

Abstract
The β-diketone moiety is commonly present in many anticancer drugs, antibiotics, and natural products. We describe a general method for radiolabeling β-diketone-bearing molecules with fluoride-18. Radiolabeling is carried out via 18F-19F isotopic exchange on non-radioactive difluoro-dioxaborinins, which are generated by minimally modifying the β-diketone as a difluoroborate. Radiochemistry is one-step, rapid (< 10 min), high-yielding (> 80%), and proceeds at room temperature to accommodate the half-life of F-18 (t1/2 = 110 min). High molar activities (7.4 Ci/μmol) were achieved with relatively low starting activities (16.4 mCi). It was found that substituents affect both the solvolytic stability and fluorescence properties of difluoro-dioxaborinins. An F-18 radiolabeled difluoro-dioxaborinin probe that is simultaneously fluorescent showed sufficient stability for in vivo PET/fluorescence imaging in mice, rabbits, and patients. These findings will guide: the design of probes with specific PET/fluorescence properties; the development of new PET/fluorescence dual-modality reporters; and accurate in vivo tracking of β-diketone molecules.
Table of Contents Graphic
INTRODUCTION
Fluoride-18 (F-18) is a widely used isotope in clinical imaging, especially in the case of 18F-FDG imaging.1 The short half-life (110 min) and small size of fluoride makes F-18 an important substituent in the context of drug discovery, development, and imaging.2 Several new methods for F-18 radiochemistry have been developed in recent years.3 These studies have sparked intense interest in discovering F-18 radiochemical strategies that are generalizable, simple, rapid, high-yielding, and convey desirable medicinal properties.4 These new strategies will allow broad and convenient investigation of new molecules in drug discovery and development programs.5
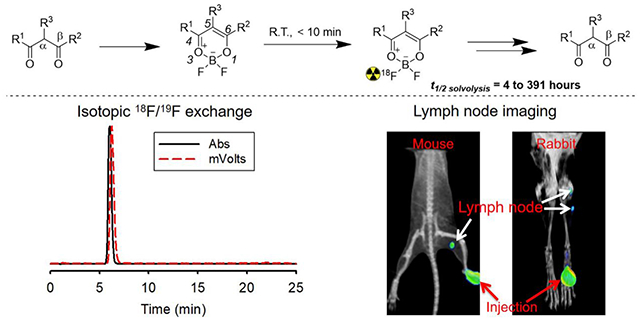
The β-diketone moiety is generally present in drugs that are in, but not limited to, anticancer drugs (Doxorubicin, Daunorubicin, etc.), antibiotics (tetracycline, glycylcycline, etc.), and natural products (emodin, curcumin, etc.). β-Diketones can be modified to bear fluorine (fluorine-19) by reacting pendant β-diketone moieties via the well-established β-diketone-boron trifluoride reaction (Figure 1a).6 The difluoroborate moiety in the generated difluoro-dioxaborinins is small and contains boron-fluoride bonds, inspiring us to exploit F-18 equivalents in pharmacodynamic and pharmacokinetic studies with Positron Emission Tomography (PET) imaging.7 Unfortunately, there has been no strategy reported to date for directly introducing the PET emitter, F-18, onto either β-diketone-containing reagents or modified difluoro-dioxaborinins.
Figure 1.
a) General scheme for converting β-diketones into difluoro-dioxaborinins. b) General scheme for 18F-19F IEX radiolabeling resulting in [18F]-difluoro-dioxaborinin. c) In water, difluoro-dioxaborinins eventually undergo solvolysis and convert back into the parent β-diketones at half-lives (t1/2 solvolysis) ranging from 4 to 391 hours depending on the substituents, R1, R2, and R3.
Recently, F-18 radiochemistry via 18F-19F isotopic exchange has been explored on other molecules that contain a silicon-fluoride bond or a boron-fluoride bond.8, 9 First introduced by the Schirrmacher group8, 18F-19F isotopic exchange radiochemistry (IEX) is not complicated, does not require HPLC product or intermediate purification with HPLC, and can proceed rapidly at room temperature with high molar activities, making it a promising strategy in F-18 radiolabeling and clinical translation10. These advantages of IEX have inspired us to explore the possibility of using 18F-19F IEX to introduce F-18 onto difluoro-dioxaborinins. To date, 18F-19F IEX has not been explored for difluoro-dioxaborinins, which also contain boron-fluoride bonds.
Herein, we report a tin (IV) chloride catalyzed IEX F-18 radiolabeling method for difluoro-dioxaborinins (Figure 1b). For one composition, the radiolabeling of difluoro-dioxaborinins (> 1 Ci/μmol) is rapid (< 10 min), one-step, and proceeds at room temperature. This method can achieve high molar activities (7.4 Ci/μmol) with relatively low starting activities (16.4 mCi) as the entry. We show that IEX F-18 radiochemistry is generalizable to many other β-diketones and that substituents can affect both the solvolytic stability and fluorescence properties of difluoro-dioxaborinins. This information will help guide the design of future stable PET/fluorescence dual-modality probes with tunable optical wavelengths (Figure 1c). Our findings indicate that tin (IV) chloride catalyzed IEX F-18 radiochemistry can be used as a general platform to radiolabel β-diketone-containing pharmaceuticals as well as other natural products for in vivo tracking. This method has implications for the development of small-molecule, fluorescence radiotracers.
RESULTS
Chemistry.
Synthesis of difluoro-dioxaborinins 1, 2, 3, 5, 6, 9, 10, 13, 14, 15, 16, and 17
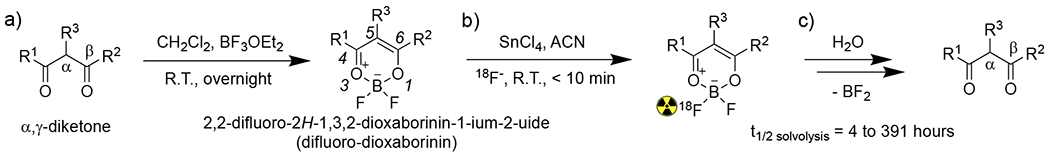
To explore the radiochemical behavior, solvolysis, and medical imaging potential of difluoro-dioxaborinins, a panel of different difluoro-dioxaborinins (1, 2, 3, 5, 6, 9, 10, 13, 14, 15, 16, and 17) were synthesized using a single, general established methodology derived from the literature (Figure 1a).6 The molecular structures for all synthesized difluoro-dioxaborinins are shown in Figure 2 (see full characterization in the supporting information). To synthesize difluoro-dioxaborinins, boron trifluoride etherate (BF3 OEt2) was added dropwise into a dry dichloromethane solution of β-diketones under magnetic stirring at room temperature for two hours. The generated difluoro-dioxaborinins were purified with preparative high-performance liquid chromatography (HPLC). General yields for all difluoro-dioxaborinins were in the range of 50%–70%. The appearance of specific chemical shifts in 1H NMR and 19F NMR generally indicate difluoro-dioxaborinin formation. In all the 1H NMR studies of difluoro-dioxaborinins shown in Figure 2, the appearance of a proton shift between 6–8 ppm corresponds to production of a deshielded difluoro-dioxaborinin α-hydrogen (position 5 in Figure 1a). 19F NMR studies for all difluoro-dioxaborinins show two peaks (with a difference of 0.1-0.2 ppm, and peak area ratios of ~ 1:4 due to the abundance of the natural boron isotope that is 20% 10B and 80% 11B), which are indicative of two fluoride atoms bound to boron. High-resolution mass spectrometry (HRMS) further confirms the synthesis of difluoro-dioxaborinins in Figure 2. Esterified products, 9 and 10, were synthesized to measure the impact more distal substituents on the solvolytic stability of difluoro-dioxaborinins. 10 was synthesized with additional intent: 10 can be readily conjugated to thiol-bearing peptides, nucleic acids, polymers, and nanoparticles in advanced applications.
Figure 2.
Synthesis (solid boxes) and attempted synthesis (dashed box) of difluoro-dioxaborinins for radiolabeling and solvolytic analysis. “X mark” denotes the chemicals in the frame that failed to be isolated in the attempted synthesis. “Check mark” denotes the chemicals in the frame that were successfully isolated.
General radiolabeling of Compounds 1, 2, 3, 5, 6, 9, 10, 13, 14, 15, 16, and 17.
All difluoro-dioxaborinins underwent a common one-step radiolabeling with F-18 (Figure 1b). This radiolabeling methodology is inspired from Lewis Acid-assisted isotopic 18F-19F exchange for [18F]-dipyrromethene precedent.9 Specifically, [18F]-fluoride ions obtained from a cyclotron were dried under nitrogen flow, without heating, in a conical vial. The drying step took 5-20 min depending on the specific concentration of the received [18F]-fluoride-ion. Nothing was added to the drying cocktail. 18F-fluoride ion solution at a quality that is sufficient for FDA approved bone scanning was received and dried as received. A freshly prepared solution of 2% SnCl4 (20 μL, neat) in anhydrous acetonitrile (1 mL, HPLC) was used to partially solubilize the dry [18F]-fluoride ion. This solution was immediately used to suspend lyophilized difluoro-dioxaborinins.
