

Abstract
Single-atom catalysts (SACs) featuring atomically dispersed metal cations covalently embedded in a carbon matrix show significant potential to achieve high catalytic performance in various electrocatalytic reactions. Although considerable advances have been achieved in their syntheses and electrochemical applications, further development and fundamental understanding are limited by a lack of strategies that can allow the quantitative analyses of their intrinsic catalytic characteristics, that is, active site density (SD) and turnover frequency (TOF). Here we show an in situ SD quantification method using a cyanide anion as a probe molecule. The decrease in cyanide concentration triggered by irreversible adsorption on metal-based active sites of a model Fe–N–C catalyst is precisely measured by spectrophotometry, and it is correlated to the relative decrease in electrocatalytic activity in the model reaction of oxygen reduction reaction. The linear correlation verifies the surface-sensitive and metal-specific adsorption of cyanide on Fe–Nx sites, based on which the values of SD and TOF can be determined. Notably, this analytical strategy shows versatile applicability to a series of transition/noble metal SACs and Pt nanoparticles in a broad pH range (1–13). The SD and TOF quantification can afford an improved understanding of the structure–activity relationship for a broad range of electrocatalysts, in particular, the SACs, for which no general electrochemical method to determine the intrinsic catalytic characteristics is available.
Keywords: Single-atom catalysts, Fe−N−C catalysts, active site density, turnover frequency, oxygen reduction reaction
Introduction
Heterogeneous catalysts are of paramount importance to the world economy and the sustainable development of our society,1,2 from chemical manufacturing to energy-related applications and environmental remediation. The overall apparent activity of a catalyst in a reaction can be expressed as a mathematical product of the active site density (SD) and turnover frequency (TOF). TOF, which is defined as the number of chemical conversions of reactant molecules per catalytic site and per unit time, is a key descriptor of the intrinsic activity of the catalytic site.3 TOF is practically estimated by the combined input of the overall catalytic reaction rate (RR) of a catalyst and its SD (i.e., TOF = RR/SD). The availability of methods to deconvolute the apparent activity of a catalyst into its TOF and SD is important not only for fundamental understanding but also for guiding the rational development of novel catalysts,4 as an increase in TOF or SD has different implications in the scientific rationale, experimental development of the next-generation catalytic materials, and their implementation in devices.
Chemisorption, titration, and other methods (e.g., X-ray diffraction line broadening analysis, small-angle X-ray scattering, etc.) have been employed to measure the SD values of heterogeneous catalysts in thermochemical reactions.5−8 Among these methods, chemisorption under gas-phase conditions is generally used to determine the SD of a catalytic material.9 However, in the field of electrocatalysis, the estimation of SD depends on the measurement of the electric charge exchanged during the interaction of the site-specific adsorbates with the catalytic surface immersed in an electrolyte. Since the adsorption/desorption events can be affected by the electrochemical potential and experimental conditions, the measurement of the SD values of electrocatalysts under the same conditions as those during the intended electrochemical reaction can prevent (or minimize) systemic errors. For example, the number of active sites probed in the gas phase would exceed that measured in the liquid phase for porous catalysts, whose active sites are located in the narrow pores that are not completely wetted by the electrolyte.10 In addition, the surface properties of the catalysts can be altered under electrochemical conditions compared to those under the gas-phase conditions (e.g., because of contact with the liquid electrolyte and ionomer/binder, the reactant gas dissolved in the electrolyte, and the electrochemical potential applied). Consequently, the SD value measured at the gas/solid interface may not be directly applicable to the electrocatalysts experiencing a different environment of (gas/)liquid/solid interface during electrocatalysis. Hence, the concept of electrochemical active surface area (ECSA) has been introduced, which refers to the SD (or surface area) measured electrochemically, implying that the probed sites are both electrically and electrolytically connected. For the metallic surfaces of noble metals, the ECSA values are typically determined by measuring the electric charge during cyclic voltammetry (CV), which results from the underpotential deposition (UPD) of either hydrogen (HUPD) or metal ions (e.g., CuUPD)11−13 or from the electrochemical oxidation of strongly adsorbed species (e.g., CO-stripping).14−16 However, these in situ electrochemical methods are restricted to the metallic surfaces of platinum-group metals (PGMs) and are not applicable to a multitude of other catalysts that do not adsorb H or CO under such conditions. In particular, the measurement of SD under electrochemical conditions remains challenging for the recent promising single-atom catalysts (SACs) featuring atomically dispersed active metal sites, which have completely different characteristics from bulk metal catalysts.17
The Me–N–C catalyst, one of the promising class of SACs comprising metal–Nx sites, has attracted significant attention for catalyzing various electrochemical reactions (e.g., oxygen reduction, hydrogen evolution, and carbon dioxide reduction reactions),18−23 but shows no H and CO adsorption under ambient conditions, thereby limiting the quantification of SD in electrochemical environments. Although X-ray absorption spectroscopy (XAS) and 57Fe or 119Sn Mössbauer spectroscopy are powerful tools for identifying the Me–Nx moieties,24−27 such techniques employing high-energy radiation probe the entire volume and are not surface specific. The measurement of the electric charge based on the redox transitions of the metal center can be an effective method to determine the SD of SACs,28,29 but is applicable only when they contain a well-defined Me–Nx site (e.g., nonpyrolyzed macrocycles or molecular complexes on carbon) and is generally not reliable for Me–N–C, where the redox feature is broad and weak because of the covalently integrated Me–Nx sites in the N-doped carbon matrix.
For studies on Me–N–C, only two methods for SD quantification employing molecular probes have been established.30,31 The first method is low-temperature CO cryo chemisorption at 193 K.30 The weakness of this method includes: (1) a possible overestimation of SD since all gas-phase accessible sites can be probed (in particular for Me–N–C with high microporosity) and (2) its limitation to the quantification of Me–Nx sites with a sufficiently strong binding of CO* (i.e., Fe and Mn).32 In addition, a thermal pretreatment up to 873 K is necessary to completely remove the preadsorbed O2 on Me–Nx sites before CO adsorption. This cleaning treatment can lead to unexpected modifications in the surface chemistry, possibly changing its SD and/or TOF for a particular reaction.33 The second method is nitrite (NO2–)-stripping voltammetry.31 This is an in situ electrochemical method performed at a specific pH (pH 5.2; 0.5 M acetate buffer), which quantifies the electric charge corresponding to the reduction of the nitrosyl ligand on the Fe–Nx sites. The conversion of this electric charge into a SD value is performed by hypothesizing that the electrochemical reduction of the Fe nitrosyl complex (generated by a well-known chemical reaction between NO2– and protons) is selective and complete toward ammonia (five-electron reduction).34,35 The NO2–-stripping method has advantages compared to the CO chemisorption as it is an in situ method. However, the application of this method for evaluating the SD of the SACs other than Fe–N–C or in a broad pH range is challenging owing to the unknown number of electrons transferred during the nitrosyl reduction under different conditions. In addition to the complete five-electron reduction product, ammonia, several other products of the nitrosyl reduction reaction can be expected, for example, hydroxylamine, nitrous oxide, and nitrogen, depending on the catalytic material and electrolyte pH.36−38 Recently, a non-negligible deviation between the SD values, measured by CO chemisorption and NO2–-stripping voltammetry on four benchmark Fe–N–C catalysts, has also been reported.39
Herein, a versatile in situ method for SD quantification is proposed by employing a cyanide anion probe. Although cyanide has been reported to poison Fe–N–C catalysts in prior studies,40,41 it has not been used to quantify their SDs owing to its competitive adsorption with O2.42 Here, a new strategy to determine the SD of Fe–N–C is developed based on the combination of (1) partial cyanide poisoning of Fe3+-Nx surface sites free of O2 ligand and (2) the ensuing relative deactivation in the model reaction of oxygen reduction reaction (ORR). The decrease in cyanide concentration due to its adsorption on Fe3+–Nx is quantified using ultraviolet–visible (UV–vis) spectrophotometry, as a photoactive compound is formed via a cascade reaction between cyanide and p-nitrobenzaldehyde, which is further correlated to the relative decay in ORR activity. This SD quantification protocol is highly surface sensitive and metal specific, allowing the determination of SD and TOF of Fe–N–C. Notably, this analytical method is applicable under various electrochemical conditions ranging from acidic to alkaline environments and shows a broad material scope ranging from Me–N–C (Me = Mn, Co, and Ni) to Pt-SAC comprising atomically dispersed Pt in sulfur-doped carbon as well as conventional Pt metallic nanoparticles supported on carbon.
Results and Discussion
A model Fe–N–C catalyst (labeled ‘Fe0.5NC’),43,44 comprising only atomically dispersed Fe–Nx moieties without metallic or oxide/carbide Fe clusters, was employed to develop the in situ SD quantification method (see details for the synthetic procedure in the Experimental Section). The Fe–N–C catalyst was chosen as a model substance primarily because previous CO chemisorption and NO2–-stripping methods for SD estimation have been developed with Fe–N–C catalyst(s), allowing us to directly compare the SD values measured by multiple methods. The detailed physicochemical characterization results of Fe0.5NC can be found in Supplementary Note 1 (Figure S1 and Tables S1 and S2).
