

Abstract
Background
Roxadustat is an orally active hypoxia-inducible factor prolyl hydroxylase inhibitor for the treatment of chronic kidney disease (CKD) anemia.
Methods
This Phase 3, multicenter, randomized, double-blind, placebo-controlled study examined patients with Stages 3–5 CKD, not on dialysis (NCT01887600). Patients were randomized (2:1) to oral roxadustat or placebo three times weekly for 52–104 weeks. This study examined two primary efficacy endpoints: European Union (European Medicines Agency)—hemoglobin (Hb) response, defined as Hb ≥11.0 g/dL that increased from baseline (BL) by ≥1.0 g/dL in patients with Hb >8.0 g/dL or ≥2.0 g/dL in patients with BL Hb ≤8.0 g/dL, without rescue therapy, during the first 24 weeks of treatment; US Food and Drug Administration—change in Hb from BL to the average Hb level during Weeks 28–52, regardless of rescue therapy. Secondary efficacy endpoints and safety were examined.
Results
A total of 594 patients were analyzed (roxadustat: 391; placebo: 203). Superiority of roxadustat versus placebo was demonstrated for both primary efficacy endpoints: Hb response [odds ratio = 34.74, 95% confidence interval (CI) 20.48–58.93] and change in Hb from BL [roxadustat – placebo: +1.692 (95% CI 1.52–1.86); both P < 0.001]. Superiority of roxadustat was demonstrated for low-density lipoprotein cholesterol change from BL, and time to first use of rescue medication (both P < 0.001). The incidences of treatment-emergent adverse events were comparable between groups (roxadustat: 87.7%, placebo: 86.7%).
Conclusions
Roxadustat demonstrated superior efficacy versus placebo in terms of both Hb response rate and change in Hb from BL. The safety profiles of roxadustat and placebo were comparable.
Keywords: anemia, chronic kidney disease, iron, non-dialysis, roxadustat
Graphical Abstract

KEY LEARNING POINTS
What is already known about this subject?
anemia is a common complication of chronic kidney disease (CKD); iron therapy (oral or intravenous) is the standard first-line treatment for CKD anemia, while erythropoiesis-stimulating agents (ESAs) are available for the treatment of CKD anemia that cannot be corrected by iron therapy alone;
although iron therapy and ESAs are mainstays of the current CKD anemia treatment paradigm, studies have highlighted shortcomings related to the convenience, adverse events and efficacy of these treatments, suggesting that novel treatments should be explored in this patient population; and
roxadustat is an orally active hypoxia-inducible factor prolyl hydroxylase inhibitor (HIF-PHI) that has shown efficacy and safety in Phases 2 and 3 trials in CKD patients with anemia who are nondialysis-dependent (NDD) and those who are dialysis-dependent.
What this study adds?
this study was a Phase 3, multicenter, randomized, double-blind, placebo-controlled study in mostly European patients with anemia who have Stage 3, 4 or 5 CKD who were not on dialysis at the time of randomization;
superiority of roxadustat versus placebo was demonstrated in terms of response rate to treatment during the first 24 weeks of treatment and hemoglobin (Hb) change from baseline to the average Hb of Weeks 28–52; sensitivity and subgroup analyses confirmed these primary analyses in European patients; and
the safety profile of roxadustat in this study was generally comparable to placebo, and markers of both lipid and iron metabolism were improved in roxadustat-treated patients.
What impact this may have on practice or policy?
roxadustat is an orally active HIF-PHI that has shown efficacy and safety in Phase 3 trials in several different cohorts of CKD patients with anemia who are NDD; and
roxadustat has also demonstrated improved iron availability and improved lipid metabolism in this pre-dialysis patient population.
INTRODUCTION
Anemia is a common complication observed in patients who have chronic kidney disease (CKD). While the pathogenesis of CKD is multifactorial, decreased synthesis of erythropoietin by the kidneys due to impaired oxygen sensing [1, 2] and an altered iron metabolism are important etiologic factors [3]. Currently, iron therapy [oral or intravenous (IV)] is the standard first-line treatment for CKD anemia, while erythropoiesis-stimulating agents (ESAs) (IV and subcutaneous) are available for the treatment of CKD anemia that cannot be corrected by iron therapy alone [4]. Although iron therapy and ESAs are mainstays of the current CKD anemia treatment paradigm, studies have highlighted shortcomings related to the convenience (e.g. mode of administration), safety [e.g. increased risk of cardiovascular (CV) complications, mainly thromboembolic] and efficacy of these treatments in some patients with nondialysis-dependent (NDD) CKD, suggesting that novel treatments may benefit this patient population [5–8]. Hypoxia-inducible factor prolyl hydroxylase inhibitors (HIF-PHIs) represent a new strategy to increase hemoglobin (Hb) levels by activating the body’s natural response to hypoxia independent of cellular oxygen levels [9–11].
Roxadustat is an orally administered HIF-PHI that has shown efficacy and safety in Phases 2 and 3 trials in patients with CKD who are either dialysis-dependent (DD) [12–18] or NDD [16, 19–23]. In recent Phase 3 studies performed in Japanese and Chinese patients with CKD, roxadustat was shown to be superior compared with placebo [21], and noninferior compared with traditional ESAs [12, 15], in correcting and maintaining Hb. Roxadustat has also been shown to increase serum iron levels and iron absorption [12, 13, 15, 21, 23], increase transferrin levels and total iron-binding capacity [12, 15, 21, 23], decrease hepcidin [12, 15, 21, 23] and reduce low-density lipoprotein (LDL) cholesterol [12, 15, 21]. Roxadustat was recently approved in China and Japan for the treatment of both DD- and NDD-CKD anemia. This global study, consisting mostly of European patients, was conducted to evaluate the efficacy and safety of roxadustat in the treatment of anemia in individuals with NDD-CKD.
MATERIALS AND METHODS
Study design
This was a Phase 3, multicenter, randomized, double-blind, placebo-controlled study in anemic patients with Stage 3, 4 or 5 CKD who were not on dialysis at the time of randomization (ALPS; ClinicalTrials.gov Identifier: NCT01887600). In this study, anemia was defined as mean Hb ≤10.0 g/dL per repeated screening measurements. Patients were randomized (2:1) to oral roxadustat or placebo three times weekly (TIW) for 52–104 weeks. Randomization was stratified by the following four factors: region (Western Europe versus rest of the world); screening Hb values (≤8.0 g/dL versus >8.0 g/dL); history of CV, cerebrovascular or thromboembolic diseases (yes versus no); and screening estimated glomerular filtration rate (eGFR) (<30 mL/min/1.73 m2 versus ≥30 mL/min/1.73 m2).