IEX F-18 radiolabeling of difluoro-dioxaborinins proceeded rapidly upon mixing at room temperature (Figure 3a). In thin-layer chromatography (TLC) analysis, the desired, radiolabeled major products run at the solvent front, while free [18F]-fluoride ions remain at the baseline. Using fluorescent difluoro-dioxaborinin compounds, we find that TLC quantitation underestimates the radiochemical yield, as minor quantities of the desired product do not move from the baseline (Figures in SI section 5–7). For this reason, HPLC equipped with in line radioactivity and absorbance detectors were used to confirm radiolabelings (Figures in SI section 3).
Figure 3.
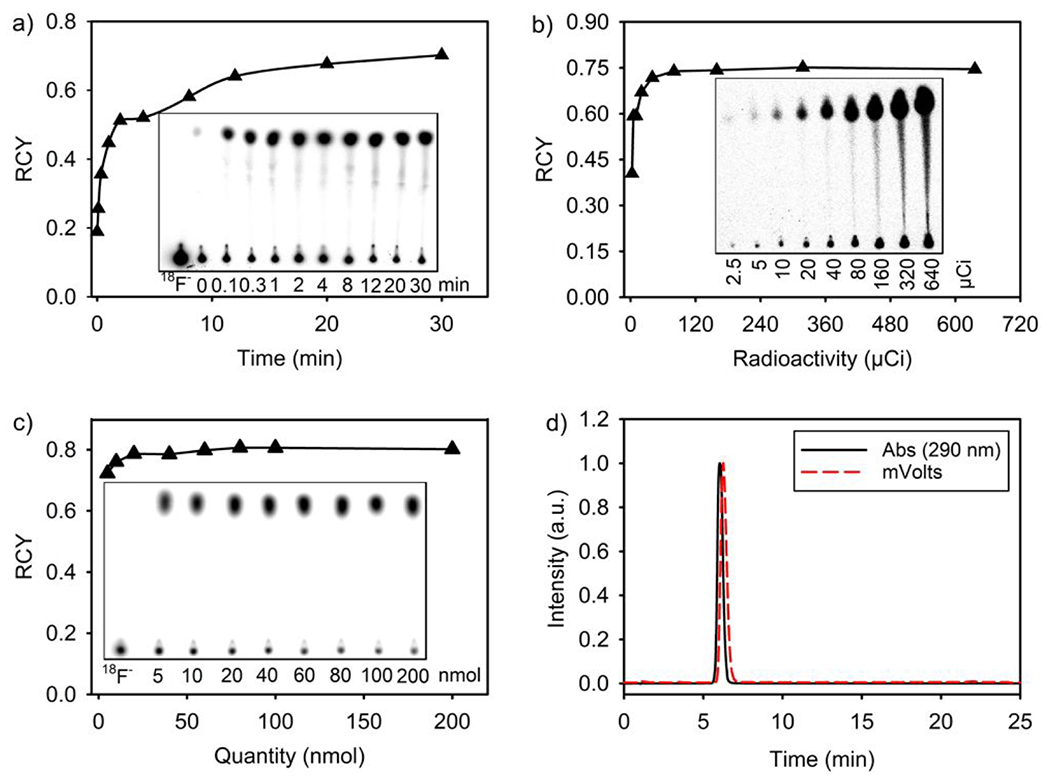
Rapid, room temperature (21 °C) 18F-19F IEX radiolabeling of 1. a) Kinetic analysis of room-temperature radiolabeling. A 0.5 mg quantity of 1 was dissolved with a SnCl4/acetonitrile solution containing 0.6 mCi [18F]-fluoride ion. At different time points, 0.2 μL aliquots of the reaction were drop cast onto silica TLC with 100% methanol as the mobile phase. (Fraction-RCY is reported where 1.0 = 100% conversion). b) Analysis of fraction-RCY as a function of starting [18F]-fluoride-ion activity. Quantities of 1 (0.5 mg) were dissolved with SnCl4/acetonitrile solutions containing increasing quantities of [18F]-fluoride ions. Fraction-RCYs were determined after 30 min. c) Determination of 1 molar activity as a function of 1 quantity. RCYs were measured after 30 min of room temperature radiolabeling with 0.32 mCi of [18F]-fluoride-ion in SnCl4-containing anhydrous acetonitrile (200 μL in total volume) and decreasing quantities of 1 (25-1000 μM in concentrations). d) HPLC characterization of 1 radiosynthetic purity, and proof of isotopic exchange. At 20 min, 10%–60% linear acetonitrile/water gradient flow and C18 Reversed Phase HPLC was used to resolve the crude 1. A peak (6.1 min) indicating radiolabeled 1 is observed. An HPLC peak at ~1 min, where free [18F]-fluoride ion generally appears, is not visible.
Low and high molar activity radiolabeling of difluoro-dioxaborinin 1 and low molar activity radiolabeling of difluoro-dioxaborinins 2, 3, 5, 6, 9, 10, 13, 14, 15, 16, and 17.
Variations of the general radiolabeling experiment were performed to determine the radiolabeling rates, efficiency, and identify the maximum achievable molar activities.
In experiments used to identify the rates of radiolabeling, 0.5 mg (3.4 μmol) 1 was dissolved in 2% SnCl4 anhydrous acetonitrile solution containing 0.32 mCi dry [18F]-fluoride-ion. At different time points (referenced to the start of radiolabeling, t = 0 min), samples undergoing radiolabeling were spot at the baseline of a silica gel plate for further analysis with thin layer chromatography (TLC). TLC spotting quenches the radiolabeling reaction, as unreacted [18F]-fluoride-ion is captured by the silica gel on the plate. After a majority of solvent had evaporated from each spot, the TLC plates were resolved with 100% methanol as the mobile phase. To rule out dissolved fluoride-ion radioactivity in the mobile phase, a control spot containing only [18F]-fluoride ion is co-run as a standard on all TLC (Figure 3a and 3c, left-most spot). [18F]-fluoride ion is not carried in the mobile phase. Radiochemical yield (RCY) was calculated by dividing the ratio of radioactivity of 1 near the solvent front by the sum of the radioactivity near the solvent front plus the baseline. [18F]-1 IEX radiochemistry was mostly completed 10 min after the start of radiolabeling, where RCY > 50% (Figure 3a). [18F]-1 radiolabeling is rapid, which is especially appreciated given that F-18 has a relatively short half-life (t1/2 = 110 min). Rapid radiolabeling is important for the maintenance of radiotracer activity amounts that are sufficiently high to account for pharmacokinetic distribution and PET scanning in patient studies.
In experiments for identifying the efficiency of radiolabeling, it was found that RCYs higher than 50% could be achieved with dry [18F]-fluoride ion at quantities that are higher than 5.0 μCi (Figure 3b). These results show that large difluoro-dioxaborinin radiolabeling efficiencies can be achieved using reduced quantities of starting radioactivity. The efficient radiosynthesis requires low-activity quantities of starting activities (fluoride-18 ion), while guaranteeing large yields (RCYs) of radiotracer. This is important in clinical production, where radiochemistries have to yield high amounts of radiotracer under clinical production.
In experiments used to identify maximum achievable molar activities, different quantities of 1 were radiolabeled with an equal radioactivity amount of 0.32 mCi dry [18F]-fluoride ion to study the impact of 1 quantity on the RCY. It was found that 1 at quantities as low as 5 nmol could be radiolabeled at an RCY as high as 70% in SnCl4-containing anhydrous acetonitrile with a total volume 200 μL (25 μM in concentration) (Figure 3c). The largest molar activity was determined to be 37.8 mCi/μmol, when 5 nmol 1 was radiolabeled with 0.32 mCi (Figures 3c and SI section 4). Encouraged with Figure 3b data suggesting that 1 radiolabeling is accommodative of low quantities of starting radioactivity, we used a higher quantity of starting [18F]-fluoride ion radioactivity, specifically a quantity of 16.4 mCi, to react 5 nmol and 0.5 nmol 1 in a SnCl4-containing anhydrous acetonitrile with a total volume 200 μL, respectively. A molar activity of ~ 1.4 Ci/μmol (not decay corrected) was achieved using 16.4 mCi dry [18F]-fluoride ion and 5 nmol 1 in a SnCl4-containing anhydrous acetonitrile with a total volume 200 μL for radiolabeling (SI section 9) and a larger molar activity of ~ 7.4 Ci/μmol (not decay corrected) was achieved using 16.4 mCi dry [18F]-fluoride ion and 0.5 nmol 1 in a SnCl4-containing anhydrous acetonitrile with a total volume 200 μL for radiolabeling (SI section 9). These results surpass the recommended molar activity for [18F]-FDG-clinical application (~ 1 Ci/μmol),11 while the starting radioactivity amount (16.4 mCi) used for radiolabeling 1 is relatively safe for a radiochemist to handle without the use of robotics.