Despite the reported poisoning effects of cyanide on the ORR activity of Fe–N–C catalysts,40,41 SD quantification with this probe anion has not yet been successfully established. This is primarily because cyanide weakly adsorbs on the Fe–Nx sites, thus showing competitive adsorption with O2.42 The relatively weak adsorption of cyanide on Fe–Nx sites compared to O2 is supported by the recovery of most of the initial ORR activity of Fe–N–C after simple rinsing with water.42,45 When the cyanide concentration in the electrolyte is significantly higher than that of O2 (ca. 1–1.5 mM O2 in aqueous electrolytes at room temperature; RT), the equilibrium between cyanide and O2 occupation of Fe–Nx sites is displaced toward the former owing to Le Chatelier’s effect.42
Hence, a SD quantification protocol based on cyanide adsorption on Fe–Nx sites demands some prerequisites. First, the electrochemical environments and active sites must be free of O2, such that cyanide can potentially bind to most of the surface active sites. Second, cyanide poisoning must be irreversible, such that the ORR activity of the cyanide-poisoned sample can be measured in a cyanide-free O2-saturated electrolyte. To quantify the amount of cyanide adsorbed (and thus the number of surface sites occupied by cyanide, assuming a 1:1 ratio) on Fe–N–C under specific conditions, the changes in the cyanide concentration in the electrolyte (CCN–electrolyte) and the ORR activity before and after cyanide adsorption by Fe0.5NC need to be quantified (Figure 1a). A highly sensitive spectrophotometric method was selected to achieve this goal. This method was based on the cascade chemical reaction of cyanide with p-nitrobenzaldehyde, followed by a reaction with tetrazolium blue to produce a photoactive diformazan compound (Figure S2).46,47 The final product yielded maximum absorbance at ca. 520 nm in the UV–vis spectrum, and the absorbance was linearly proportional to the cyanide concentration in the UV–vis cuvette (CCN–), which was 0–10 μM (Figure S3). The correlation between UV–vis absorption at 520 nm and cyanide concentration is sufficiently accurate to measure very low cyanide concentrations, providing a suitable analytical platform for detecting small changes in CCN–electrolyte before and after the cyanide poisoning of Fe0.5NC.
Figure 1.

Scheme of in situ SD quantification protocol using a cyanide probe. (a) In situ SD quantification procedure depends on the combined measurement of the decrease in the cyanide concentration (left-hand-side inset in (a)) due to its adsorption on the metal-based active sites (central scheme in (a); blue = N, gray = C) and irreversible decrease in ORR activity after cyanide poisoning (right-hand-side inset in (a)). (b) In situ cyanide poisoning protocol comprises three steps: (1) removal of predissolved O2 in the electrolyte by Ar purging, (2) electrochemical removal of preadsorbed O2 on the catalyst, and (3) cyanide poisoning of the catalyst under the controlled electrochemical potential of the working electrode.
Cyanide poisoning of Fe0.5NC was carried out in a two-compartment H-type electrochemical cell (Figure 1b). Each compartment was initially filled with 0.1 M HClO4 electrolyte, and a graphite-rod working electrode was placed in compartment 1, while the counter and reference electrodes were placed in compartment 2. The two compartments were separated using a Nafion membrane. The key reason for the separation was to prevent pollution by O2 produced at the counter electrode during the electrochemical removal of O2 in compartment 1. A known amount of Fe0.5NC powder was dispersed only in working electrode compartment 1 and mechanically stirred to prevent its sedimentation during the entire poisoning protocol. Following the addition of Fe0.5NC, the electrolyte was deaerated by Ar purging, while the working electrode was set at the open circuit potential (OCP), and all subsequent (electro)chemical treatments were conducted in an Ar-filled chamber to prevent undesirable O2 dissolution in the electrolyte (first step). In the next step, preadsorbed O2 on the Fe–Nx sites, which could not be simply removed by Ar purging, was electrochemically removed. This was performed under continuous Ar purging and mechanical stirring by applying a sufficiently low potential of 0.5 VRHE to the working electrode (second step), to ensure that the preadsorbed O2 was reduced to water when the catalyst particles collided with the working electrode through convective motion. Subsequently, an aliquot of deaerated cyanide aqueous solution was injected into both compartments (third step). The initial CCN–electrolyte was adjusted to 200 μM, resulting in an optimized balance between the irreversible poisoning of Fe–Nx sites and a significant decrease in cyanide concentration. The working electrode was polarized at 1.0 VRHE (i.e., Ethird) to ensure that the surface Fe–Nx sites were in the ferric state (Figure S4). The ferric state of Fe–Nx sites has a higher binding affinity to cyanide than the ferrous state, based on the studies on hemoproteins and Fe cyanide complexes.48,49 After cyanide injection, Ar purging was stopped, and all compartments were closed to prevent any undesirable decrease in cyanide concentration via the release of HCN in the gas-phase (Figure S5).
The decrease in CCN–electrolyte due to cyanide adsorption by Fe0.5NC was then measured by UV–vis spectrophotometry. An aliquot of the electrolyte, which was collected after filtering the solution of compartment 1 from Fe0.5NC, was diluted 20 times, and a fixed amount of p-nitrobenzaldehyde and tetrazolium blue was added (see Experimental Section). Control experiments without the addition of a catalyst in compartment 1 or with the addition of Fe-free N-doped carbon (NC) revealed a negligible decrease in CCN– after the poisoning protocol (Figure 2a,b). On the other hand, the spectrophotometric data collected after the addition of Fe0.5NC showed that CCN–electrolyte decreased from 200 to 181 μM after the cyanide poisoning of Fe0.5NC, indicating that the surface Fe–Nx moieties adsorbed cyanide ions. The poisoned Fe0.5NC powder that was collected after filtration and water rinsing showed a current density (j) of −0.060 ± 0.003 mA cm–2 at 0.85 VRHE (Figure 2c), which was significantly lower than that of pristine Fe0.5NC (j = −0.110 mA cm–2). Cyanide was hardly detected in the rinsed water (Figure S6), which indicates the irreversible adsorption of cyanide on the Fe–Nx site during the poisoning protocol.
Figure 2.
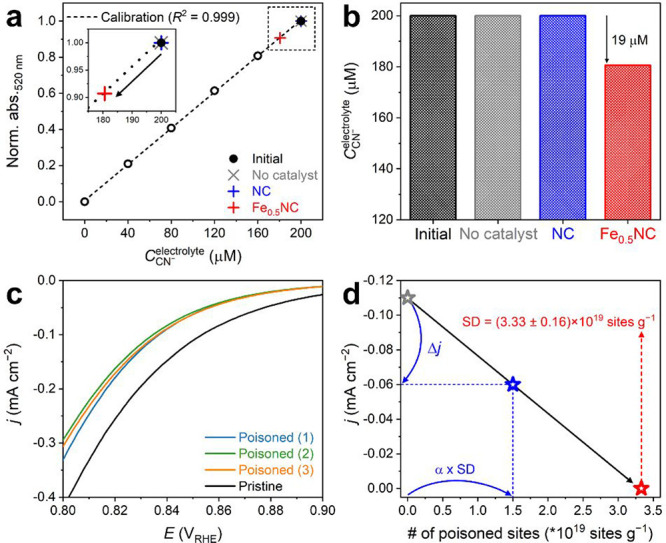
In situ SD quantification of Fe0.5NC using the cyanide probe method. (a) Determination of CCN–electrolyte using a UV–vis spectrophotometer at 520 nm before and after cyanide adsorption on Fe0.5NC. Results obtained for the two control experiments, that is, without any catalyst (gray cross) and with NC in solution (blue cross), are also compared. Calibration values are indicated by open circles, and initial CCN– is indicated by a filled circle. (b) Initial cyanide concentration (same for all three experiments) and cyanide concentration after the blank experiment without any catalyst or the cyanide poisoning of NC and Fe0.5NC in solution. (c) ORR activity of Fe0.5NC measured in O2-saturated 0.1 M HClO4 electrolyte before and after in situ SD quantification with cyanide poisoning. Three repeated experiments are presented. (d) Estimation of the SD value of Fe0.5NC (red star) by linear extrapolation to zero current of the line defined by the initial point (gray star; pristine Fe0.5NC) and second point (blue star; cyanide-poisoned Fe0.5NC). The x and y positions of each point are defined by the measured absolute amount of Fe–Nx sites poisoned and ORR activity after cyanide poisoning, respectively.
However, any O2 contamination in the electrolyte during the protocol could interrupt the cyanide poisoning of Fe0.5NC, likely due to the competitive adsorption of O2 on Fe–Nx sites (Figure S7). It is of note that the ORR activity was measured by linear sweep voltammetry (LSV) in a restricted potential range of 0.70–1.05 VRHE with a low scan rate of 1 mV s–1, which afforded high reproducibility of the ORR polarization curves for the pristine and poisoned catalysts (Supplementary Note 2 and Figures S8 and S9).
Then, SD could be derived from the change in cyanide concentration (ΔCCN–electrolyte) and the relative decrease in ORR activity (Δj/jpristine) obtained from the collected cyanide-poisoned Fe0.5NC powder. If it is assumed that the decrease in ORR activity is linear to the amount of adsorbed cyanide on the surface Fe–Nx moieties from zero to “SD” and the ORR activity reaches zero when all surface Fe–Nx sites are occupied by cyanide (Figure S10), their relationship can be expressed by the following equation:
| 1 |
where α is the fraction of poisoned sites to total sites (i.e., poisoned + nonpoisoned) of Fe–Nx species, and jpristine and Δj are the pristine catalytic activity and poisoning-induced activity decrease at a given potential, respectively. Then, SD can be expressed by the following equation:
| 2 |
where V is the electrolyte volume (40 mL), NA is the Avogadro constant, and mcat is the catalyst amount (30 mg).
However, eqs 1 and 2 are based on the linearity hypothesis (Figure S10). Hence, the above calculations are valid only when the cyanide poisoning on Fe–Nx sites occurs in a similar manner on all surface Fe–Nx sites. If there are two or more types of surface Fe–Nx sites on Fe0.5NC with different affinities for cyanide binding, the SD value estimated according to eq 2 will be erroneous (Supplementary Note 3). Considering this hypothesis using eqs 1 and 2, the estimated SD value was (3.33 ± 0.16) × 1019 sites g–1 for Fe0.5NC (Figure 2d). Based on the SD value and measured bulk metal content, the utilization factor (U) of Fe0.5NC can be calculated using eq 3, which represents the electrochemically accessible active metal sites to the total number of metal atoms in the material.
| 3 |
where m is the metal content at the surface and bulk, %Me is the weight percentage of the metal in the catalyst, and M is the atomic mass of the metal. Using eq 3 and the SD values obtained for three different repetitive experiments, U was determined to be 20–22% in Fe0.5NC.
