurell CB, Brennan SO, Owen MC, Vaughan L, et al. Structure and variation of human alpha 1‐antitrypsin. Nature. 1982;298:329–34. [DOI] [PubMed] [Google Scholar]
- 33.Huntington JA. Shape‐shifting serpins–advantages of a mobile mechanism. Trends Biochem Sci. 2006;31:427–35. [DOI] [PubMed] [Google Scholar]
- 34.Kaiserman D, Whisstock JC, Bird PI. Mechanisms of serpin dysfunction in disease. Expert Rev Mol Med. 2006;8:1–19. [DOI] [PubMed] [Google Scholar]
- 35.Wang S, Tatman M, Monteiro MJ. Overexpression of UBQLN1 reduces neuropathology in the P497S UBQLN2 mouse model of ALS/FTD. Acta Neuropathol Commun. 2020;8:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Monteiro MJ, Mical TI. Resolution of kinase activities during the HeLa cell cycle: identification of kinases with cyclic activities. Exp Cell Res. 1996;223:443–51. [DOI] [PubMed] [Google Scholar]
- 37.Xiao J, Monteiro MJ. Identification and characterization of a novel (115 kDa) neurofilament‐associated kinase. J Neurosci. 1994;14(3 Pt 2):1820–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3. [DOI] [PubMed] [Google Scholar]
- 39.Sampathu DM, Neumann M, Kwong LK, Chou TT, Micsenyi M, Truax A, et al. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin‐positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol. 2006;169:1343–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whiteley AM, Prado MA, de Poot SAH, Paulo JA, Ashton M, Dominguez S, et al. Global proteomics of Ubqln2‐based murine models of ALS. J Biol Chem. 2020. 10.1074/jbc.RA120.015960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abraham CR, Selkoe DJ, Potter H. Immunochemical identification of the serine protease inhibitor alpha 1‐antichymotrypsin in the brain amyloid deposits of Alzheimer's disease. Cell. 1988;52:487–501. [DOI] [PubMed] [Google Scholar]
- 42.Abu‐Rumeileh S, Halbgebauer S, Steinacker P, Anderl‐Straub S, Polischi B, Ludolph AC, et al. CSF SerpinA1 in Creutzfeldt‐Jakob disease and frontotemporal lobar degeneration. Ann Clin Transl Neurol. 2020;7:191–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ebbert MTW, Ross CA, Pregent LJ, Lank RJ, Zhang C, Katzman RB, et al. Conserved DNA methylation combined with differential frontal cortex and cerebellar expression distinguishes C9orf72‐associated and sporadic ALS, and implicates SERPINA1 in disease. Acta Neuropathol. 2017;134:715–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nielsen HM, Minthon L, Londos E, Blennow K, Miranda E, Perez J, et al. Plasma and CSF serpins in Alzheimer disease and dementia with Lewy bodies. Neurology. 2007;69:1569–79. [DOI] [PubMed] [Google Scholar]
- 45.von Neuhoff N, Oumeraci T, Wolf T, Kollewe K, Bewerunge P, Neumann B, et al. Monitoring CSF proteome alterations in amyotrophic lateral sclerosis: obstacles and perspectives in translating a novel marker panel to the clinic. PLoS One. 2012;7:e44401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wormser U, Mandrioli J, Vinceti M, Fini N, Sintov A, Brodsky B, et al. Reduced levels of alpha‐1‐antitrypsin in cerebrospinal fluid of amyotrophic lateral sclerosis patients: a novel approach for a potential treatment. J Neuroinflammation. 2016;13:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, et al. Human and mouse single‐nucleus transcriptomics reveal TREM2‐dependent and TREM2‐independent cellular responses in Alzheimer's disease. Nat Med. 2020;26:131–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoshii SR, Mizushima N. Monitoring and measuring autophagy. Int J Mol Sci. 2017;18:e1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–9. [DOI] [PubMed] [Google Scholar]
- 50.Gooptu B, Lomas DA. Conformational pathology of the serpins: themes, variations, and therapeutic strategies. Annu Rev Biochem. 2009;78:147–76. [DOI] [PubMed] [Google Scholar]
- 51.Lomas DA, Elliott PR, Sidhar SK, Foreman RC, Finch JT, Cox DW, et al. alpha 1‐Antitrypsin Mmalton (Phe52‐deleted) forms loop‐sheet polymers in vivo. Evidence for the C sheet mechanism of polymerization. J Biol Chem. 1995;270:16864–70. [DOI] [PubMed] [Google Scholar]
- 52.Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–4. [DOI] [PubMed] [Google Scholar]
- 53.Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, et al. Huntingtin‐encoded polyglutamine expansions form amyloid‐like protein aggregates in vitro and in vivo. Cell. 1997;90:549–58. [DOI] [PubMed] [Google Scholar]