This study consisted of three study periods (Figure 1), including screening (up to 6 weeks), a treatment period and a follow-up period. Within the treatment period, there was a correction period, in which the study drug was initially dosed for Hb correction until patients achieved a target Hb value of ≥11.0 g/dL and a Hb increase from baseline (BL) of ≥1.0 g/dL at two consecutive study visits separated by at least 5 days, and a subsequent maintenance period, where the aim was to treat to a Hb level of 11.0 g/dL by maintaining Hb levels between 10.0 and 12.0 g/dL. After the treatment period, patients proceeded to the 4-week follow-up period. Patients who stopped treatment prior to Week 104 completed the end-of-treatment (EOT) visits (EOT visit and EOT + 2-week visit) and the end-of-study (EOS) visit. Thereafter, these patients were followed for vital status and serious treatment-emergent adverse events (TEAEs), as well as CV and thromboembolic TEAEs, until their projected date of completion, until the last patient randomized reached EOS or until consent was withdrawn.
FIGURE 1.

Study design. R, randomized. *Once Hb correction was reached, the patient entered the maintenance period; correction period varied from patient to patient. **The treatment period, a minimum of 52 weeks, provides sufficient data on the long-term treatment of patients with anemia of CKD using roxadustat.
This study was conducted in accordance with the ethical principles of the declaration of Helsinki, Good Clinical Practice (GCP), the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use guidelines, and applicable laws and regulations. The protocol was approved by each Institutional Review Board and all subjects provided written informed consent. Details pertaining to protocol revisions can be found in the Supplementary Methods.
Study population
In this study, patients were ≥18 years old, had been diagnosed with CKD Stage 3, 4 or 5 (eGFR <60 mL/min/1.73 m2) and were not receiving dialysis. The mean of each patient’s three most recent Hb values during the screening period had to be ≤10.0 g/dL, with a difference of ≤1.0 g/dL between the highest and the lowest values. Patients were excluded if they had received ESA treatment or more than one dose of IV iron within 12 weeks prior to randomization, red blood cell (RBC) transfusion within 8 weeks prior to randomization, had chronic inflammatory disease that could impact erythropoiesis, or had received any prior treatment with roxadustat or an HIF-PHI. A full list of eligibility criteria, as well as information regarding compliance requirements, can be found in the Supplementary Methods.
Study drug administration
Patients were randomized via interactive response technology to receive roxadustat or placebo. The initial double-blind study drug dose for both groups was based on the tiered, weight-based dosing scheme (weight ≥45 to ≤70 kg = 70 mg; weight >70 to ≤160 kg = 100 mg). Study drug was dosed TIW during the correction period with doses administered at least 2 days apart, but no more than 4 days apart. Dose adjustments were permitted from Week 4 onward; doses had to remain stable for 4 weeks following any dose adjustment. All dose adjustments were made to maintain study patients’ Hb level within the predefined target range.
Study outcomes and assessments
Efficacy assessments of treatment with study drug were based on Hb as assessed by a central laboratory from IV blood sampling. For the European Union [EU; European Medicines Agency (EMA)], the primary efficacy endpoint was Hb response defined as Hb ≥11.0 g/dL and an Hb increase from BL by ≥1.0 g/dL in any patient with BL Hb >8.0 g/dL, or an increase from BL by ≥2.0 g/dL in any patient with BL Hb ≤8.0 g/dL at two consecutive visits separated by at least 5 days during the first 24 weeks of treatment without rescue therapy (i.e. RBC transfusion, ESA or IV iron) prior to Hb response. For the US Food and Drug Administration (FDA), the primary efficacy endpoint was the change in Hb from BL to the average Hb level during the evaluation period (defined as Weeks 28–52), regardless of rescue therapy. In the event that rescue therapy was required, rescue therapy guidelines were standardized (see Supplementary Methods). Key secondary efficacy endpoints (listed in Table 1) were analyzed using the full analysis set (FAS) (Supplementary data, Table S1).
Table 1.
Analysis of key secondary efficacy endpoints
| Number | Endpoint | Primary analysis method |
|---|---|---|
| 1 | Hb change from BL to the average Hb in Weeks 28–36, without having received rescue therapy within 6 weeks prior to and during this 8-week evaluation period |
Analysis method: MMRM Categorical variables: region, history of CV, visits and visits by treatment Continuous covariates: BL Hb, BL eGFR and BL Hb by visit |
| 2 | Change from BL in LDL cholesterol to the average LDL cholesterol of Weeks 12–28 |
Analysis method: MMRM Categorical variables: region, history of CV, visits and visits by treatment Continuous covariates: BL LDL, BL Hb and BL eGFR |
| 3 | Occurrence and time to first use of rescue therapy (composite of RBC transfusions, IV iron supplementation and rescue ESA) |
Analysis method: Cox regression + Kaplan–Meier Categorical variables: stratified by region, history of CV and adjusted for BL Hb and BL eGFR as continuous covariates |
| 4 | Change from BL in SF-36 VT subscore to the average VT subscore of Weeks 12–28 |
Analysis method: MMRM Categorical variables: region, history of CV, visits and visits by treatment Continuous covariates: BL Hb, BL SF-36 VT subscore and BL eGFR |
| 5 | Change from BL in SF-36 PF subscore to the average PF subscore of Weeks 12–28 |
Analysis method: MMRM Categorical variables: region, history of CV, visits and visits by treatment Continuous covariates: BL Hb, BL SF-36 PF subscore and BL eGFR |
MMRM, mixed model of repeated measures. Superiority was tested using a fixed sequence testing procedure.
Additional secondary efficacy endpoints included change from BL in mean arterial pressure (MAP) to the average MAP value of Weeks 20–28, occurrence and time to the first occurrence of hypertension [defined as either systolic blood pressure (SBP) ≥170 mmHg and an increase from BL ≥20 mmHg, or as diastolic blood pressure (DBP) ≥110 mmHg and an increase from BL of ≥15 mmHg], rate of progression of CKD measured by annualized eGFR slope over time in the entire study cohort, change in Hb level without rescue therapy over treatment periods, time to first Hb response, change from BL in cholesterol levels and apolipoproteins, the occurrence and time to the first hospitalization, change from BL in serum hepcidin, change from BL in soluble transferrin receptor and change from BL in health-related quality of life (HRQoL) measures. Information pertaining to the HRQoL measures can be found in the Supplementary Methods. Data pertaining to measures of iron utilization, including ferritin, transferrin saturation (TSAT) and serum iron, were also collected.