RCYs were also measured for all other synthesized difluoro-dioxaborinins (using 0.6 mCi of [18F]-fluoride ion and 0.5 mg quantities of all non-radioactive difluoro-dioxaborinins) in 30 min long radiolabeling experiments. Generally, RCYs are > 90%, as determined by radio-HPLC (Figures 3d, Table 1 and SI section 3). RCYs were corroborated by silica TLC using pure methanol as the mobile phase. Radio-HPLC analyses of the fluorescent derivatives (SI section 7) suggest that TLC-autoradiography-determined RCYs (61%–74%) underestimate the true RCYs. This is confirmed by the observation of fluorescent, radiolabeled products retained at the baseline of TLC (SI section 7).
Table 1.
A summary of the radiochemical and solvolytic properties of synthesized difluoro-dioxaborinin compounds.
| Compounds | RCY (%) by HPLC | RCY (%) by silica TLC (n = 3) | Isolated RCY | Stability (t1/2, h) 90% H2O/10% DMSO | Stability (t1/2, h) 10% H2O/90% DMSO | Fold increase in stability (90% to 10% H2O) |
|---|---|---|---|---|---|---|
| 1 | 99 | 69.6 ± 12.6 | 2.73 ± 0.02 | 7.63 ± 0.18 | 2.8 | |
| 2 | 97 | 67.0 ± 12.7 | 17.1 ± 0.7 | 53.9 ± 1.1 | 3.2 | |
| 3 | 97 | 69.9 ± 12.8 | 15.2 ± 0.5 | 53.9 ± 1.5 | 3.5 | |
| 5 | 93 | 64.1 ± 12.6 | 49.9 | - | 391 ± 4 | |
| 6 | 99 | 68.5 ± 12.7 | 5.19 ± 0.15 | 24.6 ± 0.5 | 4.7 | |
| 9 | 75 | 61.6 ± 12.6 | 37.3 | - | 11.9 ± 0.4 | |
| 10 | 80 | 64.4 ± 12.6 | - | 16.0 ± 0.2 | ||
| 13 | 98 | 68.0 ± 12.6 | - | 10.7 ± 1.8 | ||
| 14 | 98 | 67.5 ± 12.5 | - | 4.46 ± 0.05 | ||
| 15 | 98 | 66.0 ± 12.4 | 6.75 ± 0.19 | 33.7 ± 1.1 | 5.0 | |
| 16 | 88 | 67.2 ± 12.5 | - | 118 ± 12 | ||
| 17 | 92 | 73.8 ± 12.9 | - | 122 ± 3 |
Radiochemical and solvolytic measurements on the compounds shown in Figure 2. RCYs were determined after a 30 min, room-temperature radiolabeling. Solvolytic stability was determined by 19F NMR (Supporting Information Section 13). ‘–’ denote solvolytic stabilities that could not be determined due to compound insolubility.
Difluoro-dioxaborinin solvolytic stability evaluation of compounds 1, 2, 3, 5, 6, 9, 10, 13, 14, 15, 16, and 17.
Solvolysis can be a problem in the in vivo application of F-18 radiotracers, including clinical 18F-difluoro-dioxaborinin applications. In aqueous solution, free [18F]-fluoride ion is ultimately generated upon difluoro-dioxaborinin solvolysis. In a patient, generated [18F]-fluoride ion will accumulate at the bone resulting in a strong background that can interfere with 18F-PET image interpretation. Perrin et al. describe a strategy to control the solvolysis rates of aryl and alkyl trifluoroborates by modifying the electronegativity of nearby substituents.12 Fortunately, a similar strategy can be employed to control the solvolysis rates of [18F]-difluoro-dioxaborinin. Using the compounds shown in Figure 2, we measure and identify the mechanisms that contribute to the solvolytic stability of [18F]-difluoro-dioxaborinins.
Ideally, [18F]-difluoro-dioxaborinins should have a solvolytic stability that is much greater than the nuclear stability of F-18 (t1/2 = 110 min, ~2 h) for a [18F]-difluoro-dioxaborinin to be useful as clinical [18F]-PET contrast (e.g. as a lymphatic mapping agent in lumpectomy and axillary surgery). All reported, characterized compounds have this solvolytic stability. Both silica TLC autoradiography and 1H/19F NMR were used to determine the stability of the synthesized compounds shown in Figure 2 (Figure 4a, 4b and SI section 14). In the presence of water, all [18F]-difluoro-dioxaborinins underwent gradual aqueous solvolysis; however, they were not equally stable. To calculate the solvolytic half-lives of difluoro-dioxaborinins in the aqueous solution, the integration of the 19F NMR peak of the original difluoro-dioxaborinins was divided by the sum of the integrations of all 19F NMR peaks including the original difluoro-dioxaborinins and solvolytic products. Then, the data for different time points were plotted and fitted according to a single, 2-parameter exponential decay equation (f = a*exp(−k*x), R2 > 0.99 in 88% of the experiments, where f is the fraction of difluoro-dioxaborinin yet to be solvolyzed; a is the starting fraction of difluoro-dioxaborinin (1.0); k is the decay constant, rate constant, or transformation constant; x is time; and R2 is the closeness of a least-squared regression analysis fit (where 1.0 is exact correlation between empirical data and mathematical model) (SI section 13). The statistical, R-squared value (R2, Sigma plot 10.0) was > 0.99 in 88% of the experiments, which indicates that the 2-parameter exponential decay equation is likely the correct equation to predict the chemical half-life against solvolysis. In statistics, (R2) > 0.95 has been taken as sufficient evidence to back a scientific conclusion. As seen in Table 1, all difluoro-dioxaborinins, including the least stable [18F]-difluoro-dioxaborinin (14), possess solvolysis half-lives longer than the F-18 nuclear decay half-life (110 min, ~ 2h i.e., t1/2 difluoro-dioxaborinin solvolysis > t1/2 decay of atomic F-18).
Figure 4.
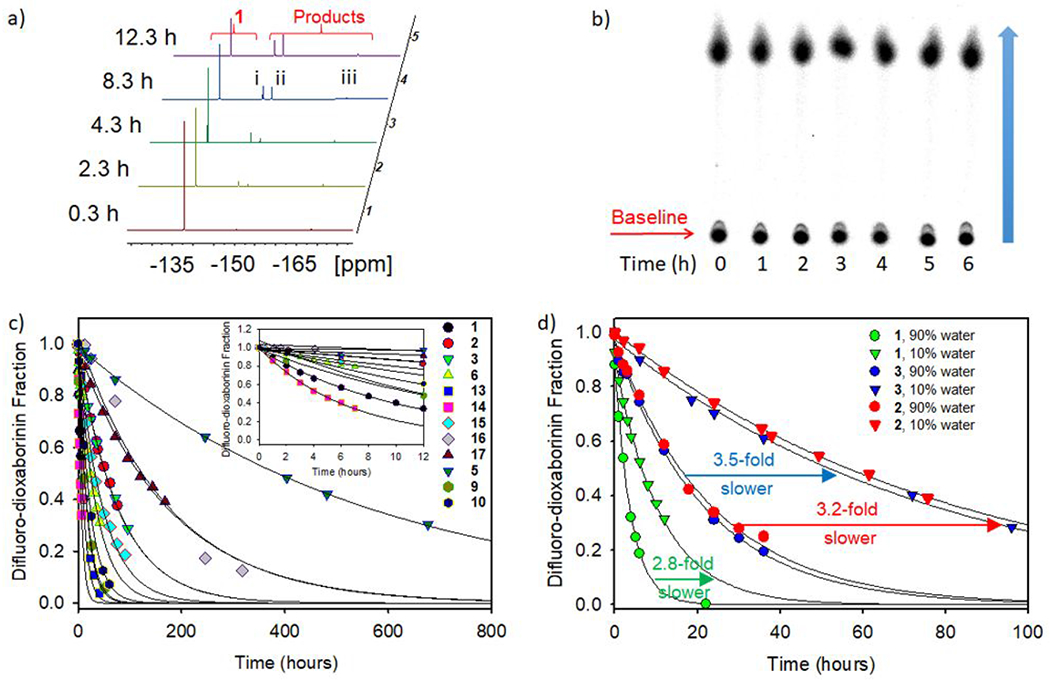
Measurement of difluoro-dioxaborinin solvolysis by 19F NMR and autoradiography. a) Representative 19F NMR stack showing solvolysis of 1 over 12 hours in 10% water/90% DMSO (5.55 M H2O) (see Figure in SI section 13 for 19F NMR analysis of all difluoro-dioxaborinins). 1 appears at −137.9 ppm and solvolyzes into three products: i) an intermediate (−147.5 ~ −148.5 ppm), ii) a fluoroborate ion, (SI section 9, −150.5 ppm), and iii) a fluoride ion (−168.6 ppm). Solvolysis products i) and ii) carry B-F bonds, as indicated by the 10B-19F/11B-19F isotopic 19F NMR shift (SI section 13.2). b) 18F-TLC phosphorimaging of 1 over a 6-hour period in 5.55 M H2O/PBS buffer. c) First order decay for all compounds shown in Figure 2 in 10% water/90% DMSO. 5 and 14 are the least and most stable compounds, respectively. d) Plot of 19F NMR solvolytic data for highly soluble 1, 2 and 3 in 10% water/90% DMSO (triangles) and 90% water/10% DMSO (circles). Generally, reducing the molarity of water 10-fold delays difluoro-dioxaborinin solvolysis by 3- to 5-fold.