To verify whether these assumptions and the results obtained using this new method were reliable, the SD and U values were estimated with different extents of cyanide poisoning of Fe0.5NC (Figure 3). The first approach to modulate the extent of cyanide poisoning comprised the application of different potentials on the working electrode during the third step of the poisoning protocol (third step in Figure 1b). In comparison to the original protocol (Ethird = 1.0 VRHE), the results showed that cyanide adsorption on Fe0.5NC was mitigated when Ethird decreased from 1.0 to 0.7 VRHE (Figure 3a). The ΔCCN–electrolyte of ca. 19 μM at 1.0 VRHE decreased to ca. 14 μM at 0.7 VRHE. In situ X-ray absorption near edge structure (XANES) measurements at different potentials suggested that the decrease in ΔCCN– is attributed to a decrease in the population of ferric species with a higher binding affinity to cyanide than the ferrous species (Figures 3b and S4b).48,49 However, with a decrease in ΔCCN–electrolyte, the ORR activity loss for Fe0.5NC also decreased simultaneously (Figure S11). This led to comparable SD ((3.20 ± 0.30) × 1019 sites g–1) and U (20 ± 2%) values, regardless of the applied Ethird values (Figure 3a), because the proportional changes in CCN– and Δj/jpristine canceled out in eq 2. In the second approach, the extent of cyanide poisoning was modified by varying the initial CCN–electrolyte values in compartment 1 from 100 to 400 μM (200 μM for the original protocol). The results showed that ΔCCN– increased from ca. 9 to 27 μM with an increase in the initial CCN–electrolyte (Figure 3c). Similar to the effects observed with the electrochemical potential modulation, ΔCCN– and Δj/jpristine changed proportionally (both increased when the initial CCN–electrolyte was increased; Figure S12), thereby resulting in similar values of SD ((3.52 ± 0.34) × 1019 sites g–1) and U (22 ± 2%) for the initial CCN– values ranging from 100 to 400 μM (Figure 3c).
Figure 3.
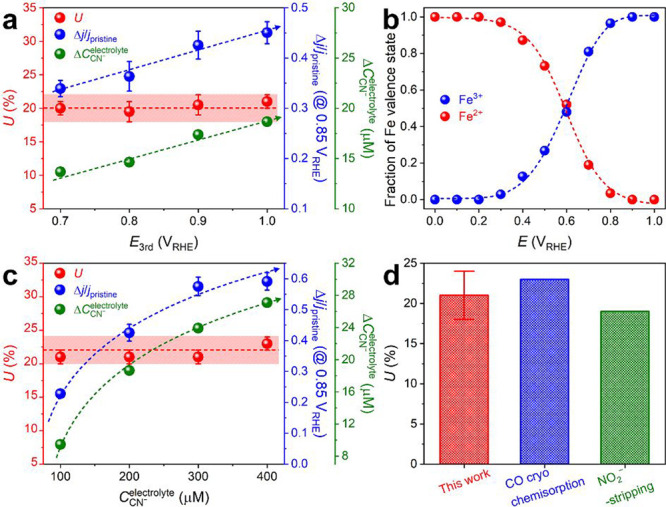
Determination of U values under various conditions of in situ SD quantification. (a) U, Δj/jpristine, and ΔCCN–electrolyte values estimated at different Ethird values applied on the working electrode. (b) Fraction of ferric and ferrous ions of Fe0.5NC at various potentials, fitted from the Fe K-edge in situ XANES spectra (Figure S4b). (c) U, Δj/jpristine, and ΔCCN– values estimated at different initial CCN–electrolyte values in the working electrode compartment. (d) Comparison of the U values of Fe0.5NC measured by the cyanide-poisoning in situ SD quantification, gas-phase CO chemisorption, and electrochemical NO2–-stripping methods.31,33
Therefore, the effects of the potential applied in the third step and initial cyanide concentration afforded almost identical SD ((3.38 ± 0.48) × 1019 sites g–1) and U (21 ± 3%) values, even though different amounts of cyanide were irreversibly adsorbed on Fe0.5NC. This observation clearly supports the random cyanide poisoning on all surface Fe–Nx sites of Fe0.5NC, verifying the validity of the assumptions in eqs 1 and 2 (Supplementary Note 3). Notably, the U values obtained by the present in situ cyanide poisoning method (21% on average) were slightly lower than those measured previously using the ex situ CO cryo chemisorption method (ca. 23%; Figure S13), but were slightly higher than those obtained by the modified in situ NO2–-stripping method (ca. 19%; Figure S14) for an identically prepared catalyst (Figure 3d). It is of note that a different hypothesis for the final product of nitrosyl ligand reduction was employed for the NO2–-stripping method. Together, these results imply that the cyanide probe in the liquid electrolyte can easily access the active sites, and this in situ protocol provides a new analytical platform for the accurate quantification of active Fe–Nx sites on Fe–N–C catalysts in acidic environments.
With the SD descriptor obtained by a combination of spectrophotometry and a decrease in catalytic activity, the TOF of Fe0.5NC in ORR could be calculated using the following equation:
| 4 |
where J is the catalytic mass activity at a given potential and F is the Faraday constant. In an acidic medium, the TOF of Fe0.5NC was determined to be 0.40 ± 0.05 e site–1 s–1 at 0.80 VRHE through control experiments performed with different Ethird and CCN–electrolyte values, which showed small deviations in the TOF values. The TOF of Fe0.5NC was calculated using the SD value, which was obtained at 0.85 VRHE owing to the non-negligible limitation of the current density by diffusion below 0.85 VRHE (Figure S9). This TOF is slightly higher (but in the same order of magnitude) than that measured by CO chemisorption using an identically prepared and other Fe–N–C catalysts (0.15–0.18 e site–1 s–1),26,33 probably due to a possible overestimation in SD value measured at the gas-phase condition.
In addition to the determination of SD and TOF for the Fe–N–C catalyst, which can also be achieved by the previous CO chemisorption and NO2–-stripping methods,31,33 the general applicability of the cyanide protocol was investigated under various experimental conditions and on other electrocatalytic materials (Figure 4). First, to examine the applicability of the cyanide protocol in conditions other than the acidic electrolyte (pH 1), the SD of Fe0.5NC was measured in neutral (pH 7; 0.1 M phosphate buffer) and alkaline (pH 13; 0.1 M KOH) electrolytes. As the SD value (in this case, the number of Fe–Nx sites) is an inherent characteristic of the catalytic material, it was expected that if the cyanide protocol is applicable under neutral and alkaline conditions and no significant demetalation of Fe–Nx sites occurs in acidic pH (as suggested by slightly lower SDs than the gas-phase CO chemisorption), the SD of the Fe0.5NC catalyst measured at pH 7 and 13 should be similar to that measured at pH 1. In practice, ΔCCN–electrolyte values in neutral and alkaline electrolytes (ca. 16 and 17 μM at pH 7 and 13, respectively; Figure S15) were similar (or slightly lower) to that measured in the acidic electrolyte (ca. 19 μM), resulting in comparable U values (19 ± 1%) regardless of the electrolyte pH (Figure 4a,f). This result indicated that the cyanide protocol could be applied to measure the SD of the Fe–N–C catalysts over a broad range of pH values. Although this does not provide additional information if the SD is pH-independent, it brings direct information regarding the TOFs of the Fe–Nx sites for ORR at different pH values.
Figure 4.

General applicability of cyanide-poisoning in situ SD quantification protocol. (a–c) Δj/jpristine and ΔCCN–electrolyte values obtained after the in situ SD quantification of Fe0.5NC at various pH values (a), Me0.5NC (Me = Mn, Co, and Ni) and Pt/HSC (b), and Pt nanoparticles with different particle sizes (i.e., 2.5, 2.9, 3.8, and 4.5 nm) (c). (d) HUPD and CO-stripping voltammograms of Pt nanoparticles. (e) Comparison between ECSA values, derived from HUPD (black y-axis) and CO-stripping (red y-axis), and ΔCCN– (blue y-axis) for Pt nanoparticles with different particle sizes. (f,g) Summary of U (f) and TOF (g) values estimated by the in situ SD quantification of Fe0.5NC at various pH values and other control SACs and Pt nanoparticles at pH 1. The values reported in the literature (green star) or obtained by ECSACO (black star) are also included for comparison.26,33
Subsequently, the applicability of the cyanide protocol to quantify the SDs of the other SACs was examined. First, other Me–N–C catalysts (Me = Mn, Co, and Ni; labeled ‘Me0.5NC’) were prepared in a similar manner as Fe0.5NC by changing only the metal. Because these transition metals have similar atomic masses (i.e., 54.9–58.7 amu) and the carbonization of ZIF-8 as well as the formation of Me–Nx moieties occur during flash pyrolysis (i.e., increase in temperature from RT to 1323 K within a few seconds), it can be reasonably surmised that the physicochemical characteristics of the Me0.5NC catalysts are similar to those of Fe0.5NC. In particular, metal content in the Me0.5NC catalysts was ca. 1.5 wt %, and most of the metal species were dispersed as Me–Nx moieties as confirmed by XAS spectra (Figures S16–S18 and Table S1). This was consistent with identical physicochemical characteristics (except for the nature of the metal) for other series of Me0.5NC catalysts prepared via the same synthetic protocol and studied for CO2RR and ORR to H2O2.50,51
In the SD quantification, the ΔCCN–electrolyte values of Mn0.5NC, Co0.5NC, and Ni0.5NC were ca. 15, 31, and 22 μM, respectively (Figure S19). This difference (under identical conditions applied during the protocol) highlighted the different binding affinities of cyanide on Me–Nx sites as a function of the metal characteristics.48 However, the similar trends of Δj/jpristine values as those of ΔCCN– resulted in U values of 21 ± 2% for all Me0.5NC catalysts, which were comparable to that of Fe0.5NC (Figure 4b,f). These results indicated that the fraction of surface Me–Nx species to total Me–Nx species was similar for all catalysts in this series, which was reasonable based on similar active site structure and surface area of this series of materials.50,51
Thereafter, a catalyst featuring single-atom Pt sites was introduced, in which atomically dispersed Pt2+ species (5 wt % Pt content) were ligated by the thiophene/thiolate functionalities of S-doped carbon (Figure S20; named “Pt/HSC”).52,53 Unlike the unitary process of Fe0.5NC (and Me0.5NC) synthesis, Pt/HSC was synthesized by the stabilization of isolated active Pt species (Pt2+–S4) via conventional wet impregnation on the prepared HSC support (see Experimental Section). This afforded the predominant presence of Pt–S4 moieties at the HSC surface and expectedly complete utilization of Pt (i.e., U = ∼100%). Consequently, with this model catalyst, the accuracy of the cyanide protocol and its extension to SD quantification of PGM-based SACs, which have attracted significant attention owing to their unique electrocatalytic characteristics,54−56 could be directly evaluated. The SD quantification results afforded ΔCCN–electrolyte values of 10 and 120 μM for HSC and Pt/HSC, respectively (Figure S21). The difference (110 μM) indicated that ΔCCN– was induced by cyanide adsorption on the isolated Pt–S4 moieties. Based on the decrease in the ORR activity following cyanide poisoning from −0.077 to −0.018 ± 0.002 mA cm–2 at 0.65 VRHE (Figure S21), the ΔCCN–electrolyte value of 110 μM corresponds to almost 100% utilization of Pt in Pt/HSC (Figure 4b,f).