- 54.Yu MH, Lee KN, Kim J. The Z type variation of human alpha 1‐antitrypsin causes a protein folding defect. Nat Struct Biol. 1995;2:363–7. [DOI] [PubMed] [Google Scholar]
- 55.Qu D, Teckman JH, Omura S, Perlmutter DH. Degradation of a mutant secretory protein, alpha1‐antitrypsin Z, in the endoplasmic reticulum requires proteasome activity. J Biol Chem. 1996;271:22791–5. [DOI] [PubMed] [Google Scholar]
- 56.Badola S, Spurling H, Robison K, Fedyk ER, Silverman GA, Strayle J, et al. Correlation of serpin‐protease expression by comparative analysis of real‐time PCR profiling data. Genomics. 2006;88:173–84. [DOI] [PubMed] [Google Scholar]
- 57.Sun Z, Brodsky JL. Protein quality control in the secretory pathway. J Cell Biol. 2019;218:3171–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016;529:326–35. [DOI] [PubMed] [Google Scholar]
- 59.Chandrasekhar K, Ke H, Wang N, Goodwin T, Gierasch LM, Gershenson A, et al. Cellular folding pathway of a metastable serpin. Proc Natl Acad Sci U S A. 2016;113:6484–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Davies MJ, Miranda E, Roussel BD, Kaufman RJ, Marciniak SJ, Lomas DA. Neuroserpin polymers activate NF‐kappaB by a calcium signaling pathway that is independent of the unfolded protein response. J Biol Chem. 2009;284:18202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kroeger H, Miranda E, MacLeod I, Perez J, Crowther DC, Marciniak SJ, et al. Endoplasmic reticulum‐associated degradation (ERAD) and autophagy cooperate to degrade polymerogenic mutant serpins. J Biol Chem. 2009;284:22793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lomas DA, Hurst JR, Gooptu B. Update on alpha‐1 antitrypsin deficiency: New therapies. J Hepatol. 2016;65:413–24. [DOI] [PubMed] [Google Scholar]
- 63.Schipanski A, Oberhauser F, Neumann M, Lange S, Szalay B, Krasemann S, et al. Lectin OS‐9 delivers mutant neuroserpin to endoplasmic reticulum associated degradation in familial encephalopathy with neuroserpin inclusion bodies. Neurobiol Aging. 2014;35:2394–403. [DOI] [PubMed] [Google Scholar]
- 64.Lim PJ, Danner R, Liang J, Doong H, Harman C, Srinivasan D, et al. Ubiquilin and p97/VCP bind erasin, forming a complex involved in ERAD. J Cell Biol. 2009;187:201–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Halloran M, Ragagnin AMG, Vidal M, Parakh S, Yang S, Heng B, et al. Amyotrophic lateral sclerosis‐linked UBQLN2 mutants inhibit endoplasmic reticulum to Golgi transport, leading to Golgi fragmentation and ER stress. Cell Mol Life Sci. 2020;77(19):3859–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee JA, Yerbury JJ, Farrawell N, Shearer RF, Constantinescu P, Hatters DM, et al. SerpinB2 (PAI‐2) modulates proteostasis via binding misfolded proteins and promotion of cytoprotective inclusion formation. PLoS One. 2015;10:e0130136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gelling CL, Brodsky JL. Mechanisms underlying the cellular clearance of antitrypsin Z: lessons from yeast expression systems. Proc Am Thorac Soc. 2010;7:363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kamimoto T, Shoji S, Hidvegi T, Mizushima N, Umebayashi K, Perlmutter DH, et al. Intracellular inclusions containing mutant alpha1‐antitrypsin Z are propagated in the absence of autophagic activity. J Biol Chem. 2006;281:4467–76. [DOI] [PubMed] [Google Scholar]
- 69.Kounnas MZ, Church FC, Argraves WS, Strickland DK. Cellular internalization and degradation of antithrombin III‐thrombin, heparin cofactor II‐thrombin, and alpha 1‐antitrypsin‐trypsin complexes is mediated by the low density lipoprotein receptor‐related protein. J Biol Chem. 1996;271:6523–9. [DOI] [PubMed] [Google Scholar]
- 70.Fra A, Cosmi F, Ordonez A, Berardelli R, Perez J, Guadagno NA, et al. Polymers of Z alpha1‐antitrypsin are secreted in cell models of disease. Eur Respir J. 2016;47:1005–9. [DOI] [PubMed] [Google Scholar]
- 71.Huntington JA, Sendall TJ, Yamasaki M. New insight into serpin polymerization and aggregation. Prion. 2009;3:12–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chou SM, Taniguchi A, Wang HS, Festoff BW. Serpin=serine protease‐like complexes within neurofilament conglomerates of motoneurons in amyotrophic lateral sclerosis. J Neurol Sci. 1998;160(Suppl 1):S73–S79. [DOI] [PubMed] [Google Scholar]