It was found that some [18F]-difluoro-dioxaborinins had a water solubility that allowed them to be resolved on silica TLC with water as the mobile phase (1, 2, 3, 6, 15) Figure in SI section 8). All solvolytic studies of water-soluble difluoro-dioxaborinins (SI section 13) were performed in 50 M H2O (90% water/10% DMSO). We additionally chose to study compounds in 90% water/10% DMSO, as this media approaches physiologically appropriate conditions that allow the stability of these difluoro-dioxaborinins to be compared with rates of aryl/alkyl trifluoroborate solvolysis in the literature.11 Difluoro-dioxaborinins solvolysis was not observed in pure (100%) DMSO (SI section 13.2.4).
Unfortunately, some difluoro-dioxaborinins were insoluble in water; therefore, solvolytic studies of all difluoro-dioxaborinins could only be performed in 10% H2O/90% DMSO (5.55 M H2O, deionized). All 19F NMR solvolysis data closely follow a single, 2-parameter exponential decay (f = a*exp(−k*x), R2 > 0.99 in 88% of the experiments) (SI section 13). This result suggests that, under the reported conditions, aqueous [18F]-difluoro-dioxaborinin solvolysis is irreversible, and that solvolysis is dependent on a single, rate-determining, [H2O] dependent first-order reaction.
1 is the lowest molecular weight compound and the second most rapid difluoro-dioxaborinin to undergo aqueous solvolysis. In the absence of water (100% DMSO), 1 solvolysis was not observed (SI section 13.2.4). A 9-fold reduction in water concentration to 5.5 M H2O resulted in a 2.8-fold reduction in the 1 solvolysis rate (Figure 4d). These data suggest that 1 is less useful in PET imaging, as 1 would solvolyze at a half-life that is nearly on par with the decay of F-18. Assuming the recorded in vitro solvolysis rate translates in vivo, 37% of 1 will have decomposed at 110 min (one F-18 half-life) post i.v. injection, which will result in a serious background in PET imaging.
2, 3, 6 and 15 were synthesized and evaluated in an attempt to design more solvolytically stable [18F]-difluoro-dioxaborinins. 2, 3, 6, and 15 all bear substituents with a C-H σ-bond at position 5 of difluoro-dioxaborinin (Figure 1a). It is hypothesized that a position-5 substituent will stabilize the 4, 5, 6-dioxaborinin π-system through hyperconjugation. Position-5 modification was mildly successful in stabilizing the difluoro-dioxaborinin towards solvolysis. Position-5 methylated (2), ethylated (3), proprionated (ethyl ester, 6), or cyclohexylated (15) difluoro-dioxaborinins, were 6.2, 5.6, 1.9, and 2.5 times more resistant to solvolysis than 1 in 50 M H2O, respectively (Figure 4c and table 1). Therefore, 2, 3, 6, and 15 would be more stable for clinical 18F-PET imaging. Solvolysis rates of 2, 3, 6 and 15 were delayed 3.2-, 3.5-, 4.7-, and 5.0-fold, respectively, in 5.5 M H2O vs. 50 M H2O (Table 1). These data demonstrate that [H2O] is an important factor in accelerating the solvolysis of difluoro-dioxaborinins.
To evaluate the effect of extending the difluoro-dioxaborinin 4,5,6 π-system on solvolysis, compounds that bear an extended π-conjugation through the difluoro-dioxaborinin moiety (positions 4 and 6) were synthesized. 13 and 14 bear moderately electron-poor π-substituents at the R1 and R2 positions and underwent more rapid solvolysis than 1 (Figure 4c and Table 1). We attempted to test the corollary, by synthesizing difluoro-dioxaborinins (4, 7, 8 and 12) bearing even more electron-poor π-substituents at the R1 and R2 positions. Unfortunately, we failed to purify these compounds by C18 reverse-phase preparative HPLC. We hypothesize that these difluoro-dioxaborinins are unstable and cannot be isolated because of the electron poor groups. We hypothesize that the very electron-poor R1 and R2 π-substituents lead to rapid solvolysis preventing isolation.
Difluoro-dioxaborinins with electron-rich, extended difluoro-dioxaborinin 4,5,6 π-systems proved to be the most solvolytically stable. 5, 16 and 17 bear para-phenolic alcohols and ether functionality, allowing resonance structures to be contemplated showing electron donation through positions 4 and 6 in the difluoro-dioxaborinins. 5, 16 and 17 underwent solvolysis 51-,15-, and 16-fold more slowly than 1, and 7.3-, 2.2-, and 2.3-fold more slowly than 2, respectively.
Among all Table 1 compounds, 5 was most resistant to solvolysis. We hypothesized that the pendant phenol-alcohols on 5 contribute electron density to the π-system of 4,5,6-difluoro-dioxaborinin, thus delaying solvolytic decomposition. To confirm this hypothesis, 5 phenolic alcohols were esterified to generate 9 and 10. Ester functionality was less electron-rich than a phenolic alcohol functionality. As hypothesized, 9 and 10 underwent solvolysis 33- and 24-fold more rapidly than 5 (determined in 5.55 M H2O) (Table 1 and Figure 4c).
Difluoro-dioxaborinin fluorescent property determination of compounds 1, 2, 3, 5, 6, 13, 14, 15, 16, and 17.
The placement of electron-rich functionality at R1 and R2 positions of difluoro-dioxaborinin not only improved the compounds’ resistance to solvolysis but also induced donor-acceptor-pair enhanced redshifted optical properties (Figure 5). Both absorption peaks and emission peaks shifted to a longer wavelength with stronger electron-rich functionality at the R1 and R2 positions (Table 2). This redshift was visible in the four derivatives (Table 2 and SI section 7). 5-backbone-bearing derivatives with strong-rich functional groups at the R1 and R2 positions have been reported in the literature. These compounds emit in the near-infrared region (> 700 nm).13 Based on these data, it should be possible to transform near-infrared fluorescent, difluoro-dioxaborinin probes,14 which are specific for amyloid-beta deposits or brown adipose tissue,15 into near-infrared optical/18F-PET dual modality contrast using our described radiochemistry. Interestingly, the modification of a β-diketone as a difluoro-dioxaborinin induces an optical redshift compared with the original β-diketones (Table 2). This finding supports the minimal modification of β-diketones into difluoro-dioxaborinins as a way for developing more useful, redshifted fluorescent/18F-PET dual modality probes.
Figure 5.