After demonstrating the validity of the cyanide protocol for SD measurements of various SACs, the applicability of this protocol to classical catalysts featuring metal nanoparticles supported on carbon was investigated. We focused on Pt nanoparticles on carbon (Figure S22) because they are one of the most widely used active materials in electrocatalysis and their SD can be estimated by the conventional voltammetric ECSA measurement methods (i.e., HUPD and CO-stripping). Four commercial Pt catalysts with different particle sizes (ca. 2.5, 2.9, 3.8, and 4.5 nm estimated by transmission electron microscopy; TEM) were introduced to evaluate the precision of the cyanide adsorption method (Figures S23 and S24 and Table S3). The SD variations caused by different sizes of Pt nanoparticles were distinguishable by the ECSA measurements (Figure 4d), which showed a decrease in the ECSA as the particle size increased (at fixed Pt loading). Similar to the voltammetric methods, the spectrophotometry method, in which Ethird was set to 0.6 VRHE (instead of 1.0 VRHE used for the SACs) to prevent the surface oxide formation of Pt nanoparticles, also successfully differentiated the SDs of the Pt nanoparticle catalysts (Figure 4e). The ΔCCN–electrolyte value decreased from 173 to 56 μM with an increase in the particle size, a trend similar to that observed for ECSA.
Despite the non-negligible uncertainty in the SD calculation for the Pt nanoparticles with ΔCCN–electrolyte, which was caused by the different cyanide adsorption modes depending on the Pt sites,57 their U values were estimated with an assumption of the coverage (i.e., CN–/Pt) to be 0.5, based on the previous measurements for a model Pt(111) surface.58 The calculated U value was 67% for 2.5 nm Pt nanoparticles, which decreased to 56, 41, and 22% with an increase in the particle size to 2.9, 3.8, and 4.5 nm, respectively (Figure 4c,f). This assumption seemingly led to an overestimation of the estimated U values in comparison to those obtained by CO-stripping voltammetry (i.e., ECSACO; U = 53, 39, 21, and 14%, respectively), probably due to the non-negligible contributions of other sites (e.g., Pt(100) or edge) with different adsorption modes of cyanide.57 However, rough consideration of the different surface coverages of the heterogeneous Pt sites, for instance, CN–/Pt = 0.5, 1.0, and 1.0 for Pt(111), Pt(100), and edge, respectively59 (Supplementary Note 4), could alleviate the deviations between the U values obtained with different probe molecules (i.e., cyanide and CO; Figure S25), providing a clue for the precise measurement of the U value of Pt nanoparticles if an accurate coverage of each facet is determined.
Based on the case studies with various SACs and Pt nanoparticles, the precise quantification of SD can be achieved by spectrophotometry using a cyanide probe under electrochemical operating conditions. Then, the TOF values for the Me0.5NC catalysts, Pt/HSC, and Pt nanoparticles in ORR were estimated at 0.80, 0.65, and 0.90 VRHE, respectively (Figure 4g). The intrinsic ORR activity of Fe0.5NC (0.40 ± 0.05 e site–1 s–1 at 0.80 VRHE) surpassed that of all other Me0.5NC catalysts (0.06 ± 0.03 e site–1 s–1 at 0.80 VRHE), and in the alkaline medium, the TOF of Fe0.5NC (2.00 ± 0.05 e site–1 s–1 at 0.80 VRHE) considerably exceeded that obtained in the acidic medium. These results are in good agreement with previous reports on the center metal and pH effects on the ORR catalysis of the Me–N–C catalysts,60−62 and experimentally demonstrate for the first time that the higher ORR activity of Fe–N–C in comparison to those of other Me–N–C catalysts is due to the higher TOF of Fe–Nx sites, not due to the changes in SD. In Pt/HSC, the full atomic dispersion of Pt (i.e., U = 100%) and its selective two-electron ORR pathway in an acidic medium (Figure S21d) resulted in a low TOF value of 0.80 × 10–2 e site–1 s–1, even at a low potential of 0.65 VRHE. The TOF of the 2.5 nm Pt nanoparticle was determined to be 2.08 e site–1 s–1 at 0.90 VRHE, which decreased to 0.67, 0.26, and 0.13 e site–1s –1 with an increase in the particle size to 2.9, 3.8, and 4.5 nm, respectively. These TOF values were slightly lower than those calculated from ECSACO (2.63, 0.96, 0.51, and 0.20 e site–1s–1, respectively) because of the overestimation of the U values with the cyanide probe. However, consideration of the contributions of different heterogeneous Pt sites (i.e., CN–/Pt = 0.5, 1.0, and 1.0 for Pt(111), Pt(100), and edge, respectively) reduced the deviations between the TOF values determined with the cyanide and CO probes (Figure S25c), and the values were similar to those reported in the literature (10–1–100 e site–1 s–1 estimated by ECSA).63,64 Therefore, the versatile applicability of this analytical strategy is demonstrated for the SD and TOF quantifications of a series of atomically dispersed transition/noble metal catalysts and Pt nanoparticles in a wide pH range (1–13) of electrolytes.
Conclusions and Perspectives
In summary, the in situ SD quantification method provides a new experimental approach to evaluate the key descriptors of catalytic activity, that is, SD and TOF. The introduction of cyanide as an ionic probe and its titration by UV–vis spectrophotometry enabled the determination of these descriptors of the Fe–N–C catalyst over a wide range of electrolyte pH values. In addition, this analytical methodology is widely applicable to a series of atomically dispersed transition/noble metal catalysts and metallic Pt nanoparticles. However, the developed method has an important safety issue owing to the toxic release of HCN(g) in an acidic environment. Extreme caution is needed during the application of this method (please read the Experimental Section carefully before performing the experiments), and it is strongly recommended that the experiments are performed with appropriate safety precautions (e.g., fume hood, HCN gas detector, etc.) or in neutral/alkaline environments. Considering the safety concerns, its scalable generality in SD and TOF evaluations can likely afford a fundamental atomic-level understanding of the electrocatalytic activity, particularly for the SACs (except Fe–N–C), whose trends in site-specific catalytic activity have been evaluated exclusively by density functional theory calculations without direct experimental confirmation.65−67 Experimental knowledge of the SD and TOF that can be acquired for a broad range of SACs and under various synthesis conditions using this method can provide important insights and ultimately allow the establishment of structure–SD and structure–TOF relationships, allowing the rational development of novel catalysts with improved SD and TOF in the future. Specifically, this method can allow the understanding of the detailed ORR-promoting effect observed via codoping of Me–N–C catalysts with other p-block elements (e.g., sulfur) or post-treatment of Me–N–C with final pyrolysis in ammonia.26,68,69 Currently, it is unclear whether the promoting effect is due to an increased SD or TOF value.70,71
Experimental Section
Catalyst Preparation
The Me0.5NC catalysts were prepared using Me2+ acetate salts (95–98%, Sigma-Aldrich), phen (≥99%, Sigma-Aldrich), and ZIF-8 (Basolite Z1200, BASF). The precursor powders (1 g) containing Me/phen/ZIF-8 with a mass ratio of 0.5/20/80 were placed in a ZrO2 crucible with 100 ZrO2 balls (5 mm in diameter) and homogenized using a ball mill with four cycles of 30 min each at 400 rpm. The powder mixture was then pyrolyzed at 1323 K in N2 for 1 h, and Me0.5NC catalysts were collected after cooling to RT.20,44,72 The Me0.5NC catalysts contained ca. 1.5 wt % of Me content, as confirmed by inductively coupled plasma-atomic emission spectroscopy (ICP-AES). Recently, simple and general strategies enabling the controlled synthesis of a series of SACs have also been developed.73,74 For the synthesis of the Fe-free NC catalyst, a precursor mixture comprising phen and ZIF-8 was dry ball-milled and pyrolyzed, similar to the method for Me0.5NC. Owing to the presence of trace amounts of Fe impurities in the commercial ZIF-8 (>100 ppm),75 NC was individually synthesized using Fe-free ZIF-8, which was prepared by mixing 2-methylimidazole (2-MeIm; 99%, Sigma-Aldrich) and Zn nitrate hexahydrate (Zn salt; 98%, Sigma-Aldrich) in an aqueous solution (Zn salt/2-MeIm/water molar ratio of 1/60/2228).76 Carbon with high sulfur content (HSC) was synthesized by chemical vapor deposition (CVD) of acetylene/H2S mixed gas (1.4/1.4% in He) on NaX zeolite (5 g) at 823 K for 24 h.52,53 After CVD, the resulting carbon/zeolite composite was further treated at 1073 K under 5% H2S/He flow (80 mL min–1) for 3 h. HSC was collected after etching the zeolite template with an aqueous HF/HCl solution (1.1/0.8 wt %). Thereafter, 5 wt % Pt was supported on the HSC by conventional wet impregnation and subsequent H2 reduction. HSC (0.3 g) and H2PtCl6-5·5H2O (0.04 g; 99%, Kojima Chemicals) were dispersed/dissolved in 100 mL of deionized water, and the solvent was evaporated at 353 K. The resultant powder sample was reduced at 523 K for 3 h under H2 flow (200 mL min–1). Commercial Pt nanoparticles, HiSPEC 2000 (2.5 nm), HiSPEC 3000 (2.9 nm), HiSPEC 4000 (3.8 nm), and HiSPEC 9000 (4.5 nm), were purchased from Thermo Fisher Scientific.