- 73.McCauley ME, Baloh RH. Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. 2019;137:715–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thonhoff JR, Simpson EP, Appel SH. Neuroinflammatory mechanisms in amyotrophic lateral sclerosis pathogenesis. Curr Opin Neurol. 2018;31:635–9. [DOI] [PubMed] [Google Scholar]
- 75.Arecco N, Clarke CJ, Jones FK, Simpson DM, Mason D, Beynon RJ, et al. Elastase levels and activity are increased in dystrophic muscle and impair myoblast cell survival, proliferation and differentiation. Sci Rep. 2016;6:24708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Trias E, King PH, Si Y, Kwon Y, Varela V, Ibarburu S, et al. Mast cells and neutrophils mediate peripheral motor pathway degeneration in ALS. JCI Insight. 2018;3:e123249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gong D, Farley K, White M, Hartshorn KL, Benarafa C, Remold‐O'Donnell E. Critical role of serpinB1 in regulating inflammatory responses in pulmonary influenza infection. J Infect Dis. 2011;204:592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yasumatsu R, Altiok O, Benarafa C, Yasumatsu C, Bingol‐Karakoc G, Remold‐O'Donnell E, et al. SERPINB1 upregulation is associated with in vivo complex formation with neutrophil elastase and cathepsin G in a baboon model of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2006;291:L619–L627. [DOI] [PubMed] [Google Scholar]
- 79.Gould TW, Dominguez B, de Winter F, Yeo GW, Liu P, Sundararaman B, et al. Glial cells maintain synapses by inhibiting an activity‐dependent retrograde protease signal. PLoS Genet. 2019;15:e1007948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Amara U, Rittirsch D, Flierl M, Bruckner U, Klos A, Gebhard F, et al. Interaction between the coagulation and complement system. Adv Exp Med Biol. 2008;632:71–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang HA, Lee JD, Lee KM, Woodruff TM, Noakes PG. Complement C5a–C5aR1 signalling drives skeletal muscle macrophage recruitment in the hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. Skelet Muscle. 2017;7:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. [DOI] [PubMed] [Google Scholar]
- 83.Ramachandran R, Noorbakhsh F, Defea K, Hollenberg MD. Targeting proteinase‐activated receptors: therapeutic potential and challenges. Nat Rev Drug Discov. 2012;11:69–86. [DOI] [PubMed] [Google Scholar]
- 84.Thakker‐Varia S, Elkabes S, Schick C, Silverman GA, Peng L, Sherwood AC, et al. Gene expression in activated brain microglia: identification of a proteinase inhibitor that increases microglial cell number. Brain Res Mol Brain Res. 1998;56(1–2):99–107. [DOI] [PubMed] [Google Scholar]
- 85.Zhang J, Velmeshev D, Hashimoto K, Huang YH, Hofmann JW, Shi X, et al. Neurotoxic microglia promote TDP‐43 proteinopathy in progranulin deficiency. Nature. 2020;588:459–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S12
FIGURE S1 Immunoblots of HeLa cell lysates transfected with Myc‐tagged Serpin A1, B1, C1 and I1 expression cDNAs to validate the specificity of serpin antibodies used in this study. All antibodies detect only their cognate recombinant protein and the correct size endogenous protein
FIGURE S2 Age‐dependent alteration of UBQLN2 and serpin staining in P497S mutant mice. (A and B) Immunofluorescence staining of UBQLN2 and Serpin A1, B1, C1, or I1 in the hippocampal region of sagittal brain sections from 8‐week‐old (A) and 52‐weekold (B) P497S animals. Scale bars shown = 100 μm for all images
FIGURE S3 Colocalization of serpin and UBQLN2 proteins in different brain regions of P497S animals. Confocal microscopy images of 52‐week‐old P497S animals similar to Fig 2, but showing the UBQLN2 and Serpin A1, C1, and I1 protein staining in the CA1 (top panel), CA3 (middle panel), and cortex (bottom panel) of the brain. Scale bars shown = 20 μm for all images
FIGURE S4 Line scan analysis showing colocalization of serpins with UBQLN2 staining in the dentate gyrus and spinal cord sections of P497S animals. Line scans were performed through representative regions of the magnified images shown in Figs 2 and 3. Plots depict pixel intensity of the individual UBQLN2 (red) and serpin (green) image channels for the lines analyzed in the images shown to the right of each plot. Lines are 2 drawn through the same region of interest in each image to determine the extent of overlap between the UBQLN2 and serpin staining