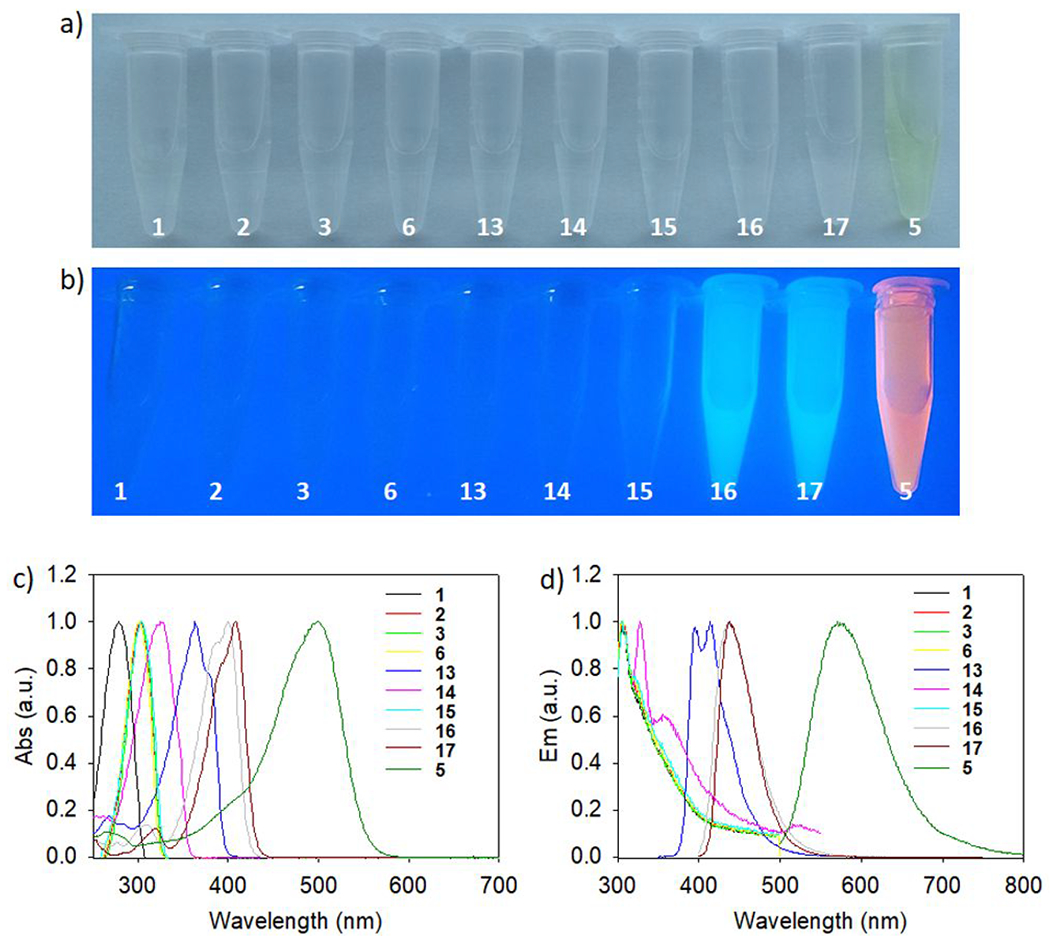
Optical properties of selected Figure 2 difluoro-dioxaborinins. a) A photo of 1, 2, 3, 6, 13, 14, 15, 16, 17, and 5 methanol solutions under a) ambient light and b) 365 nm lamp irradiation. c) The absorption spectra for the compounds shown in Figure 2 in methanol. d) The emission spectra for the compounds shown in Figure 2 in methanol.
Table 2.
A summary of the optical properties of synthesized difluoro-dioxaborinin compounds.
| Compounds | Abs (nm) | Em (nm) | Extinction Coefficient (cm−1M−1) | Quantum yield (%, in Methanol) | Stokes Shift (nm) |
|---|---|---|---|---|---|
| 1 | 279 | 305 | 9,180 | * | 26 |
| 2 | 303 | 307 | 12,200 | * | 4 |
| 3 | 303 | 306 | 12,000 | * | 3 |
| 5 | 500 | 571 | 73,000 | 1.3 | 71 |
| 5’ | 434 | 535 | 56,500 | 2.4 | 101 |
| 6 | 301 | 307 | 12,600 | * | 6 |
| 9 | 430 | 512 | 50,800 | 3.0 | 82 |
| 10 | 434 | 509 | 54,000 | 3.1 | 75 |
| 13 | 363 | 413 | 38,000 | 11.2 | 50 |
| 13’ | 343 | 390 | 29,000 | * | 47 |
| 14 | 325 | 328 | 19,800 | * | 3 |
| 15 | 303 | 307 | 12,000 | * | 4 |
| 16 | 400 | 439 | 62,000 | 67.3 | 39 |
| 16’ | 358 | 388 | 44,400 | * | 30 |
| 17 | 408 | 438 | 63,600 | 65.8 | 30 |
| 17’ | 363 | 391 | 33,800 | * | 28 |
Optical measurements for the compounds shown in Figure 2. Optical properties were determined in methanol. Quantum yields were determined by using quinine bisulfate (QY = 0.546) as the reference.16
Could not be determined due to short excitation wavelengths. Entries 5’, 13’, 16’, and 17’ are β-diketone precursors to 5, 13, 16, and 17, respectively and are shown to illustrate redshifted optical properties that are conveyed onto difluoro-dioxaborinin products.
[18F]-difluoro-dioxaborinin PET imaging of mice digestive track using compound 5
Esophageal, intestinal and small bowel perforation or obstruction are diseases impacting normal digestive system flow that may induce intestinal necrosis, consequent infection, and even result in death. Quick and precise diagnosis can facilitate localization of the event for surgical treatment. Herein, normal digestive system imaging was conducted with [18F]-5. Figure 6a and videos 1 and 2 show maximum intensity projection (MIP) of X-ray CT or PET/CT images of mice mouse fed with clinically available BaSO4 suspension. Small animal CT is not sensitive enough to observe the digestive system. Both soft tissue and bone contribute signal to a CT image, making it difficult to recognize digestive system quickly with BaSO4. In comparison, an 18F-PET signal is exogenous, there is no background or endogenous signal to subtract, making PET a higher contrast for quick recognition. PET signal is clearly seen in the intestines in Figure 6b and video 2, suggesting that [18F]-5 could be used to image esophageal, intestinal, and small bowel perforation or obstruction are diseases impacting normal digestive system.
Figure 6.
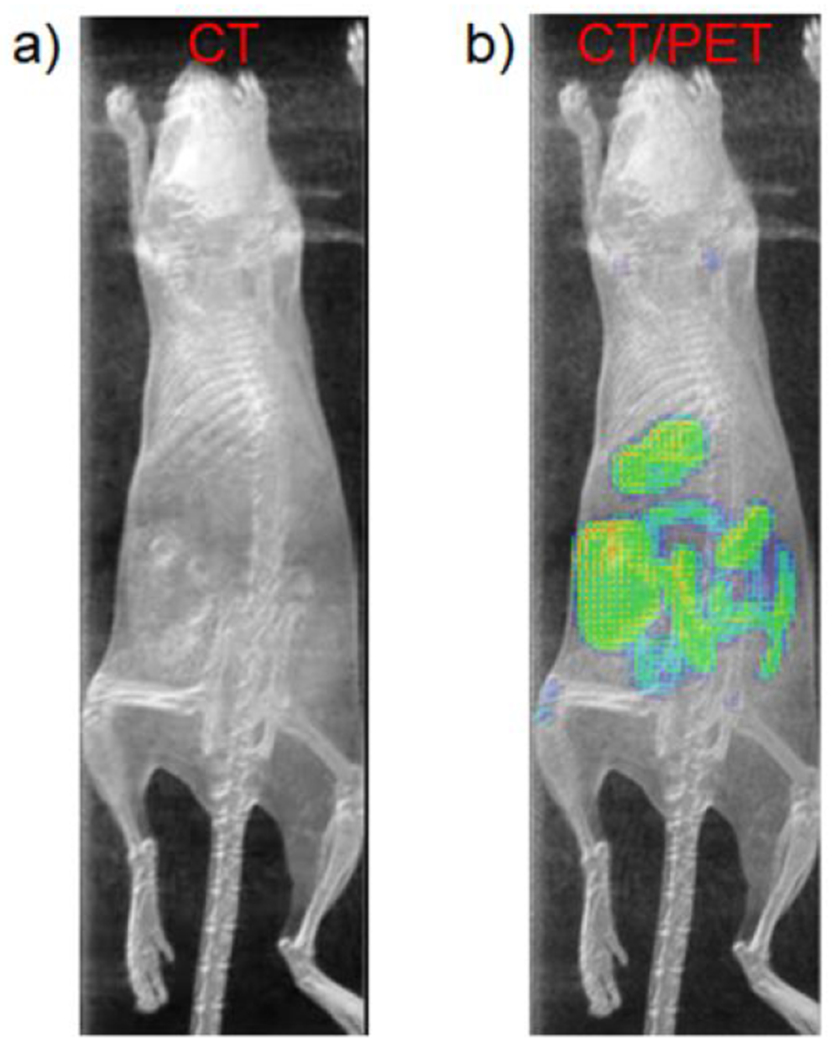
a) The MIP CT image of the healthy mouse 1.5 hours post being fed with clinical BaSO4 suspension (Bracco diagnostics product #450104 Redi-Cat-2). b) The overlay of MIPs of CT and PET images in healthy mice 1.5 hours post being fed with a mixed suspension of clinically available BaSO4 suspension and [18F]-5 (~ 30 μCi, 400 μL mixed suspension for each mouse). CT signal is presented in gray. PET signal is presented in the NIH color table. Intestines are more clearly delineated by PET/CT vs. CT (See Video in Supporting information).
[18F]-difluoro-dioxaborinin PET imaging of mice, rabbits and patient lymph nodes using compound 9.