Physicochemical Characterization
X-ray diffraction (XRD) patterns were obtained using a high-resolution X-ray diffractometer (X’Pert PRO MPD, PANalytical) equipped with a Cu Kα X-ray source at an accelerating voltage of 60 kV and a current of 55 mA with a scan rate of 10° min–1 and a step-size of 0.02°. Raman spectra were obtained using an NRS-5000 series Raman spectrometer (JASCO) with 633 nm laser excitation. ICP-AES analysis was carried out using a POLYSCAN 61E system (Hewlett-Packard). X-ray photoelectron spectroscopy (XPS) measurements were performed with a K-Alpha+ (Thermo Scientific) instrument equipped with a microfocused monochromator X-ray source. The binding energies were calibrated using the adventitious C1s signal at 284.5 eV as a reference. The XPS data were analyzed using XPSPEAK41 software with a ± 0.1 eV deviation in the binding energy. The XPS-N1s spectra were fitted to four N species: pyridinic-N (398.5 eV), pyrrolic-N (400.1 eV), graphitic-N (401.1 eV), and pyridinic-oxide (403.2 eV).77 The binding energy used for the peak deconvolution of the XPS-Pt spectra (for 4f7/2) was 72.2 eV for Pt2+ with a spin–orbit splitting of 3.33 eV.52 XAS measurements were performed with a synchrotron radiation light source in the transmission mode at Pohang Accelerator Laboratory (8C, Nano XAFS). The XAS energy scale was calibrated using each metal foil before the measurements to correct any energy shift during data acquisition. The 57Fe Mössbauer spectrum was acquired using a 57Co source in Rh. The measurement was performed by maintaining both the source and absorber at 5 K. The spectrometer was operated using a triangular velocity waveform, and a NaI scintillation detector was used to detect γ-rays. TEM and high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) analyses were carried out using a JEM-2100 LaB6 (Jeol) and Titan 80–300 (FEI), respectively.
Electrochemical Characterization
The electrochemical properties were investigated using a VMP3 potentiostat (Bio-Logic) in a three-electrode cell equipped with a graphite rod counter electrode and a saturated Ag/AgCl reference electrode (RE-1A for acidic/neutral media and RE-16 for alkaline medium, EC-Frontier). The reference electrode was doubly separated using glass and polyetheretherketone bridge tubes for acidic/neutral and alkaline media, respectively. The electrolyte was prepared with ultrapure water (>18.2 MΩ, Sartorius) and HClO4 (70%, Sigma-Aldrich), phosphoric acid (≥85%, Sigma-Aldrich), or KOH (99.99%, trace metal basis, Sigma-Aldrich). The reference electrode was calibrated against a Pt electrode in a H2-saturated electrolyte, and all potentials were reported on a reversible hydrogen electrode (RHE) scale. The Me0.5NC catalyst inks were prepared by dispersing 10 mg of the catalyst in an aqueous solution (3472 μL of deionized water, 267 μL of isopropanol, and 80 μL of 5 wt % Nafion solution). After ultrasonication of the suspension for 30 min, working electrodes were prepared by dropping 15 μL of catalyst ink onto a glassy carbon disk (0.196 cm2) of the rotating disk electrode (01169, ALS). The catalyst loadings were set to 200 μg cm–2 for Me0.5NC and 20 μgPt cm–2 for Pt/HSC and Pt nanoparticle catalysts. To prevent (or minimize) any cyanide desorption from the catalytic sites, the ORR polarization curves were recorded by LSV without a typical electrochemical cleaning step (potential cycling in general).78 The LSV curves were measured from 0.70 VRHE to 1.05 VRHE with a scan rate of 1 mV s–1 and a rotation speed of 900 rpm in an O2-saturated electrolyte. Electrochemical CO-stripping was conducted to determine the ECSACO values of the Pt nanoparticle catalysts. After CO adsorption at 0.05 VRHE in a CO-saturated electrolyte and subsequent removal of dissolved CO from the electrolyte, two cycles of CV were conducted in the potential range of 0.05–1.2 VRHE at a scan rate of 50 mV s–1.
In situ SD Quantification Method
Caution: For safety reasons, the sequence of experiments should be done very carefully when performing the same or similar tests elsewhere. We strongly emphasize that all experiments with cyanide must be conducted in a fume hood or an isolated place such as a glovebox. The researcher(s) must be equipped with an HCN detector during the experiments to avoid any inhalation of HCN. The addition of KCN powder directly to the acid solution is strictly prohibited. Please refer toSupplementary Note 5for detailed information on the laboratory safety guidelines for cyanide.
In situ SD quantification was performed in a two-compartment electrochemical H-type cell contacted by a Nafion 115 membrane (DuPont), which separated the working electrode from the counter and reference electrodes. Two graphite rods (6.5 mm in diameter) were used as the working and counter electrodes, and the Ag/AgCl electrode was used as the reference electrode. The aqueous cyanide solution was prepared with deaerated ultrapure water and KCN (≥96%, Sigma-Aldrich).
Cyanide poisoning was conducted in an Ar-filled chamber to prevent any undesirable O2 dissolution in the electrolyte and subsequent competitive adsorption of O2 on the samples. First, the catalyst powder (30 mg) was dispersed in the electrolyte and mechanically stirred with a magnetic bar at a rotation speed of ca. 300 rpm to prevent sedimentation. Prior to the analysis, predissolved O2 in the electrolyte was degassed by Ar purging for 30 min at the OCP. Thereafter, with continuous Ar purging and mechanical stirring, a potential of 0.5 VRHE was applied to the working electrode until the measured j reached zero (ca. for 20 min), where the catalyst particles collided and electrified during convective motion. After stabilization for 10 min at a specific potential (typically 1.0 VRHE, but 0.6 VRHE for Pt nanoparticles to prevent the formation of surface oxide), aqueous cyanide solution (8 mM) was injected into both compartments, and the initial CCN–electrolyte value was adjusted to 200 μM. Catalyst poisoning was carried out for 15 min at an identical potential for the stabilization step, and the compartment was fully closed without any gas purging to prevent cyanide loss via the degasification of HCN(aq). The ΔCCN– value in the filtered electrolyte was measured after dilution with deionized water (electrolyte/water = 1/19 volume ratio). For spectrophotometric titration of ΔCCN–electrolyte, the diluted electrolyte (400 μL) was mixed with 30 mM p-nitrobenzaldehyde solution (400 μL; ≥95%, TGI), 0.6 mM tetrazolium blue chloride solution (400 μL; Sigma-Aldrich), methanol (760 μL; ≥99.8%, Sigma-Aldrich), and 0.2 M NaOH (40 μL; 99.99%, trace metals basis, Sigma-Aldrich). It is of note that a fixed amount of solution added for titration was not considered in the dilution ratio. Subsequently, the color change to purple was monitored at 520 nm using an UV–vis spectrophotometer (Genesis 400, Thermo-Fisher Scientific) with reference to a blank electrolyte.46,47 The cyanide-poisoned catalyst filtered after the poisoning protocol was rinsed with deionized water, and its ORR activity was measured to calculate the difference in j values between the pristine and poisoned catalysts (i.e., Δj).
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT; NRF-2019M3D1A1079309 and 2020R1A2C4002233) and by the Korea Institute of Science and Technology (KIST) institutional program. Experiments at PLS-II were supported in part by MSIT and POSTECH.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.1c00074.