FIGURE S5 Colocalization of UBQLN2 and serpins proteins in puncta within spinal MN. Confocal microscopy images of 32‐week‐old P497S animals similar to Fig 3, but showing higher magnification images of MN, identified by ChAT‐positive staining, in the SC of age‐matched P497S (top panel), WT356 (middle panel), and Non‐Tg (bottom panel) animals. Scale bars shown = 20 μm for all images
FIGURE S6 Line scans showing the degree of overlap between UBQLN2, serpin, and LC3 staining in P497S and Non‐Tg SC sections shown in Fig 4. Plots depict pixel intensity for UBQLN2 (red), serpin (green), and the autophagosome marker LC3 (magenta) for the lines shown in images below each plot. Lines are drawn through the same region of interest containing representative puncta for either the larger, extracellular puncta in P497S animals (left column) or the smaller puncta within motor neurons of P497S and Non‐Tg animals (middle and right columns, respectively)
FIGURE S7 Line scans showing the degree of overlap between UBQLN2, serpins and LAMP1 in P497S and Non‐Tg spinal cord sections shown in Fig 5. Similar to Fig S6, but measuring colocalization between UBQLN2 (red), serpins (green), the LAMP1 lysosomal marker (magenta)
FIGURE S8 The UBQLN2 and serpin double‐positive puncta in the SC are more closely juxtaposed with microglia than with astrocytes. Confocal microscopy images of the staining of UBQLN2, Serpin A1 or C1 proteins with either GFAP (top panels) or IBA1 (bottom panels) in the SC of 32‐week‐old P497S animals. Scale bars shown = 100 μm and 20 μm for 1× and 5× zoom images, respectively
FIGURE S9 Comparison of the UBQLN2, Serpin A1 and C1 staining patterns in the dentate gyrus of P497S, WT 356 and Non‐Tg animals. Similar to Fig 6, but showing staining for all three genotypes, as indicated. The P497S staining panels are the same as Fig 6. Scale bars shown = 100 μm and 20 μm for 1× and 5× zoom images, respectively
FIGURE S10 Comparison of the UBQLN2, Serpin A1 and C1 staining patterns in the SC of P497S, WT 356 and Non‐Tg animals. Similar to Fig S8, but showing staining in the SC for all three genotypes, as indicated. The P497S staining panels are the same as Fig 6. Scale bars shown = 100 μm and 20 μm for 1× and 5× zoom images, respectively. Scale bars shown = 100 μm and 20 μm for 1× and 5× zoom images, respectively
FIGURE S11 Decreased solubility of UBQLN2 and Serpin A1 in P497S mutant animals. (A) Immunoblots of equal portions of low salt (LS), Triton‐X100 (TX‐100) sarkosyl (SARK) and urea fractions following sequential biochemical extraction of equal weight of cortical brain tissue from 32‐week‐old Non‐Tg, WT356, and P497S mice. The fractions analyzed were obtained by progressive extraction of the tissues with low salt, Triton X4 100, sarkosyl, and urea buffers. The Triton X‐100 fraction was not included because of aberrant migration of proteins extracted with the buffer. (B) Quantification of penultimate SARK fractions for UBQLN2 and serpin proteins. (C) Quantification of ultimate urea fractions for each serpin protein. (D) Ubiquitin (UBQTN) immunoblots of urea fractions from 8‐ and 32‐week‐old animals. (E) Quantification of 32 week urea fractions for UBQLN2 and UBQTN
FIGURE S12 Quantification of serpin protein levels in mouse tissues by SDS‐PAGE analysis. (A) Immunoblots of hippocampus (left) and lumbar SC tissue (right) of 3 WT356, Non‐Tg, and P497S animals per genotype analyzed after separation by SDSPAGE. (B) Quantification of immunoblots in A shows a significant decrease in Serpin I1 in the hippocampus and a significant decrease in Serpin C1 in the spinal cord of 32‐ week‐old P497S animals. Other serpins trended toward a decrease in P497S animals though not to a level of significance. As expected, levels of the UBQLN2 transgene in both the hippocampus and spinal cord were significantly increased in P497S animals
Data Availability Statement
All of the data in contained in either the body of the manuscript or included in the Supplemental material provided. Any additional data used in the study are available from the corresponding author upon reasonable request.