The safety of roxadustat was assessed by monitoring the occurrence of TEAEs. Severity of TEAEs was graded according to National Cancer Institute—Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 4.0. Additional safety assessments included findings from laboratory tests, vital signs, physical examinations and 12-lead electrocardiograms (ECGs). The safety-emergent period was defined as the evaluation period from the analysis date of first drug intake up to 28 days after the Analysis Last Dose date. The schedule of assessments is reported in Supplementary data, Table S2.
Statistical methods
All statistical comparisons were made using two-sided tests at the α = 0.05 significance level, unless stated otherwise. Null hypotheses for superiority testing were of no treatment difference and corresponding alternative hypotheses were two-sided. Null hypotheses for noninferiority testing were of the inferiority of roxadustat treatment and were one-sided at the α = 0.025 significance level.
The EU (EMA) primary efficacy endpoint was a binary variable—Hb response (yes/no)—and was analyzed using the FAS. The proportion of responders in the primary efficacy variable was analyzed using a Cochran–Mantel–Haenszel (CMH) test adjusting for covariates (region; history of CV, cerebrovascular or thromboembolic diseases; BL Hb; and BL eGFR), comparing roxadustat with placebo. The CMH adjusted odds ratio (roxadustat versus placebo) and its 95% confidence interval (CI) were calculated. Superiority of roxadustat versus placebo was declared if the lower bound of the two-sided 95% CI of the CMH odds ratio was higher than one (>1.00). In addition, a 95% CI for the proportion of each treatment group (roxadustat and placebo) based on the exact method of Clopper–Pearson was calculated.
The primary efficacy endpoint for the US FDA was Hb change from BL to the average Hb of Weeks 28–52 regardless of rescue therapy and was analyzed using all randomized patients. The change from BL to the average Hb of Weeks 28–52 was analyzed using analysis of covariance (model with multiple imputations, adjusting for covariates (categorical: region and history of CV; continuous: BL Hb and BL eGFR), comparing roxadustat with placebo. Difference of least square means (LSMs; roxadustat minus placebo) and its 95% CI were estimated for the change from BL to the average of Weeks 28–52. Superiority of roxadustat versus placebo was declared if the lower bound of the two-sided 95% CI of the difference between treatment arms (roxadustat minus placebo) was higher than zero (>0.00).
Key secondary endpoints were tested using a fixed sequence testing procedure in order to maintain the overall two-sided type I error rate at 0.05; if the null hypothesis was rejected for a test, the claim of superiority was considered successful and the test progressed to the next comparison in the sequence (Table 1). The additional secondary efficacy endpoints—change from BL in MAP to the average MAP value of Weeks 20–28 and time to the first occurrence of hypertension—were tested for noninferiority using the per-protocol set, and remaining endpoints were tested for superiority using the FAS. The treatment effect results for inferential analyses were presented as roxadustat versus placebo.
Demographic and other BL characteristics for each treatment group were summarized using descriptive statistics and frequency tabulations; the number and percentage of patients with TEAEs, as well as the incidence rate (per 100 patient-years at risk) and event rate (per 100 patient-years) of TEAEs, were summarized for each treatment group. These data are underpowered to assess and compare safety outcomes across groups. All data processing, summarization and analyses were performed using SAS® version 9.3. A detailed description of sample size calculations and analysis populations can be found in the Supplementary Methods.
RESULTS
Patient disposition and demographics
A total of 1051 patients signed the informed consent form and were screened. Of these patients, 597 met inclusion criteria and were randomized to receive treatment. Three randomized patients were excluded due to GCP violations. Therefore, a total of 594 randomized patients were analyzed: 391 in the roxadustat treatment group and 203 in the placebo group.
A total of 334 (56.2%) patients received study treatment for 2 years: 245/391 (62.7%) in the roxadustat treatment group and 89/203 (43.8%) in the placebo treatment group (Figure 2). At 2 years, the incidence of treatment discontinuations was lower for patients in the roxadustat treatment group (146/391, 37.3%) compared with the placebo treatment group (114/203, 56.2%), with the most pronounced difference in patients with lower eGFR at BL (Supplementary data, Figure S1). Patient demographics and BL characteristics were similar between treatment groups (Table 2;Supplementary data, Table S3). Details regarding treatment exposure can be found in the Supplementary Results.
FIGURE 2.

Patient disposition.
Table 2.
Demographics and BL characteristics (safety analysis set)
| Parameter | Category/statistic | Roxadustat (n = 391) | Placebo (n = 203) |
|---|---|---|---|
| Sex | Male | 169 (43.2 %) | 99 (48.8 %) |
| Female | 222 (56.8 %) | 104 (51.2 %) | |
| Age (years) | Median (range) | 62.0 (20–89) | 63.0 (26–90) |
| Race | White | 335 (85.7 %) | 182 (89.7 %) |
| Black or African American | 10 (2.6 %) | 3 (1.5 %) | |
| Asian | 9 (2.3 %) | 0 | |
| Other | 37 (9.5 %) | 18 (8.9 %) | |
| Region | Western Europe | 28 (7.2 %) | 16 (7.9 %) |
| Rest of World (mostly Eastern European) | 363 (92.8 %) | 187 (92.1 %) | |
| Hb (g/dL) | Mean (SD) | 9.08 (0.76) | 9.10 (0.72) |
| ≤8.0 g/dL | 32 (8.2 %) | 20 (9.9 %) | |
| >8.0 g/dL | 359 (91.8 %) | 183 (90.1 %) | |
| LDL cholesterol (mmol/L) | Mean (SD) | 2.99 (1.29) | 2.88 (1.14) |
| eGFR (mL/min/1.73 m2) | Mean (SD) | 16.5 (10.2) | 17.2 (11.7) |
| Median | 13.1 | 13.4 | |
| eGFR (mL/min/1.73 m2) categories | <10 | 119 (30.4 %) | 57 (28.1 %) |
| 10 to <15 | 102 (26.1 %) | 61 (30.0 %) | |
| 15 to <30 | 128 (32.7 %) | 58 (28.6 %) | |
| 30 to <45 | 34 (8.7 %) | 19 (9.4 %) | |
| 45 to <60 | 8 (2.0 %) | 7 (3.4 %) | |
| ≥60 | 0 | 1 (0.5)c | |
| CKD etiology | Diabetic nephropathy | 109 (27.9 %) | 66 (32.5) |
| Hypertensive nephropathy | 116 (29.7 %) | 58 (28.6) | |
| Glomerulonephritis, unspecified | 52 (13.3 %) | 23 (11.3) | |
| Pyelonephritis | 49 (12.5 %) | 24 (11.8) | |
| Polycystic kidney disease | 36 (9.2 %) | 21 (10.3) | |
| Other | 84 (21.5 %) | 41 (20.2) | |
| Weight, kg | Mean (SD) | 73.86 (16.49) | 76.50 (16.51) |
| Iron repletion at BL | Ferritin ≥100 ng/mL and TSAT ≥20% | 204 (52.2 %) | 109 (53.7 %) |
| hs-CRP, nmol/La | Mean (SD)b | 92.02 (228.72) | 87.37 (149.36) |
| Medianb | 29.15 | 29.25 | |
| ≤ULN | 245 (63.1 %) | 135 (66.8 %) | |
| >ULN | 143 (36.9 %) | 67 (33.2 %) | |
| Missing | 3 | 1 | |
| SBP, mmHg | Mean (SD) | 135 (13.62) | 134 (12.30) |
| DBP, mmHg | Mean (SD) | 77 (8.83) | 77 (8.68) |
| Cardiac and vascular disorders | Angina pectoris | 40 (10.2 %) | 26 (12.8 %) |
| Cardiac failure, chronic | 68 (17.4 %) | 44 (21.7 %) | |
| Hypertension | 379 (96.9 %) | 194 (95.6 %) | |
| Diabetes mellitus | Present | 131 (33.5 %) | 76 (37.4 %) |
| Statin use at BL | Yes | 119 (30.4 %) | 61 (30.0 %) |
hs-CRP, high-sensitivity C-reactive protein; ULN, upper limit of normal.