Physicochemical characteristics of Fe0.5NC; ORR activity measurement protocol for in situ SD quantification using cyanide probe; non-site-specific adsorption of cyanide on Fe–Nx moieties; different cyanide adsorption modes of heterogeneous Pt sites with different surface coverages; spectrophotometric titration results; ORR polarization curves before and after cyanide poisoning; CO chemisorption and NO2–-stripping results; laboratory safety guidelines (PDF)
Author Contributions
○ G.B. and H.K. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Ertl G. Reactions at Surfaces: From Atoms to Complexity (Nobel Lecture). Angew. Chem., Int. Ed. 2008, 47 (19), 3524–3535. 10.1002/anie.200800480. [DOI] [PubMed] [Google Scholar]
- Schauermann S.; Nilius N.; Shaikhutdinov S.; Freund H.-J. Nanoparticles for Heterogeneous Catalysis: New Mechanistic Insights. Acc. Chem. Res. 2013, 46 (8), 1673–1681. 10.1021/ar300225s. [DOI] [PubMed] [Google Scholar]
- Kozuch S.; Martin J. M. L. “Turning Over” Definitions in Catalytic Cycles. ACS Catal. 2012, 2 (12), 2787–2794. 10.1021/cs3005264. [DOI] [Google Scholar]
- Zagal J. H.; Specchia S.; Atanassov P. Mapping Transition Metal-MN4 Macrocyclic Complex Catalysts Performance for the Critical Reactivity Descriptors. Curr. Opin. Electrochem. 2021, 27, 100683. 10.1016/j.coelec.2020.100683. [DOI] [Google Scholar]
- Burcham L. J.; Briand L. E.; Wachs I. E. Quantification of Active Sites for the Determination of Methanol Oxidation Turn-Over Frequencies Using Methanol Chemisorption and In Situ Infrared Techniques. 2. Bulk Metal Oxide Catalysts. Langmuir 2001, 17 (20), 6175–6184. 10.1021/la010010t. [DOI] [Google Scholar]
- Li X.; Wei S.; Zhang Z.; Zhang Y.; Wang Z.; Su Q.; Gao X. Quantification of the Active Site Density and Turnover Frequency for Soot Combustion with O2 on Cr Doped CeO2. Catal. Today 2011, 175 (1), 112–116. 10.1016/j.cattod.2011.03.057. [DOI] [Google Scholar]
- Sullivan M. M.; Held J. T.; Bhan A. Structure and Site Evolution of Molybdenum Carbide Catalysts Upon Exposure to Oxygen. J. Catal. 2015, 326, 82–91. 10.1016/j.jcat.2015.03.011. [DOI] [Google Scholar]
- Bergeret G.; Gallezot P.. Particle Size and Dispersion Measurements. In Handbook of Heterogeneous Catalysis; Ertl G., Knözinger H., Schüth F., Weitkamp J., Eds.; Wiley-VCH: Weinheim, Germany, 2008; Vol. 1, pp 738–765. [Google Scholar]
- Goodwin J. G. Jr.; Kim S.; Rhodes W. D.. Turnover Frequencies in Metal Catalysis: Meanings, Functionalities and Relationships. In Catalysis; Spivey J. J., Roberts G. W., Eds.; The Royal Society of Chemistry: London, 2004; Vol. 17, pp 320–348. [Google Scholar]
- Rudi S.; Cui C.; Gan L.; Strasser P. Comparative Study of the Electrocatalytically Active Surface Areas (ECSAs) of Pt Alloy Nanoparticles Evaluated by Hupd and CO-Stripping Voltammetry. Electrocatalysis 2014, 5 (4), 408–418. 10.1007/s12678-014-0205-2. [DOI] [Google Scholar]
- Lorenz W. J.; Hermann H. D.; Wüthrich N.; Hilbert F. The Formation of Monolayer Metal Films on Electrodes. J. Electrochem. Soc. 1974, 121 (9), 1167. 10.1149/1.2402005. [DOI] [Google Scholar]
- Herrero E.; Buller L. J.; Abruña H. D. Underpotential Deposition at Single Crystal Surfaces of Au, Pt, Ag and Other Materials. Chem. Rev. 2001, 101 (7), 1897–1930. 10.1021/cr9600363. [DOI] [PubMed] [Google Scholar]
- Woods R. Hydrogen Adsorption on Platinum, Iridium and Rhodium Electrodes at Reduced Temperatures and the Determination of Real Surface Area. J. Electroanal. Chem. Interfacial Electrochem. 1974, 49 (2), 217–226. 10.1016/S0022-0728(74)80229-9. [DOI] [Google Scholar]
- Binninger T.; Fabbri E.; Kötz R.; Schmidt T. J. Determination of the Electrochemically Active Surface Area of Metal-Oxide Supported Platinum Catalyst. J. Electrochem. Soc. 2014, 161 (3), H121–H128. 10.1149/2.055403jes. [DOI] [Google Scholar]
- Brightman E.; Hinds G.; O’Malley R. In Situ Measurement of Active Catalyst Surface Area in Fuel Cell Stacks. J. Power Sources 2013, 242, 244–254. 10.1016/j.jpowsour.2013.05.046. [DOI] [Google Scholar]
- Dao D. V.; Adilbish G.; Le T. D.; Lee I.-H.; Yu Y.-T. Triple Phase Boundary and Power Density Enhancement in PEMFCs of a Pt/C Electrode with Double Catalyst Layers. RSC Adv. 2019, 9 (27), 15635–15641. 10.1039/C9RA01741K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou H.; Wang D.; Li Y. How to Select Effective Electrocatalysts: Nano or Single Atom?. Nano Select 2021, 2 (3), 492–511. 10.1002/nano.202000239. [DOI] [Google Scholar]
- Proietti E.; Jaouen F.; Lefèvre M.; Larouche N.; Tian J.; Herranz J.; Dodelet J.-P. Iron-Based Cathode Catalyst with Enhanced Power Density in Polymer Electrolyte Membrane Fuel Cells. Nat. Commun. 2011, 2 (1), 416. 10.1038/ncomms1427. [DOI] [PubMed] [Google Scholar]
- Chung H. T.; Cullen D. A.; Higgins D.; Sneed B. T.; Holby E. F.; More K. L.; Zelenay P. Direct Atomic-Level Insight into the Active Sites of a High-Performance PGM-Free ORR Catalyst. Science 2017, 357 (6350), 479. 10.1126/science.aan2255. [DOI] [PubMed] [Google Scholar]
- Li J.; Chen M.; Cullen D. A.; Hwang S.; Wang M.; Li B.; Liu K.; Karakalos S.; Lucero M.; Zhang H.; Lei C.; Xu H.; Sterbinsky G. E.; Feng Z.; Su D.; More K. L.; Wang G.; Wang Z.; Wu G. Atomically Dispersed Manganese Catalysts for Oxygen Reduction in Proton-Exchange Membrane Fuel Cells. Nat. Catal. 2018, 1 (12), 935–945. 10.1038/s41929-018-0164-8. [DOI] [Google Scholar]
- Shi Z.; Yang W.; Gu Y.; Liao T.; Sun Z. Metal-Nitrogen-Doped Carbon Materials as Highly Efficient Catalysts: Progress and Rational Design. Adv. Sci. 2020, 7 (15), 2001069. 10.1002/advs.202001069. [DOI] [Google Scholar]
- Lei C.; Wang Y.; Hou Y.; Liu P.; Yang J.; Zhang T.; Zhuang X.; Chen M.; Yang B.; Lei L.; Yuan C.; Qiu M.; Feng X. Efficient Alkaline Hydrogen Evolution on Atomically Dispersed Ni–Nx Species Anchored Porous Carbon with Embedded Ni Nanoparticles by Accelerating Water Dissociation Kinetics. Energy Environ. Sci. 2019, 12 (1), 149–156. 10.1039/C8EE01841C. [DOI] [Google Scholar]
- Gu J.; Hsu C.-S.; Bai L.; Chen H. M.; Hu X. Atomically Dispersed Fe3+ Sites Catalyze Efficient CO2 Electroreduction to CO. Science 2019, 364 (6445), 1091. 10.1126/science.aaw7515. [DOI] [PubMed] [Google Scholar]
- Li J.; Ghoshal S.; Liang W.; Sougrati M.-T.; Jaouen F.; Halevi B.; McKinney S.; McCool G.; Ma C.; Yuan X.; Ma Z.-F.; Mukerjee S.; Jia Q. Structural and Mechanistic Basis for the High Activity of Fe–N–C Catalysts toward Oxygen Reduction. Energy Environ. Sci. 2016, 9 (7), 2418–2432. 10.1039/C6EE01160H. [DOI] [Google Scholar]
- Jia Q.; Ramaswamy N.; Tylus U.; Strickland K.; Li J.; Serov A.; Artyushkova K.; Atanassov P.; Anibal J.; Gumeci C.; Barton S. C.; Sougrati M.-T.; Jaouen F.; Halevi B.; Mukerjee S. Spectroscopic Insights into the Nature of Active Sites in Iron–Nitrogen–Carbon Electrocatalysts for Oxygen Reduction in Acid. Nano Energy 2016, 29, 65–82. 10.1016/j.nanoen.2016.03.025. [DOI] [Google Scholar]
- Luo F.; Roy A.; Silvioli L.; Cullen D. A.; Zitolo A.; Sougrati M. T.; Oguz I. C.; Mineva T.; Teschner D.; Wagner S.; Wen J.; Dionigi F.; Kramm U. I.; Rossmeisl J.; Jaouen F.; Strasser P. P-Block Single-Metal-Site Tin/Nitrogen-Doped Carbon Fuel Cell Cathode Catalyst for Oxygen Reduction Reaction. Nat. Mater. 2020, 19, 1215–1223. 10.1038/s41563-020-0717-5. [DOI] [PubMed] [Google Scholar]