Sentinel node mapping and resection are clinically important in cancer management and staging. To show clinical utility of difluoro-dioxaborinins, [18F]-9 was tested in sentinel node mapping in murine and leporine models. [18F]-9 (0.185 MBq for mice and 7.4 MBq for rabbits, respectively) was injected as a 2% DMSO/98% 1× PBS solution (pH 7.4) into the right foot fat pad of mice and rabbits. [18F]-9 contrast imaging of sentinel nodes was performed according to the literature (Figures 7a, 7b, 7c, and SI sections 19, 20).17 The models are based on healthy Bal b/c mice and rabbits, which are pretty simple. The lymph node close to the paw of a rodent animal is in the armpit, which we regard as the sentinel node serving the paw fat pad. Injected contrast penetrates the lymphatic vessels and flow to the lymph node at the armpit, as is reported in many publications.17 Only sentinel nodes (and not distal nodes) show uptake by PET in all models. Sentinel lymph nodes and lymph tracks are visible in rabbit PET images. Flow-through to distal (lumbar) nodes was not observed. These findings indicate that [18F]-9 contrast could serve as a competitive alternative to non-targeted fluorescence contrast including indocyanine green (ICG) and lymphazurin (isosulfan blue),18 which travel through sentinel nodes to delineate multiple non-sentinel nodes. Unlike [18F]-9, ICG and lymphazurin does not allow for PET imaging. In mouse models, [18F]-9 fluorescence was also visible in exposed sentinel nodes. Fluorescence imaging was captured using excitation and emission wavelengths of 430 nm and 520 nm, respectively. These wavelengths approach the absorption and emission peaks of 9 (see table 2). The fluorescence imaging further confirmed the accumulation of [18F]-9 in murine sentinel nodes. To visualize [18F]-9, it was necessary to remove the skin immediately overlying each node, as the skin shows a high degree of autofluorescence (Figure 7). Fluorescence signals in the collected nodes were visible by histological examination (SI section 19.2), where fluorescence imaging is useful for confirming the accumulation of [18F]-9 in murine sentinel nodes. In the clinical setting, [18F]-9 is potentially useful for intraoperative navigation during clinical lymphatic dissection, where [18F]-9 fluorescence can be visualized with an optical or gamma camera in real-time to guide the sentinel node resection.
Figure 7.
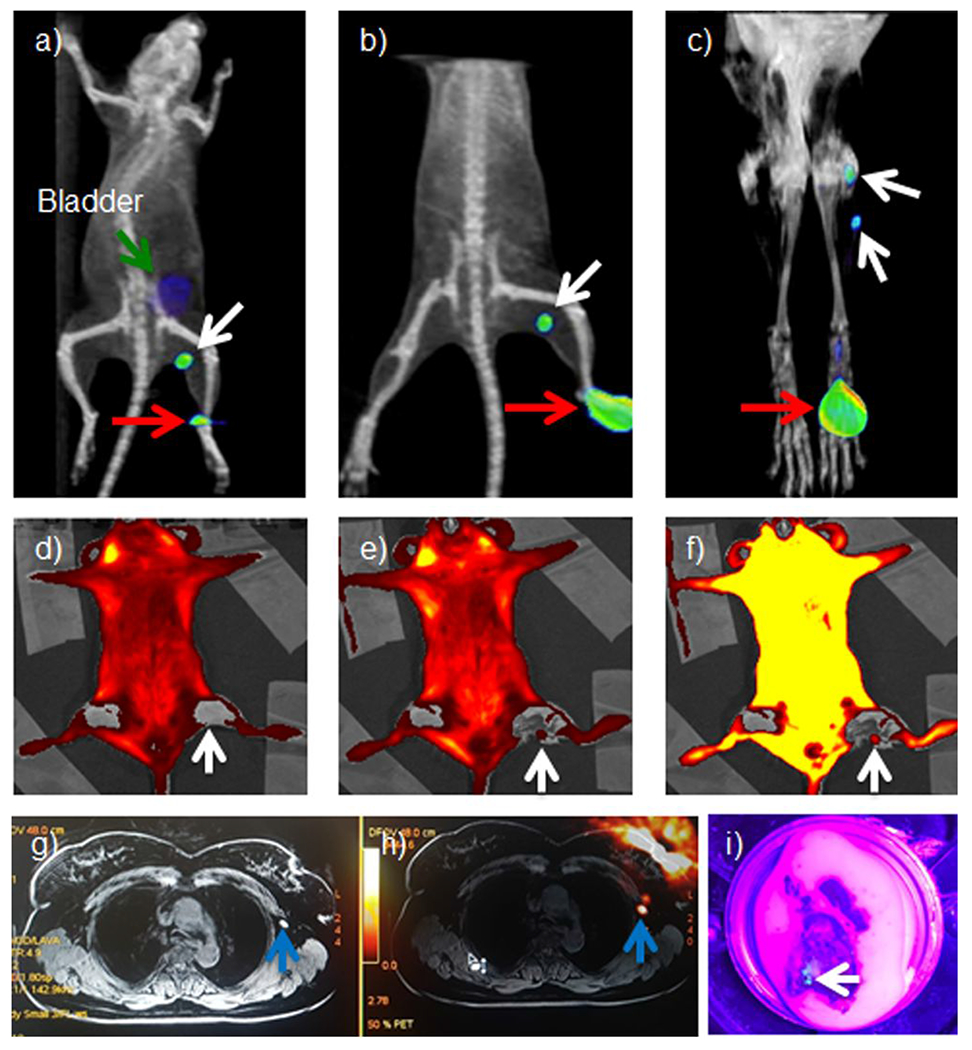
Sentinel lymph node imaging using [18F]-9. A 0.185 MBq radioactivity amount of [18F]-9 solution was injected into the right rear footpad of mice (n = 3). The radiotracer penetrates into lymphatic vessels and flows to the sentinel lymph nodes automatically, as previously described in many reports.17 The injection site (red arrow), sentinel lymph node (popliteal, white arrow), and bladder (green arrow) are visible at 2 hours (a) and 4 hours (b) post-injection. c) PET/MRI imaging of rabbit sentinel (popliteal) lymph nodes (there are two nodes) using [18F]-9. A 7.4 MBq injection of [18F]-9 was made into the right rear paw of a rabbit. The injection site (red arrow) and sentinel lymph node (popliteal, white arrow) are visible 1h post-injection. d-f) Fluorescence confirmation of [18F]-9 in murine sentinel nodes was carried out using 430/520 nm excitation/emission optical filtration, i.e., wavelengths that approach the absorption and emission peaks of 9 (see table 2). d) Skin on the predissection fluorescent image (node is not visible). e) Fluorescent image where skin over the sentinel lymph node is removed (node is visible). f) Overexposed Figure (e) (node is more visible). g) An MR image of an enlarged lymph node (1.5-2 cm) in a breast cancer patient. A blue arrow indicates the lymph node. h) A PET/MR image of a breast cancer patient where the lymph node is confirmed by [18F]-9 PET. (1.2 GBq (32 mCi) of 18F− was used to synthesize [18F]-6F-Cur-BF2. A 0.45 GBq (12 mCi) quantity of product was isolated). A blue arrow indicates the lymph node. i) The [18F]-9 fluorescence image of the same resected lymph node within the lumpectomy tissue-specimen excised from the breast cancer patient. The tissue was imaged under 380-400 nm light illumination. The lymph node is cyan-blue in coloration and is the same node indicated by the blue arrow in figures 7g–h (See experimental details in supporting information).
In a healthy physician volunteer, we highlight [18F]-DK utility in identifying of trochlear sentinel nodes from interdigital injection. As [18F]-9 travels through lymphatic channels and sentinel nodes, [18F]-9 is deposited in the lymph tracks and nodes that immediately serve the injection site in mice and rabbits (SI sections 21.1 and 21.2). In a healthy physician volunteer who received [18F]-9 utility in an interdigital injection, the sentinel (trochlear) sentinel nodes are clear in PET/MR (SI section 21.3). This promising clinical data suggests that [18F]-DKs would be highly useful in lymphatic mapping scenarios, e.g. partial mastectomy.
[18F]-9 was used to image sentinel nodes in patient that was a candidate for partial mastectomy-lumpectomy due to breast cancer (SI Section 21.4). As seen in Figure 7g, a lymph node is visible in the MR image. It is not 100% clear that this signal is a lymph node as the white coloration in the MR is easily confused with the surrounding tissue signals. In comparison, the PET signal of the lymph node is clearly visible in the PET/MR image (Figure 7h). The lymph-node containing tissue was recovered after surgical resected and confirmed under fluorescent 380-400 nm light. This node is indicated by the white arrow (Figure 7i).
DISCUSSION
We report a methodology for creating wavelength-tunable, [18F]-positron-emitting difluoro-dioxaborinins.6,14 A 6–8 ppm α-hydrogen shift in the 1H-NMR of all synthesized difluoro-dioxaborinin suggests a deshielded proton similar to the shift of phenyl C-H protons, suggesting that a difluoro-dioxaborinin could substitute for phenyl-moiety-aromaticity in diagnostic drug design and natural product modification.