ULN = 47.6 nmol/L.
Based on data from FAS.
Patient met study criteria at screening.
Efficacy outcomes
Primary endpoints
Following analysis of the primary endpoint for the EU (EMA), superiority of roxadustat versus placebo was demonstrated in terms of response rate to treatment during the first 24 weeks of treatment without patients having received rescue therapy in the FAS, as the lower bound of the two-sided 95% CI of the odds ratio was higher than one (>1.00) (Table 3). In the FAS, 79.2% of patients in the roxadustat treatment group were responders compared with 9.9% in the placebo group [odds ratio = 34.74 (95% CI 20.48–58.93)]. The odds ratio was statistically significant in favor of roxadustat (P < 0.001) (Table 3). Sensitivity and subgroup analyses of the primary EU (EMA) efficacy analysis confirmed the primary analysis (Supplementary data, Figures S2 and S3).
Table 3.
Hb response without rescue therapyg [EU (EMA) primary efficacy endpoint; FAS]
| Parameter/statistic | Roxadustat (n = 389) |
Placebo (n = 203) |
|---|---|---|
| Number of patients who met Hb response criteria at Week 24,an (%) | 308 (79.2) | 20 (9.9) |
| 95% CI (%) | 74.8–83.1 | 6.1–14.8 |
| Number of nonresponders per definition,an (%) | 81 (20.8) | 183 (90.1) |
| Patients failing to meet criteria for Hb response,bn (%) | 77 (19.8) | 177 (87.2) |
| Patients under rescue therapy,cn (%) | 4 (1.0) | 6 (3.0) |
| Patients who discontinued treatment prior to Hb responsed | 0 | 0 |
| Difference of proportions (roxadustat – placebo),e % | 69.3 | |
| 95% CI of difference (%) | 63.6–75.1 | |
| Odds ratio (roxadustat – placebo)f | 34.74 | |
| 95% CI | 20.48–58.93 | |
| P-value | P < 0.001 | |
Response is defined as Hb ≥11.0 g/dL and change ≥1.0 g/dL if BL Hb >8.0 g/dL; or change ≥2.0 g/dL if BL Hb ≤8.0 g/dL at two consecutive visits (dates) (with available data) separated by at least 5 days during the first 24 weeks of treatment without having received rescue therapy (RBC transfusion, ESA or IV iron), or having discontinued prior to Hb response. The proportions and 95% CI are unadjusted for covariates, and the exact method of Clopper–Pearson is used for 95% CI.
Patient who did not meet the Hb criteria detailed above during the first 24 weeks.
Patient who met the Hb criteria detailed above but started rescue therapy between the two consecutive visits.
Patient who met the Hb criteria detailed above but discontinued prior to the day of the second consecutive visit.
95% CI of the difference in proportions is calculated using Wald’s method.
CMH test is adjusted by region, history of CV disease, BL Hb and BL eGFR.
Rescue therapy is defined as RBC transfusion, ESA or IV iron.
Following analysis of the primary endpoint for the US FDA, superiority of roxadustat versus placebo was demonstrated because the lower bound of the two-sided 95% CI of the difference between treatment arms (roxadustat – placebo) was higher than zero (>0.00). BL Hb was comparable between the treatment groups (Table 4). The LSM change from BL was 1.992 (95% CI 1.82–2.16) g/dL for patients in the roxadustat group and 0.300 (95% CI 0.09–0.51) g/dL for patients in the placebo group. The LSM of the treatment difference for roxadustat versus placebo was +1.692 (95% CI 1.52–1.86); this difference was statistically significant (P < 0.001) (Table 4; Figure 3; Supplementary data, Figure S4). Sensitivity and subgroup analyses of the primary US FDA efficacy analysis confirmed the primary analysis (Supplementary data, Figures S5 and S6).
Table 4.
Change from BL to the average Hb in Weeks 28–52 regardless of rescue therapy use (US FDA primary efficacy endpoint; all randomized patients)
| Parameter/statistic | Roxadustat (n = 391) |
Placebo (n = 203) |
|---|---|---|
| BL Hb, mean (SD), g/dL | 9.078 (0.761) | 9.095 (0.721) |
| Hb change from BL to the average Hb in Weeks 28–52 (g/dL) | ||
| N | 312 | 146 |
| Mean | 1.988 | 0.406 |
| SD | 0.953 | 0.979 |
| Min | −1.19 | −2.09 |
| Median | 1.938 | 0.209 |
| Max | 4.43 | 3.80 |
| Analysis using analysis of covariance with multiple imputations for Hb change from BL to Weeks 28–52 | ||
| LSM | 1.992 | 0.300 |
| 95% CI | 1.82–2.16 | 0.09–0.51 |
| LSM difference (roxadustat—placebo) | 1.692 | |
| 95% CI | 1.52, 1.86 | |
| P-value | P < 0.001 | |
The model includes treatment as fixed factor, region and history of CV disease as class factors, and BL Hb and BL eGFR as continuous covariates.
BL Hb is defined as the mean of four latest central laboratory Hb values prior to or on the same date as first study drug intake (pre-dose).
FIGURE 3.