- Li J.; Sougrati M. T.; Zitolo A.; Ablett J. M.; Oğuz I. C.; Mineva T.; Matanovic I.; Atanassov P.; Huang Y.; Zenyuk I.; Di Cicco A.; Kumar K.; Dubau L.; Maillard F.; Dražić G.; Jaouen F. Identification of Durable and Non-Durable FeNx Sites in Fe–N–C Materials for Proton Exchange Membrane Fuel Cells. Nat. Catal. 2021, 4 (1), 10–19. 10.1038/s41929-020-00545-2. [DOI] [Google Scholar]
- Sato S.; Namba K.; Hara K.; Fukuoka A.; Murakoshi K.; Uosaki K.; Ikeda K. Kinetic Behavior of Catalytic Active Sites Connected with a Conducting Surface through Various Electronic Coupling. J. Phys. Chem. C 2016, 120 (4), 2159–2165. 10.1021/acs.jpcc.5b09364. [DOI] [Google Scholar]
- Toyama T.; Sato S.; Motobayashi K.; Uosaki K.; Ikeda K. A Rotating Disk Electrode Study on Catalytic Activity of Iron(II) Phthalocyanine-Modified Electrodes for Oxygen Reduction in Acidic Media. J. Solid State Electrochem. 2021, 25 (1), 141–147. 10.1007/s10008-019-04461-9. [DOI] [Google Scholar]
- Sahraie N. R.; Kramm U. I.; Steinberg J.; Zhang Y.; Thomas A.; Reier T.; Paraknowitsch J.-P.; Strasser P. Quantifying the Density and Utilization of Active Sites in Non-Precious Metal Oxygen Electroreduction Catalysts. Nat. Commun. 2015, 6 (1), 8618. 10.1038/ncomms9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malko D.; Kucernak A.; Lopes T. In Situ Electrochemical Quantification of Active Sites in Fe–N/C Non-Precious Metal Catalysts. Nat. Commun. 2016, 7 (1), 13285. 10.1038/ncomms13285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju W.; Bagger A.; Hao G.-P.; Varela A. S.; Sinev I.; Bon V.; Roldan Cuenya B.; Kaskel S.; Rossmeisl J.; Strasser P. Understanding Activity and Selectivity of Metal-Nitrogen-Doped Carbon Catalysts for Electrochemical Reduction of CO2. Nat. Commun. 2017, 8 (1), 944. 10.1038/s41467-017-01035-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo F.; Choi C. H.; Primbs M. J. M.; Ju W.; Li S.; Leonard N. D.; Thomas A.; Jaouen F.; Strasser P. Accurate Evaluation of Active-Site Density (SD) and Turnover Frequency (TOF) of PGM-Free Metal–Nitrogen-Doped Carbon (MNC) Electrocatalysts using CO cryo Adsorption. ACS Catal. 2019, 9 (6), 4841–4852. 10.1021/acscatal.9b00588. [DOI] [Google Scholar]
- Rosca V.; Duca M.; de Groot M. T.; Koper M. T. M. Nitrogen Cycle Electrocatalysis. Chem. Rev. 2009, 109 (6), 2209–2244. 10.1021/cr8003696. [DOI] [PubMed] [Google Scholar]
- Einsle O.; Messerschmidt A.; Huber R.; Kroneck P. M. H.; Neese F. Mechanism of the Six-Electron Reduction of Nitrite to Ammonia by Cytochrome c Nitrite Reductase. J. Am. Chem. Soc. 2002, 124 (39), 11737–11745. 10.1021/ja0206487. [DOI] [PubMed] [Google Scholar]
- de Groot M. T.; Merkx M.; Koper M. T. M. Heme Release in Myoglobin–DDAB Films and Its Role in Electrochemical NO Reduction. J. Am. Chem. Soc. 2005, 127 (46), 16224–16232. 10.1021/ja0546572. [DOI] [PubMed] [Google Scholar]
- de Groot M. T.; Merkx M.; Wonders A. H.; Koper M. T. M. Electrochemical Reduction of NO by Hemin Adsorbed at Pyrolitic Graphite. J. Am. Chem. Soc. 2005, 127 (20), 7579–7586. 10.1021/ja051151a. [DOI] [PubMed] [Google Scholar]
- de Groot M. T.; Merkx M.; Koper M. T. M. Bioinspired Electrocatalytic Reduction of Nitric Oxide by Immobilized Heme Groups. C. R. Chim. 2007, 10 (4), 414–420. 10.1016/j.crci.2006.11.005. [DOI] [Google Scholar]
- Primbs M.; Sun Y.; Roy A.; Malko D.; Mehmood A.; Sougrati M.-T.; Blanchard P.-Y.; Granozzi G.; Kosmala T.; Daniel G.; Atanassov P.; Sharman J.; Durante C.; Kucernak A.; Jones D.; Jaouen F.; Strasser P. Establishing Reactivity Descriptors for Platinum Group Metal (PGM)-Free Fe–N–C Catalysts for PEM Fuel Cells. Energy Environ. Sci. 2020, 13 (8), 2480–2500. 10.1039/D0EE01013H. [DOI] [Google Scholar]
- Li Y.; Zhou W.; Wang H.; Xie L.; Liang Y.; Wei F.; Idrobo J.-C.; Pennycook S. J.; Dai H. An Oxygen Reduction Electrocatalyst Based on Carbon Nanotube–Graphene Complexes. Nat. Nanotechnol. 2012, 7 (6), 394–400. 10.1038/nnano.2012.72. [DOI] [PubMed] [Google Scholar]
- Tylus U.; Jia Q.; Strickland K.; Ramaswamy N.; Serov A.; Atanassov P.; Mukerjee S. Elucidating Oxygen Reduction Active Sites in Pyrolyzed Metal–Nitrogen Coordinated Non-Precious-Metal Electrocatalyst Systems. J. Phys. Chem. C 2014, 118 (17), 8999–9008. 10.1021/jp500781v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung M. W.; Chon G.; Kim H.; Jaouen F.; Choi C. H. Electrochemical Evidence for Two Sub-Families of FeNxCy Moieties with Concentration-Dependent Cyanide Poisoning. ChemElectroChem 2018, 5 (14), 1880–1885. 10.1002/celc.201800067. [DOI] [Google Scholar]
- Choi C. H.; Lim H.-K.; Chung M. W.; Chon G.; Ranjbar Sahraie N.; Altin A.; Sougrati M.-T.; Stievano L.; Oh H. S.; Park E. S.; Luo F.; Strasser P.; Dražić G.; Mayrhofer K. J. J.; Kim H.; Jaouen F. The Achilles’ Heel of Iron-Based Catalysts During Oxygen Reduction in an Acidic Medium. Energy Environ. Sci. 2018, 11 (11), 3176–3182. 10.1039/C8EE01855C. [DOI] [Google Scholar]
- Zitolo A.; Goellner V.; Armel V.; Sougrati M.-T.; Mineva T.; Stievano L.; Fonda E.; Jaouen F. Identification of Catalytic Sites for Oxygen Reduction in Iron- and Nitrogen-Doped Graphene Materials. Nat. Mater. 2015, 14 (9), 937–942. 10.1038/nmat4367. [DOI] [PubMed] [Google Scholar]
- Gupta S.; Fierro C.; Yeager E. The Effects of Cyanide on the Electrochemical Properties of Transition Metal Macrocycles for Oxygen Reduction in Alkaline Solutions. J. Electroanal. Chem. Interfacial Electrochem. 1991, 306 (1), 239–250. 10.1016/0022-0728(91)85233-F. [DOI] [Google Scholar]
- Pitschmann V.; Kobliha Z.; Tušarová I. A Simple Spectrophotometric Determination of Cyanides by p-Nitrobenzaldehyde and Tetrazolium Blue. Adv. Mil. Technol. 2011, 6, 19–27. [Google Scholar]
- Guilbault G. G.; Kramer D. N. Ultra Sensitive, Specific Method for Cyanide Using p-Nitrobenzaldehyde and o-Dinitrobenzene. Anal. Chem. 1966, 38 (7), 834–836. 10.1021/ac60239a009. [DOI] [Google Scholar]
- Kumar R.; Saha S.; Dhaka S.; Kurade M. B.; Kang C. U.; Baek S. H.; Jeon B.-H. Remediation of Cyanide-Contaminated Environments Through Microbes and Plants: A Review of Current Knowledge and Future Perspectives. Geosyst. Eng. 2017, 20 (1), 28–40. 10.1080/12269328.2016.1218303. [DOI] [Google Scholar]
- Yoshikawa S.; O’Keeffe D. H.; Caughey W. S. Investigations of Cyanide as an Infrared Probe of Hemeprotein Ligand Binding Sites. J. Biol. Chem. 1985, 260 (6), 3518–3528. 10.1016/S0021-9258(19)83653-0. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Silvioli L.; Sahraie N. R.; Ju W.; Li J.; Zitolo A.; Li S.; Bagger A.; Arnarson L.; Wang X.; Moeller T.; Bernsmeier D.; Rossmeisl J.; Jaouen F.; Strasser P. Activity–Selectivity Trends in the Electrochemical Production of Hydrogen Peroxide over Single-Site Metal–Nitrogen–Carbon Catalysts. J. Am. Chem. Soc. 2019, 141 (31), 12372–12381. 10.1021/jacs.9b05576. [DOI] [PubMed] [Google Scholar]
- Li J.; Pršlja P.; Shinagawa T.; Martín Fernández A. J.; Krumeich F.; Artyushkova K.; Atanassov P.; Zitolo A.; Zhou Y.; García-Muelas R.; López N.; Pérez-Ramírez J.; Jaouen F. Volcano Trend in Electrocatalytic CO2 Reduction Activity over Atomically Dispersed Metal Sites on Nitrogen-Doped Carbon. ACS Catal. 2019, 9 (11), 10426–10439. 10.1021/acscatal.9b02594. [DOI] [Google Scholar]