Resulting difluoro-dioxaborinins undergo aqueous-solution solvolysis at rates that are substituent dependent, where electron-poor substituents result in faster solvolysis. Most organic fluorophores owe their fluorescent properties to electron-rich systems with extended, π-conjugated functionality including fluorescein, rhodamines, and cyanines.19 The incorporation of difluoro-dioxaborinins into these advanced systems should give rise to solvolysis resistant, highly fluorescent/18F-PET dual modality contrast agents.
In general, difluoro-dioxaborinins undergo [H2O]-dependent first-order solvolysis. In the compounds that we report, the time required for solvolysis is generally longer than the atomic decay half-life of F-18 (110 min). It was observed that different difluoro-dioxaborinins underwent different rates of solvolysis. Solvolysis can be delayed by: 1) adding hyperconjugative σ-electron pair donating substitutions at position 5, 2) extending π-conjugation through electron-rich moieties at positions 4 and 6, and 3) reducing the water content. Conversely, solvolysis can be made more rapid by placing extended, electron-poor π-conjugation at positions 4 and 6.
To demonstrate in vivo utility in sentinel node mapping, we exploited the fact that [18F]-difluoro-dioxaborinins does not undergo solvolysis in pure DMSO. We also exploited the aqueous insolubility of [18F]-9 twice, once to allow rapid radiochemical purification by precipitation and a second time during in vivo sentinel node mapping. We speculate that as [18F]-9 travels through lymphatic channels, minor quantities of DMSO in the solution are separated from [18F]-9 and that [18F]-9 is deposited in the lymph tracks and nodes that immediately serve the injection site. We demonstrate the clinical potential of [18F]-9 by using [18F]-9 for PET/CT and fluorescent sentinel node mapping during breast cancer surgery to facilitate the intraoperative sentinel node resection in a patient.
CONCLUSIONS
In conclusion, we report a general method for F-18 radiolabeling β-diketone-bearing structures. Some [18F]-difluoro-dioxaborinins are solvolytically stable enough for in vivo PET application. Difluoro-dioxaborinins can be radiolabeled in a rapid, one-step, room-temperature, SnCl4-mediated 18F/19F IEX reaction. We report general formulae for the design of stable, fluorescent 18F-PET difluoro-dioxaborinins that can be used to transform β-diketone containing pharmaceuticals and natural products into 18F-PET contrast agents. This work impacts the design of small-molecule, fluorescent radiotracers, and describes a general F-18 radiochemistry for future PET drug development and drug discovery.
EXPERIMENTAL SECTION
General Procedures
Chemicals were purchased from Oakwood Chemical, Sigma-Aldrich, Combi-blocks, Strem, and Alfa Aesar and used without further purification. Reaction progress and determination of purity were conducted via analytical, reverse phase UPLC on a Waters Acquity H class HPLC/SQD2 mass spectrometer and a Phenomenex Kinetex 1.7μm C18 100Å, 50 cm x 2.1 mm I.D. column (00B-4475-AN), with a 1.5 min, a 10-90% H2O:acetonitrile (ACN) (0.05% TFA) gradient and a flow rate of 0.6 mL/min (unless stated otherwise). Preparative HPLC was performed on an Agilent 1200 Series HPLC equipped on a Phenomenex Luna C18(2) 100Å, 250 cm x 21.20 mm I.D. 10 μm reverse phase column (00G-4253-P0 AX), with a flow rate of 12 mL/min. 1H/13C/19F NMR were performed on a 500 MHz Bruker spectrometer at the temperature of 25 °C. HRMS was obtained on an Agilent 6550 QToF with a dual sprayer ESI source, coupled to an Agilent 1290 Infinity LC system. Samples were analyzed by either FIA (flow injection analysis) using a mobile phase of 50% acetonitrile in water (0.1% formic acid) with a flow rate of 0.4 mL/min, or LCMS with an Agilent Poroshell 120 SB-C18 column (2.7um, 2.1x50mm) at 45 °C, using a linear gradient of 5-95% acetonitrile in water (0.1% formic acid) and a flow rate of 0.4 mL/min. Mass spectra were obtained in either positive or negative mode, and acquired using the MassHunter Acquisition Software (version B.05.01); then analyzed by MassHunter Qualitative Analysis (version B.06.00).
General synthesis of difluoro-dioxaborinins (DKs).
The general synthesis of difluoro-dioxaborinins (DKs) was performed as follows: the 1,3-diketoalkane (3 mmol) was dissolved in 10 mL of dry CH2Cl2. BF3 . (OEt)2 (6 mmol) was added to the solution dropwise, under magnetic stirring at room temperature. The reaction was monitored by UPLC. After two hours of reaction, 5 mL of deionized water was added to quench the reaction. The mixture was dried by rotary evaporation. Dried material was re-dissolved in DMF and purified by preparative chromatography (Agilent, 1260 Infinity) equipped with a C18 Reversed Phase LC Column (Luna® 10 μm C18, 100 Å, LC Column 250 × 21.2 mm), water/acetonitrile: 90/10 to 10/90, linear gradient, 20 min, with a flow rate of 12 mL/min, ultrapure water and HPLC acetonitrile (Sigma, St. Louis, MO, USA)). For all reported compounds, purity is >95% as verified by 1H, 19F-NMR and UV-absorbance HPLC (215-900 nm). Silica gel 60 F254 from milllipore sigma (#100390) was used for TLC analysis.
Evidence of 4, 7, 8 and 12 product formation were not found by analytical, reverse phase UPLC. The reaction conditions described are not suitable for the synthesis of 4, 7, 8 and 12.
2,2-Difluoro-4,6-dimethyl-2H-1λ3,3,2λ4-dioxaborinine (1).
Pentane-2,4-dione (300 mg, 3 mmol) was dissolved in 10 mL of dry CH2Cl2. BF3 · (OEt)2 (46.5%, 916 mg, 6 mmol) was added to the solution dropwise, under magnetic stirring at room temperature. The reaction was monitored with UPLC. After two hours of reaction, 5 mL of deionized water was added to quench the reaction. The mixture was dried by rotary evaporation. Dried material was re-dissolved in DMF (HPLC) and purified by preparative C18 Reversed Phase LC chromatography. The gradient was water/acetonitrile at a flow rate of 12 mL/min (100/0 to 40/60 linear gradient, 20 min, and then isocratic water/acetonitrile 40/60 for another 25 min). Ultrapure water and HPLC acetonitrile (Sigma, St. Louis, MO, USA) were used. The fractions of 21 min to 22.5 min were collected, kept at −80 °C overnight and then lyophilized into white color powder. Yields: 242 mg (54.5%). 1H NMR (500 MHz, d6-DMSO, 25 °C, TMS): δ 6.40 (s, 1H), 2.34 (s, 6H); 13C NMR (125 MHz, d6-DMSO, 25 °C, TMS): δ 193.32, 102.72, 24.19; 19F NMR (470 MHz, d6-DMSO, 25 °C, CFCl3): [10B]-BF2: δ −137.987, [11B]-BF2: δ −138.049; HRMS (ESI) calc’d for [M]= [C5H7BF2O2]: 148.0507, [M-H]− = [C5H6BF2O2]−: 147.0429, found [M-H]−: 147.0435.
2,2-Difluoro-4,5,6-trimethyl-2H-1λ3,3,2λ4-dioxaborinine (2).
3-Methylpentane-2,4-dione (342 mg, 3 mmol) was dissolved in 10 mL of dry CH2Cl2. BF3 · (OEt)2 (46.5%, 916 mg, 6 mmol) was added to the solution dropwise, under magnetic stirring at room temperature. The reaction was monitored with UPLC. After two hours of reaction, 5 mL of deionized water was added to quench the reaction. The mixture was dried by rotary evaporation. Dried material was re-dissolved in DMF and purified by preparative C18 Reversed Phase LC chromatography. The gradient was water/acetonitrile at a flow rate of 12 mL/min (90/10 to 10/90 linear gradient, 20 min, and then isocratic water/acetonitrile 10/90 for another 25 min). Ultrapure water and HPLC acetonitrile (Sigma, St. Louis, MO, USA) were used. The fraction of 17 min to 18 min was collected, kept at −80 °C overnight and then lyophilized into white color powder. Yields: 283 mg (58.2%). 1H NMR (500 MHz, d6-DMSO, 25 °C, TMS): δ 2.36 (s, 6H), 1.90 (s, 3H); 13C NMR (125 MHz, d6-DMSO, 25 °C, TMS): δ 191.18, 108.07, 23.25, 11.93; 19F NMR (470 MHz, d6-DMSO, 25 °C, CFCl3): [10B]-BF2: δ −139.053 (18.3%), [11B]-BF2: δ −139.116 (81.7%); HRMS (ESI) calc’d for [M]=[C6H9BF2O2]: 162.0664, [M-H]−=[C6H8BF2O2]−: 161.0585, found [M-H]−: 161.0590.