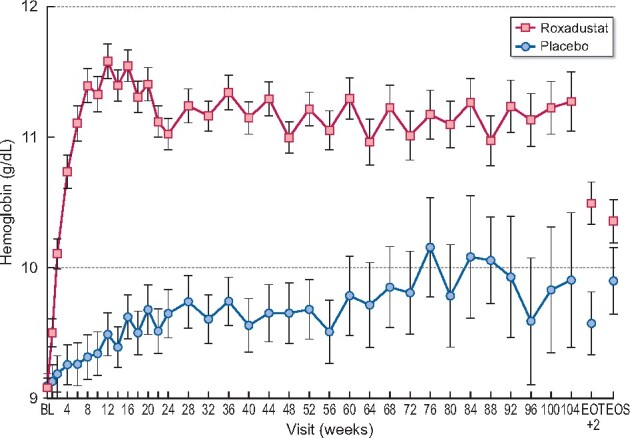
Mean Hb over time regardless of rescue therapy use (all randomized patients).
Secondary endpoints
In the sequentially tested key secondary endpoints, superiority of roxadustat versus placebo was demonstrated for Hb change from BL to the average of Weeks 28–36 (Supplementary data, Figure S7), LDL cholesterol change from BL to the average of Weeks 12–28 (mean change in LDL cholesterol from BL, average of Weeks 12–28: −0.602 mmol/L for roxadustat; 0.151 mmol/L for placebo) and time to first use of rescue medication (Supplementary data, Tables S1, S4 and S5; Figure 4). Decreases in LDL cholesterol in the roxadustat group were statistically significant compared with the placebo group. Slight decreases in high-density lipoprotein (HDL) cholesterol and, subsequently, in the LDL/HDL cholesterol ratio were also seen in roxadustat treated patients (Supplementary data, Table S6 and Figures S8 and S9). In this study, rates of statin use were comparable between treatment groups. Furthermore, LDL and HDL assessments by patient subgroup—with or without use of concomitant statin treatment at BL—were consistent with the overall population (data not shown). These findings, paired with the fact that decreases in the LDL/HDL cholesterol ratio were slightly more pronounced in roxadustat-treated patients, suggest roxadustat may have an additional effect on statins.
FIGURE 4.
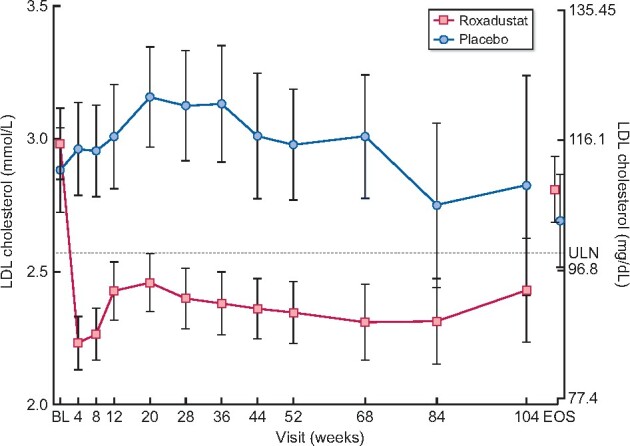
Mean (±95% CI) plot of LDL cholesterol regardless of fasting status by time (FAS). ULN, upper limit of normal.
No difference was observed between the roxadustat and placebo treatment groups in terms of change from BL in Short Form 36 (SF-36) vitality (VT) and physical functioning (PF) subscores at each timepoint assessed (Weeks 12–28) (Supplementary data, Table S1).
There was no apparent difference between the roxadustat and placebo treatment groups in terms of change from BL in MAP, cumulative incidence of first occurrence of hypertension, decrease in eGFR with time or mean changes by visit in ferritin, TSAT or serum iron. Details regarding secondary endpoints are presented in the Supplementary Results as well as Supplementary data, Figure S10 (ferritin) and Supplementary data, Figure S11 (TSAT).
Safety
An overview of TEAE incidence by category of TEAE is presented in Table 5. The overall incidence of TEAEs was comparable between treatment groups. Patients in the roxadustat treatment group had a total of 476.7 events/100 patient exposure years (PEY) TEAEs compared with 514.7 events/100 PEY events in the placebo group (Table 6). Common (≥5% in either treatment group) TEAEs in either treatment group included end-stage renal disease, hypertension, peripheral edema and decreased GFR. Noteworthy differences between treatment groups include greater incidences and event rates in the roxadustat treatment group for hypertension, nausea and diarrhea, and lower incidences and event rates in the roxadustat treatment group for anemia. Overall, 241 (61.6%) patients in the roxadustat treatment group had a total of 515 (103.6/100 PEY) serious TEAEs compared with 115 (56.7%) patients who had a total of 250 (119.0/100 PEY) events in the placebo group (Supplementary data, Table S7). The time to occurrence of serious TEAEs, TEAEs leading to patient death and TEAEs leading to withdrawal was comparable in both treatment groups.
Table 5.
Overview of TEAEs and death (safety analysis set)
| Type of Event | Roxadustat (n = 391), n (%) |
Placebo (n = 203), n (%) |
|---|---|---|
| TEAE | 343 (87.7) | 176 (86.7) |
| Serious TEAE | 241 (61.6) | 115 (56.7) |
| TEAE leading to death | 40 (10.2) | 19 (9.4) |
| TEAE leading to withdrawal of treatment | 23 (5.9) | 8 (3.9) |
| TEAE NCI-CTC Grade 3 or higher | 185 (47.3) | 88 (43.3) |
| Death during the safety- emergent period | 37 (9.5) | 16 (7.9) |
TEAEs were defined as adverse events that started during the safety emergent period, i.e. those starting after first administration of the study drug, to up to 28 days after last study drug intake.
Table 6.