- Choi C. H.; Kim M.; Kwon H. C.; Cho S. J.; Yun S.; Kim H.-T.; Mayrhofer K. J. J.; Kim H.; Choi M. Tuning Selectivity of Electrochemical Reactions by Atomically Dispersed Platinum Catalyst. Nat. Commun. 2016, 7 (1), 10922. 10.1038/ncomms10922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H. C.; Kim M.; Grote J.-P.; Cho S. J.; Chung M. W.; Kim H.; Won D. H.; Zeradjanin A. R.; Mayrhofer K. J. J.; Choi M.; Kim H.; Choi C. H. Carbon Monoxide as a Promoter of Atomically Dispersed Platinum Catalyst in Electrochemical Hydrogen Evolution Reaction. J. Am. Chem. Soc. 2018, 140 (47), 16198–16205. 10.1021/jacs.8b09211. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Zhao Y.; Guo X.; Chen C.; Dong C.-L.; Liu R.-S.; Han C.-P.; Li Y.; Gogotsi Y.; Wang G. Single Platinum Atoms Immobilized on an MXene as an Efficient Catalyst for the Hydrogen Evolution Reaction. Nat. Catal. 2018, 1 (12), 985–992. 10.1038/s41929-018-0195-1. [DOI] [Google Scholar]
- Liu J.; Jiao M.; Lu L.; Barkholtz H. M.; Li Y.; Wang Y.; Jiang L.; Wu Z.; Liu D.-j.; Zhuang L.; Ma C.; Zeng J.; Zhang B.; Su D.; Song P.; Xing W.; Xu W.; Wang Y.; Jiang Z.; Sun G. High Performance Platinum Single Atom Electrocatalyst for Oxygen Reduction Reaction. Nat. Commun. 2017, 8 (1), 15938. 10.1038/ncomms15938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng N.; Stambula S.; Wang D.; Banis M. N.; Liu J.; Riese A.; Xiao B.; Li R.; Sham T.-K.; Liu L.-M.; Botton G. A.; Sun X. Platinum Single-Atom and Cluster Catalysis of the Hydrogen Evolution Reaction. Nat. Commun. 2016, 7 (1), 13638. 10.1038/ncomms13638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B.; Wu D.-Y.; Mao B.-W.; Tian Z.-Q. Surface-Enhanced Raman Study of Cyanide Adsorption at the Platinum Surface. J. Phys. Chem. B 2003, 107 (12), 2752–2758. 10.1021/jp0258107. [DOI] [Google Scholar]
- Stuhlmann C.; Villegas I.; Weaver M. J. Scanning Tunneling Microscopy and Infrared Spectroscopy as Combined In Situ Probes of Electrochemical Adlayer Structure. Cyanide on Pt(111). Chem. Phys. Lett. 1994, 219 (3), 319–324. 10.1016/0009-2614(94)87064-0. [DOI] [Google Scholar]
- Shao M.; Peles A.; Shoemaker K. Electrocatalysis on Platinum Nanoparticles: Particle Size Effect on Oxygen Reduction Reaction Activity. Nano Lett. 2011, 11 (9), 3714–3719. 10.1021/nl2017459. [DOI] [PubMed] [Google Scholar]
- Merzougui B.; Hachimi A.; Akinpelu A.; Bukola S.; Shao M. A Pt-Free Catalyst for Oxygen Reduction Reaction Based on Fe–N Multiwalled Carbon Nanotube Composites. Electrochim. Acta 2013, 107, 126–132. 10.1016/j.electacta.2013.06.016. [DOI] [Google Scholar]
- Sgarbi R.; Kumar K.; Jaouen F.; Zitolo A.; Ticianelli E. A.; Maillard F. Oxygen Reduction Reaction Mechanism and Kinetics on M-NxCy and M@N-C Active Sites Present in Model M-N-C Catalysts under Alkaline and Acidic Conditions. J. Solid State Electrochem. 2021, 25 (1), 45–56. 10.1007/s10008-019-04436-w. [DOI] [Google Scholar]
- Meng H.; Jaouen F.; Proietti E.; Lefèvre M.; Dodelet J.-P. pH-Effect on Oxygen Reduction Activity of Fe-Based Electro-Catalysts. Electrochem. Commun. 2009, 11 (10), 1986–1989. 10.1016/j.elecom.2009.08.035. [DOI] [Google Scholar]
- Chen D.; Li Y.; Liao S.; Su D.; Song H.; Li Y.; Yang L.; Li C. Ultra-High-Performance Core–Shell Structured Ru@Pt/C Catalyst Prepared by a Facile Pulse Electrochemical Deposition Method. Sci. Rep. 2015, 5 (1), 11604. 10.1038/srep11604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaouen F.; Jones D.; Coutard N.; Artero V.; Strasser P.; Kucernak A. Toward Platinum Group Metal-Free Catalysts for Hydrogen/Air Proton-Exchange Membrane Fuel Cells. Johnson Matthey Technol. Rev. 2018, 62 (2), 231–255. 10.1595/205651318X696828. [DOI] [Google Scholar]
- Patel A. M.; Ringe S.; Siahrostami S.; Bajdich M.; Nørskov J. K.; Kulkarni A. R. Theoretical Approaches to Describing the Oxygen Reduction Reaction Activity of Single-Atom Catalysts. J. Phys. Chem. C 2018, 122 (51), 29307–29318. 10.1021/acs.jpcc.8b09430. [DOI] [Google Scholar]
- Vijay S.; Gauthier J. A.; Heenen H. H.; Bukas V. J.; Kristoffersen H. H.; Chan K. Dipole-Field Interactions Determine the CO2 Reduction Activity of 2D Fe–N–C Single-Atom Catalysts. ACS Catal. 2020, 10 (14), 7826–7835. 10.1021/acscatal.0c01375. [DOI] [Google Scholar]
- Yao M.; Shi Z.; Zhang P.; Ong W.-J.; Jiang J.; Ching W.-Y.; Li N. Density Functional Theory Study of Single Metal Atoms Embedded into MBene for Electrocatalytic Conversion of N2 to NH3. ACS Appl. Nano Mater. 2020, 3 (10), 9870–9879. 10.1021/acsanm.0c01922. [DOI] [Google Scholar]
- Wang Y.-C.; Lai Y.-J.; Song L.; Zhou Z.-Y.; Liu J.-G.; Wang Q.; Yang X.-D.; Chen C.; Shi W.; Zheng Y.-P.; Rauf M.; Sun S.-G. S-Doping of an Fe/N/C ORR Catalyst for Polymer Electrolyte Membrane Fuel Cells with High Power Density. Angew. Chem., Int. Ed. 2015, 54 (34), 9907–9910. 10.1002/anie.201503159. [DOI] [PubMed] [Google Scholar]
- Hou Y.; Qiu M.; Kim M. G.; Liu P.; Nam G.; Zhang T.; Zhuang X.; Yang B.; Cho J.; Chen M.; Yuan C.; Lei L.; Feng X. Atomically Dispersed Nickel–Nitrogen–Sulfur Species Anchored on Porous Carbon Nanosheets for Efficient Water Oxidation. Nat. Commun. 2019, 10 (1), 1392. 10.1038/s41467-019-09394-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrandon M.; Kropf A. J.; Myers D. J.; Artyushkova K.; Kramm U.; Bogdanoff P.; Wu G.; Johnston C. M.; Zelenay P. Multitechnique Characterization of a Polyaniline–Iron–Carbon Oxygen Reduction Catalyst. J. Phys. Chem. C 2012, 116 (30), 16001–16013. 10.1021/jp302396g. [DOI] [Google Scholar]
- Ramaswamy N.; Tylus U.; Jia Q.; Mukerjee S. Activity Descriptor Identification for Oxygen Reduction on Nonprecious Electrocatalysts: Linking Surface Science to Coordination Chemistry. J. Am. Chem. Soc. 2013, 135 (41), 15443–15449. 10.1021/ja405149m. [DOI] [PubMed] [Google Scholar]
- Zitolo A.; Ranjbar-Sahraie N.; Mineva T.; Li J.; Jia Q.; Stamatin S.; Harrington G. F.; Lyth S. M.; Krtil P.; Mukerjee S.; Fonda E.; Jaouen F. Identification of Catalytic Sites in Cobalt-Nitrogen-Carbon Materials for the Oxygen Reduction Reaction. Nat. Commun. 2017, 8 (1), 957. 10.1038/s41467-017-01100-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin P.; Yao T.; Wu Y.; Zheng L.; Lin Y.; Liu W.; Ju H.; Zhu J.; Hong X.; Deng Z.; Zhou G.; Wei S.; Li Y. Single Cobalt Atoms with Precise N-Coordination as Superior Oxygen Reduction Reaction Catalysts. Angew. Chem., Int. Ed. 2016, 55 (36), 10800–10805. 10.1002/anie.201604802. [DOI] [PubMed] [Google Scholar]
- Wei H.; Huang K.; Zhang L.; Ge B.; Wang D.; Lang J.; Ma J.; Wang D.; Zhang S.; Li Q.; Zhang R.; Hussain N.; Lei M.; Liu L.-M.; Wu H. Ice Melting to Release Reactants in Solution Syntheses. Angew. Chem., Int. Ed. 2018, 57 (13), 3354–3359. 10.1002/anie.201711128. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Chenitz R.; Lefèvre M.; Sun S.; Dodelet J.-P. Is Iron Involved in the Lack of Stability of Fe/N/C Electrocatalysts Used to Reduce Oxygen at the Cathode of PEM Fuel Cells?. Nano Energy 2016, 29, 111–125. 10.1016/j.nanoen.2016.02.038. [DOI] [Google Scholar]
- Kida K.; Okita M.; Fujita K.; Tanaka S.; Miyake Y. Formation of High Crystalline ZIF-8 in an Aqueous Solution. CrystEngComm 2013, 15 (9), 1794–1801. 10.1039/c2ce26847g. [DOI] [Google Scholar]
- Guo D.; Shibuya R.; Akiba C.; Saji S.; Kondo T.; Nakamura J. Active Sites of Nitrogen-Doped Carbon Materials for Oxygen Reduction Reaction Clarified Using Model Catalysts. Science 2016, 351 (6271), 361. 10.1126/science.aad0832. [DOI] [PubMed] [Google Scholar]
- Kocha S. S.; Shinozaki K.; Zack J. W.; Myers D. J.; Kariuki N. N.; Nowicki T.; Stamenkovic V.; Kang Y.; Li D.; Papageorgopoulos D. Best Practices and Testing Protocols for Benchmarking ORR Activities of Fuel Cell Electrocatalysts Using Rotating Disk Electrode. Electrocatalysis 2017, 8 (4), 366–374. 10.1007/s12678-017-0378-6. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.