Common (≥5% patients in any treatment group) TEAE (safety analysis set)
| MedDRA version 20.0 preferred term | Roxadustat (n = 391; PEY = 496.9) |
Placebo (n = 203; PEY = 210.0) |
||
|---|---|---|---|---|
| n (%) | #E (event rate/100 PEY) | n (%) | #E (event rate/100 PEY) | |
| Overall | 373 (87.7) | 2369 (476.7) | 176 (86.7) | 1081 (514.7) |
| End-stage renal disease | 135 (34.5) | 135 (27.2) | 62 (30.5) | 63 (30.0) |
| Hypertension | 87 (22.3) | 142 (28.6) | 28 (13.8) | 46 (21.9) |
| Edema peripheral | 45 (11.5) | 54 (10.9) | 21 (10.3) | 22 (10.5) |
| GFR decreased | 43 (11.0) | 48 (9.7) | 23 (11.3) | 28 (13.3) |
| Hyperkalemia | 39 (10.0) | 52 (10.5) | 15 (7.4) | 21 (10.0) |
| Viral upper respiratory tract infection | 38 (9.7) | 50 (10.1) | 9 (4.4) | 15 (7.1) |
| Nausea | 37 (9.5) | 47 (9.5) | 6 (3.0) | 6 (2.9) |
| Diarrhea | 33 (8.4) | 41 (8.3) | 7 (3.4) | 10 (4.8) |
| Pneumonia | 28 (7.2) | 35 (7.0) | 14 (6.9) | 17 (8.1) |
| Iron deficiencya | 26 (6.6) | 26 (5.2) | 8 (3.9) | 10 (4.8) |
| Anemia | 24 (6.1) | 27 (5.4) | 37 (18.2) | 54 (25.7) |
| Headache | 21 (5.4) | 22 (4.4) | 11 (5.4) | 12 (5.7) |
| Arteriovenous fistula thrombosis | 20 (5.1) | 27 (5.4) | 2 (1.0) | 3 (1.4) |
| Pruritus | 20 (5.1) | 22 (4.4) | 2 (1.0) | 2 (1.0) |
| Asthenia | 19 (4.9) | 23 (4.6) | 12 (5.9) | 15 (7.1) |
| Hyperuricemia | 9 (2.3) | 9 (1.8) | 11 (5.4) | 11 (5.2) |
Event rate per 100 PEY is defined as (number of events) x 100 divided by PEY during safety-emergent period.
Sorting order: incidence by preferred term in the roxadustat treatment group.
#E, number of events; PEY, patient exposure years.
Based on ferritin and TSAT.
There were no apparent differences between treatment groups in the occurrence of potentially clinically significant liver assessments, other laboratory assessments, vital signs or 12-lead ECG assessments. Incidence rates of potentially clinically significant SBP values (SBP ≥170 mmHg in combination with an increase of ≥20 mmHg) per 100 PEY were 12.7 and 10.4 for roxadustat- and placebo-treated patients, respectively. Time-course data for SBP and DBP are presented in Supplementary Figures S12 and S13, respectively. No difference was observed between treatment groups in the incidence rate of deaths/100 PEY [7.4 roxadustat versus 7.6 placebo, hazard ratio of 0.96 (95% CI 0.53–1.74); P = 0.902] during the safety-emergent period.
DISCUSSION
This study, conducted in a mostly European population, was a Phase 3 randomized, double-blind, placebo-controlled study designed to evaluate the efficacy of roxadustat in the treatment of anemia in patients with NDD-CKD. Patients with Stage 3, 4 or 5 CKD (with eGFR <60 mL/min/1.73 m2) not receiving dialysis were randomized in a 2:1 ratio to receive roxadustat or placebo. Treatment was to continue for at least 52 weeks and up to 104 weeks. In this study, superiority of roxadustat versus placebo was demonstrated for both primary efficacy endpoints: Hb response [odds ratio = 34.74 (95% CI 20.48–58.93)] and change in Hb from BL [roxadustat – placebo: +1.692 (95% CI 1.52–1.86); both P < 0.001]. Superiority of roxadustat was also demonstrated for LDL cholesterol change from BL, and time to first use of rescue medication (both P < 0.001). The incidences of TEAEs were comparable between groups (roxadustat: 87.7%, placebo: 86.7%).
In this study, demographics and BL disease characteristics, including disease and anemia treatment history, were comparable between treatment groups and consistent with the expected study population [12, 13, 15, 21]. It is worth noting that, in this study, more patients with advanced CKD were part of the study cohort relative to other anemia management studies that have examined ESAs in patients with NDD-CKD. For instance, in the PEARL (Safety & Efficacy of Peginesatide for the Treatment of Anemia in Participants With Chronic Renal Failure Not on Dialysis) [24] and TREAT (Trial to Reduce Cardiovascular Events with Aranesp Therapy) [5] studies, BL eGFR was roughly 30 mL/min/1.73 m2 (or higher) whereas, in this study, mean [standard deviation (SD)] eGFR was 16.5 (10.2) mL/min/1.73 m2 and 17.2 (11.7) mL/min/1.73 m2 in the roxadustat and placebo groups, respectively. This difference is likely due to differences in inclusion criteria between studies, in particular the lower maximum Hb value qualifying for inclusion.
This study met its primary objective by demonstrating superiority of roxadustat in efficacy versus placebo in terms of both response rate and Hb change from BL at Weeks 28–52. These findings are supported by previous work performed in patients with NDD-CKD [16, 19–22]. Although the studies in this program were conducted by different companies in different geographic regions where treatment practices may differ, efficacy results were consistent between trials, suggesting that roxadustat is effective in this population.
Superiority of roxadustat versus placebo was also demonstrated for LDL cholesterol change from BL to Weeks 12–28. This finding is similar to previous studies performed in patients with NDD-CKD [16, 21, 22]. Moreover, in roxadustat-treated patients, decreases in LDL cholesterol (mean change in LDL cholesterol from BL, average of Weeks 12–28: −0.602 mmol/L) were comparable to decreases seen with low-dose statins [25] and decreases in LDL cholesterol exceeded those observed in HDL cholesterol, leading to a favorable reduction in the LDL/HDL cholesterol ratio. Likewise, cholesterol levels and apolipoproteins showed a decrease at each timepoint in the roxadustat treatment group compared with a slight increase in the placebo treatment group. It may be possible that a reduction in LDL cholesterol, as well as improvements to other blood lipids, may provide clinical benefit considering that dyslipidemia is an established risk factor for CV disease in patients with CKD [26–28]. These effects may be mediated, at least in part, by HIF-dependent effects on acetyl coenzyme A that are required for the first step of cholesterol synthesis and on the degradation of 3-hydroxy-3-methylglutaryl coenzyme A reductase, the rate-limiting enzyme in cholesterol synthesis [29–31].
In this study, mean iron repletion status at BL was comparable between treatment groups (roxadustat: 52.2%; placebo: 53.7%) but changes in markers of iron status were greater in roxadustat-treated patients relative to placebo-treated patients. Also, the proportion of responders in the roxadustat treatment group was slightly higher in iron-replete patients (84.3%) compared with noniron-replete patients (73.5%). Also, when evaluating Hb change from BL to the average in Weeks 28–36, the Hb change in roxadustat-treated patients was comparable in both subgroups. However, the treatment difference in placebo-treated patients in the noniron-replete subgroup was smaller due to a larger Hb change observed for placebo. Roxadustat-related decreases in hepcidin and increases in soluble transferrin receptor levels, as seen in this study and previous studies [16–21], help provide insight into the effects of roxadustat on iron metabolism. For instance, decreases in hepcidin, a negative regulator of iron absorption and mobilization that impedes erythropoiesis, may mediate the stability of iron stores, thereby leading to improved iron bioavailability via enhanced intestinal iron absorption and iron mobilization from macrophages of the reticuloendothelial system [13, 21, 32]. Likewise, increases in soluble transferrin receptor levels improve iron availability given its role as a carrier protein for transferrin that is required for the import of iron into the cell, which results in improved iron transport to tissues and to developing erythrocytes [18]. In this study, initial increases in soluble transferrin receptor (a consequence of HIF stimulation) appears most pronounced in the first 12 weeks of treatment before stabilizing at levels higher than seen with placebo, which did not show a change from BL.
Overall, the safety profile of roxadustat in this study was generally comparable to placebo. While there was a slightly greater incidence of potentially clinically significant SBP values in the roxadustat treatment group compared with the placebo group, there was no apparent difference in the incidence of potentially clinically significant DBP values or 12-lead ECG values, and there was no apparent difference in terms of MAP with time or effect on overall BP between treatment groups. Furthermore, a confirmatory assessment of deaths revealed no difference between groups. The incidence of arteriovenous fistula thrombosis was greater in the roxadustat treatment group; an explanation of this difference is outside the scope of this study and could be related to the treatment but not necessarily, considering the many factors that contribute to this complication. However, it is worth noting that the majority of arteriovenous fistula thrombosis events in roxadustat-treated patients (67%; 18/27 events) occurred after the start of chronic dialysis.
In this study, there were fewer treatment discontinuations in the roxadustat treatment group compared with the placebo group. As expected, the incidence of discontinuation due to ‘lack of efficacy’ was very low in roxadustat (0.8%) compared with placebo (12.8%), and withdrawals due to TEAEs were low overall: 5.9% with roxadustat versus 3.9% with placebo. Overall treatment exposure and PEY were notably higher in the roxadustat treatment group compared with placebo; this is likely due to the planned 2:1 randomization of patients and the greater proportion of patients in the placebo treatment group discontinuing treatment prematurely. The majority of TEAEs in both treatment groups was nonserious, Grade 2 or 3 in severity, and considered unrelated to treatment by the investigator. The safety profile in both treatment groups was generally consistent with that expected in this study population.
One possible limitation to this study may be the homogeneity of the study population (>85% White in both treatment groups), which may limit the generalizability of the current findings. However, it should be noted that participants in this study were from a wide range of different countries, mainly from Europe, which may help increase heterogeneity. This study also observed unequal discontinuation rates between treatment groups, which may complicate the interpretation of adverse event data. This study was not powered to show significant differences in secondary endpoints. Lastly, this analysis did not consider ophthalmological data, data related to the development or worsening of renal cysts, or data related to the development or worsening of pulmonary hypertension. It should be noted, however, that ophthalmological considerations have been evaluated in other studies [12] and will be further addressed, in detail, in a forthcoming dedicated analysis.
It is also worth noting that, because this study used a placebo comparator, an evaluation of the CV safety of roxadustat versus ESAs is not presented here; CV safety is a pertinent concern based on previous data that suggest an increased risk of CV events is associated with ESA use [5, 33]. A comparison of CV-related safety between roxadustat and ESAs is a subject outside of the scope of the current analysis. However, pooled analyses of CV-related events in roxadustat- and ESA-treated patients are the focus of a forthcoming manuscript.
In conclusion, this study met its primary objective by demonstrating superiority of roxadustat in efficacy versus placebo in terms of both response rate and Hb change from BL at Weeks 28–52 in mainly European patients. In the sequentially tested key secondary endpoints, superiority versus placebo was demonstrated for Hb change from BL at Weeks 28–36, LDL cholesterol change, decreases in hepcidin, increases in soluble transferrin receptor levels and time to use of rescue medication. The safety profile observed in this study is in line with the expected event profile in NDD-CKD patients and was generally comparable between roxadustat and placebo over 104 weeks.
SUPPLEMENTARY DATA
Supplementary data are available at ndt online.
Supplementary Material
ACKNOWLEDGEMENTS
Roxadustat is being developed by FibroGen, AstraZeneca and Astellas. Medical writing/editorial support was provided by Patrick Tucker, PhD, and Elizabeth Hermans, PhD (OPEN Health Medical Communications, Chicago, IL, USA), and funded by the study sponsor. We would like to sincerely thank the investigators who participated in this trial, as well as the patients and their family members for their support.
FUNDING
This study was funded by Astellas Pharma, Inc.
AUTHORS’ CONTRIBUTIONS
M.R. was responsible for conception and study design. E.S., W.S., C.E., A.T., B.A., M.R. and N.D. were involved in acquisition of data. M.R. and U.V. were involved in analysis and interpretation of the data. Drafting and critical revision of the article for important intellectual content done by E.S., W.S., C.E., A.T., B.A., M.R., U.V. and N.D.
CONFLICT OF INTEREST STATEMENT
U.V. is an employee of Astellas Pharma Global Development, Inc. M.R. is an employee of Astellas Pharma Europe B.V. All other authors have nothing to disclose. The results presented in this paper have not been published previously in whole or part, except in abstract format.
(See related article by Locatelli and Vecchio. A new paradigm in treating patients with chronic kidney disease and anaemia after a journey lasting more than 35 years. Nephrol Dial Transplant 2021; 36: 1559–1563)
DATA AVAILABILITY STATEMENT
Researchers may request access to anonymized participant-level data, trial-level data and protocols from Astellas sponsored clinical trials at www.clinicalstudydatarequest.com. For the Astellas criteria on data sharing see: https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Astellas.aspx.
REFERENCES
- 1.Shih HM, Wu CJ, Lin SL.. Physiology and pathophysiology of renal erythropoietin-producing cells. J Formos Med Assoc 2018; 117: 955–963 [DOI] [PubMed] [Google Scholar]
- 2.Haase VH.Therapeutic targeting of the HIF oxygen-sensing pathway: lessons learned from clinical studies. Exp Cell Res 2017; 356: 160–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Babitt JL, Lin HY.. Mechanisms of anemia in CKD. J Am Soc Nephrol 2012; 23: 1631–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andrassy KM.Comments on ‘KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease’. Kidney Int 2013; 84: 622–623 [DOI] [PubMed] [Google Scholar]
- 5.Pfeffer MA, Burdmann EA, Chen CY. et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med 2009; 361: 2019–2032 [DOI] [PubMed] [Google Scholar]
- 6.Agarwal R, Kusek JW, Pappas MK.. A randomized trial of intravenous and oral iron in chronic kidney disease. Kidney Int 2015; 88: 905–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eriguchi R, Taniguchi M, Ninomiya T. et al. Hyporesponsiveness to erythropoiesis-stimulating agent as a prognostic factor in Japanese hemodialysis patients: the Q-Cohort study. J Nephrol 2015; 28: 217–225 [DOI] [PubMed] [Google Scholar]
- 8.Luo J, Jensen DE, Maroni BJ. et al. Spectrum and burden of erythropoiesis-stimulating agent hyporesponsiveness among contemporary hemodialysis patients. Am J Kidney Dis 2016; 68: 763–771 [DOI] [PubMed] [Google Scholar]
- 9.Kaplan JM, Sharma N, Dikdan S.. Hypoxia-inducible factor and its role in the management of anemia in chronic kidney disease. Int J Mol Sci 2018; 19: 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Locatelli F, Fishbane S, Block GA. et al. Targeting hypoxia-inducible factors for the treatment of anemia in chronic kidney disease patients. Am J Nephrol 2017; 45: 187–199 [DOI] [PubMed] [Google Scholar]
- 11.Joharapurkar AA, Pandya VB, Patel VJ. et al. Prolyl hydroxylase inhibitors: a breakthrough in the therapy of anemia associated with chronic diseases. J Med Chem 2018; 61: 6964–6982 [DOI] [PubMed] [Google Scholar]
- 12.Akizawa T, Iwasaki M, Yamaguchi Y. et al. Phase 3, randomized, double-blind, active-comparator (darbepoetin alfa) study of oral roxadustat in CKD patients with anemia on hemodialysis in Japan. J Am Soc Nephrol 2020; 31: 1628–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akizawa T, Otsuka T, Reusch M. et al. Intermittent oral dosing of roxadustat in peritoneal dialysis chronic kidney disease patients with anemia: a randomized, phase 3, multicenter, open-label study. Ther Apher Dial 2020; 24: 115–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Besarab A, Chernyavskaya E, Motylev I. et al. Roxadustat (FG-4592): correction of anemia in incident dialysis patients. J Am Soc Nephrol 2016; 27: 1225–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen N, Hao C, Liu BC. et al. Roxadustat treatment for anemia in patients undergoing long-term dialysis. N Engl J Med 2019; 381: 1011–1022 [DOI] [PubMed] [Google Scholar]
- 16.Chen N, Qian J, Chen J. et al. Phase 2 studies of oral hypoxia-inducible factor prolyl hydroxylase inhibitor FG-4592 for treatment of anemia in China. Nephrol Dial Transplant 2017; 32: 1373–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Provenzano R, Besarab A, Wright S. et al. Roxadustat (FG-4592) versus epoetin alfa for anemia in patients receiving maintenance hemodialysis: a phase 2, randomized, 6- to 19-week, open-label, active-comparator, dose-ranging, safety and exploratory efficacy study. Am J Kidney Dis 2016; 67: 912–924 [DOI] [PubMed] [Google Scholar]
- 18.Akizawa T, Ueno M, Shiga T. et al. Oral roxadustat three times weekly in ESA-naive and ESA-converted patients with anemia of chronic kidney disease on hemodialysis: Results from two phase 3 studies. Ther Apher Dial 2020; 24: 628–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akizawa T, Iwasaki M, Otsuka T. et al. Roxadustat treatment of chronic kidney disease-associated anemia in Japanese patients not on dialysis: a phase 2, randomized, double-blind, placebo-controlled trial. Adv Ther 2019; 36: 1438–1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Besarab A, Provenzano R, Hertel J. et al. Randomized placebo-controlled dose-ranging and pharmacodynamics study of roxadustat (FG-4592) to treat anemia in nondialysis-dependent chronic kidney disease (NDD-CKD) patients. Nephrol Dial Transplant 2015; 30: 1665–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen N, Hao C, Peng X. et al. Roxadustat for anemia in patients with kidney disease not receiving dialysis. N Engl J Med 2019; 381: 1001–1010 [DOI] [PubMed] [Google Scholar]
- 22.Provenzano R, Besarab A, Sun CH. et al. Oral hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat (FG-4592) for the treatment of anemia in patients with CKD. Clin J Am Soc Nephrol 2016; 11: 982–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akizawa T, Yamaguchi Y, Otsuka T. et al. A phase 3, multicenter, randomized, two-arm, open-label study of intermittent oral dosing of roxadustat for the treatment of anemia in Japanese erythropoiesis-stimulating agent-naive chronic kidney disease patients not on dialysis. Nephron 2020; 144: 372–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Macdougall IC, Provenzano R, Sharma A. et al. Peginesatide for anemia in patients with chronic kidney disease not receiving dialysis. N Engl J Med 2013; 368: 320–332 [DOI] [PubMed] [Google Scholar]
- 25.Grundy SM, Stone NJ, Bailey AL. et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American college of cardiology/American heart association task force on clinical practice guidelines. Circulation 2019; 139: e1082–e1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McFarlane SI, McCullough PA, Sowers JR. et al. Comparison of the CKD epidemiology collaboration (CKD-EPI) and modification of diet in renal disease (MDRD) study equations: prevalence of and risk factors for diabetes mellitus in CKD in the Kidney Early Evaluation Program (KEEP). Am J Kidney Dis 2011; 57: S24–S31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCullough PA, Verrill TA.. Cardiorenal interaction: appropriate treatment of cardiovascular risk factors to improve outcomes in chronic kidney disease. Postgrad Med 2010; 122: 25–34 [DOI] [PubMed] [Google Scholar]
- 28.Baigent C, Landray MJ, Reith C. et al. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. Lancet 2011; 377: 2181–2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hwang S, Nguyen AD, Jo Y. et al. Hypoxia-inducible factor 1alpha activates insulin-induced gene 2 (Insig-2) transcription for degradation of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase in the liver. J Biol Chem 2017; 292: 9382–9393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen AD, McDonald JG, Bruick RK. et al. Hypoxia stimulates degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase through accumulation of lanosterol and hypoxia-inducible factor-mediated induction of insigs. J Biol Chem 2007; 282: 27436–27446 [DOI] [PubMed] [Google Scholar]
- 31.Kim JW, Tchernyshyov I, Semenza GL. et al. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006; 3: 177–185 [DOI] [PubMed] [Google Scholar]
- 32.Korolnek T, Hamza I.. Macrophages and iron trafficking at the birth and death of red cells. Blood 2015; 125: 2893–2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seliger SL, Zhang AD, Weir MR. et al. Erythropoiesis-stimulating agents increase the risk of acute stroke in patients with chronic kidney disease. Kidney Int 2011; 80: 288–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Researchers may request access to anonymized participant-level data, trial-level data and protocols from Astellas sponsored clinical trials at www.clinicalstudydatarequest.com. For the Astellas criteria on data sharing see: https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Astellas.aspx.