5-Ethyl-2,2-difluoro-4,6-dimethyl-2H-1λ3,3,2λ4-dioxaborinine (3).
3-Ethylpentane-2,4-dione (384 mg, 3 mmol) was dissolved in 10 mL of dry CH2Cl2. BF3 · (OEt)2 (46.5%, 916 mg, 6 mmol) was added to the solution dropwise, under magnetic stirring at room temperature. The reaction was monitored with UPLC. After two hours of reaction, 5 mL of deionized water was added to quench the reaction. The mixture was dried by rotary evaporation. Dried material was re-dissolved in DMF and purified by preparative C18 Reversed Phase LC chromatography. The gradient was water/acetonitrile at a flow rate of 12 mL/min (90/10 to 10/90 linear gradient, 20 min, and then isocratic water/acetonitrile 10/90 for another 25 min). Ultrapure water and HPLC acetonitrile (Sigma, St. Louis, MO, USA) were used. The fractions of 18 min to 19.5 min were collected, kept at −80 °C overnight and then lyophilized into white color powder. Yields: 312 mg (59.1%) 1H NMR (500 MHz, d6-DMSO, 25 °C, TMS): δ 2.39 (J = 1.7 Hz, 6H), 2.36 (J = 7.5 and 1.7 Hz, 2H), 1.04 (J = 7.5 Hz, 3H); 13C NMR (125 MHz, d6-DMSO, 25 °C, TMS): δ 191.52, 114.08, 22.63, 19.56, 13.95; 19F NMR (470 MHz, d6-DMSO, 25 °C, CFCl3): [10B]-BF2: δ −138.365 (18.9%), [11B]-BF2: δ −138.427 (81.1%); HRMS (ESI) calc’d for [M]=[C7H11BF2O2]: 176.0820, calc’d for [M-H]-=[C7H10BF2O2]−: 175.0742, found [M-H]−: 175.0748.
Ethyl 3-(2,2-difluoro-4,6-dimethyl-2H-1λ3,3,2λ4-dioxaborinin-5-yl)propanoate (6).
Ethyl 4-acetyl-5-oxohexanoate (600 mg, 3 mmol) was dissolved in 10 mL of dry CH2Cl2. BF3 · (OEt)2 (46.5%, 916 mg, 6 mmol) was added to the solution dropwise, under magnetic stirring at room temperature. The reaction was monitored with UPLC. After two hours of reaction, 5 mL of deionized water was added to quench the reaction. The mixture was dried by rotary evaporation. Dried material was re-dissolved in DMF and purified by preparative C18 Reversed Phase LC chromatography. The gradient was water/acetonitrile at a flow rate of 12 mL/min (90/10 to 10/90 linear gradient, 20 min, and then isocratic water/acetonitrile 10/90 for another 25 min). Ultrapure water and HPLC acetonitrile (Sigma, St. Louis, MO, USA) were used. The fractions of 18.2 min to 20 min were collected, kept at −80 °C overnight and then lyophilized into white color powder. Yields: 377 mg (50.7%). 1H NMR (500 MHz, d6-DMSO, 25 °C, TMS): δ 4.09 (q, J = 7.1 Hz, 2H), 2.64 (t, J = 7.8 Hz, 2H), 2.53(t, J = 7.8 Hz, 2H), 2.42 (s, 6H), 1.20 (t, J = 7.1, 3H); 13C NMR (125 MHz, d6-DMSO, 25 °C, TMS): δ 191.99, 172.51, 111.41, 60.51, 33.07, 32.98, 21.68, 14.52; 19F NMR (470 MHz, d6-DMSO, 25 °C, CFCl3): [10B]-BF2: δ −138.494 (20.1%), [11B]-BF2: δ −138.556 (79.9%); HRMS (ESI) calc’d for [M]=[C10H16BF2O4]: 248.1031, calc’d for [M-H]−=[C10H15BF2O4]−: 247.0953, found [M-H]−: 247.0958.
2,2-Difluoro-4,6-diphenyl-2H-1λ3,3,2λ4-dioxaborinine (13).
1,3-Diphenylpropane-1,3-dione (673 mg, 3 mmol) was dissolved in 10 mL of dry CH2Cl2. BF3 · (OEt)2 (46.5%, 916 mg, 6 mmol) was added to the solution dropwise, under magnetic stirring at room temperature. The reaction was monitored with UPLC. After two hours of reaction, 5 mL of deionized water was added to quench the reaction. The mixture was dried by rotary evaporation. Dried material was re-dissolved in DMF and purified by preparative C18 Reversed Phase LC chromatography. A water/acetonitrile gradient at a flow rate of 12 mL/min (90/10 to 10/90 linear gradient, 20 min, and then isocratic water/acetonitrile 10/90 for another 25 min). Ultrapure water and HPLC acetonitrile (Sigma, St. Louis, MO, USA) were used. The fractions of 22.5 min to 24 min were collected, kept at −80 °C overnight and then lyophilized into white color powder. Yields: 452 mg (55.4%). 1H NMR (500 MHz, d6-DMSO, 25 °C, TMS): δ 8.40 (d, J = 7.7 Hz, 4H), 7.97 (s, 1H), 7.84 (t, J = 7.4 Hz, 2H), 7.68 (t, J = 7.7 Hz, 4H); 13C NMR (125 MHz, d6-DMSO, 25 °C, TMS): δ 183.19, 136.39, 131.75, 129.89, 129.84, 95.02; 19F NMR (470 MHz, d6-DMSO, 25 °C, CFCl3): [10B]-BF2: δ −139.304 (19.9%), [11B]-BF2: δ −139.367 (80.1%); HRMS (ESI) calc’d for [M]=[C15H11BF2O2]: 272.0820, found [M]−: 272.0823.
2,2-Difluoro-4-methyl-6-phenyl-2H-1λ3,3,2λ4-dioxaborinine (14).
1-Phenylbuane-1,3-dione (486 mg, 3 mmol) was dissolved in 10 mL of dry CH2Cl2. BF3 · (OEt)2 (46.5%, 916 mg, 6 mmol) was added to the solution dropwise, under magnetic stirring at room temperature. The reaction was monitored with UPLC. After two hours of reaction, 5 mL of deionized water was added to quench the reaction. The mixture was dried by rotary evaporation. Dried material was re-dissolved in DMF and purified by preparative C18 Reversed Phase LC chromatography. A water/acetonitrile gradient at a flow rate of 12 mL/min (90/10 to 10/90 linear gradient, 20 min, and then isocratic water/acetonitrile 10/90 for another 25 min). Ultrapure water and HPLC acetonitrile (Sigma, St. Louis, MO, USA) were used. The fractions of 19.7 min to 20.7 min were collected, kept at −80 °C overnight and then lyophilized into white color powder. Yields: 352 mg (55.9%). 1H NMR (500 MHz, d6-DMSO, 25 °C, TMS): δ 8.19 (d, J = 8.1 Hz, 2H), 7.82 (t, J = 7.4 Hz, 1H), 7.66 (t, J = 7.7 Hz, 2H), 7.29 (s, 1H), 2.49 (s, 3H); 13C NMR (125 MHz, d6-DMSO, 25 °C, TMS): δ 194.96, 181.72, 136.27, 131.12, 129.98, 129.43, 98.78, 24.97; 19F NMR (470 MHz, d6-DMSO, 25 °C, CFCl3): [10B]-BF2: δ −138.460 (20.4%), [11B]-BF2: δ −138.523 (79.6%); HRMS (ESI) calc’d for [M]=[C10H9BF2O2]: 210.0664, calc’d for [M-H]−=[C10H8BF2O2]−: 209.0585, found [M-H]−: 209.0591.
2,2-Difluoro-4-methyl-5,6,7,8-tetrahydro-2H-1λ3,2λ4-benzo[e][1,3,2]dioxaborinine (15).
2-Acetylcyclohexan-1-one (420 mg, 3 mmol) was dissolved in 10 mL of dry CH2Cl2. BF3 · (OEt)2 (46.5%, 916 mg, 6 mmol) was added to the solution dropwise, under magnetic stirring at room temperature. The reaction was monitored with UPLC. After two hours of reaction, 5 mL of deionized water was added to quench the reaction. The mixture was dried by rotary evaporation. Dried material was re-dissolved in DMF and purified by preparative C18 Reversed Phase LC chromatography. The gradient was water/acetonitrile at a flow rate of 12 mL/min (90/10 to 10/90 linear gradient, 20 min, and then isocratic water/acetonitrile 10/90 for another 25 min). Ultrapure water and HPLC acetonitrile (Sigma, St. Louis, MO, USA) were used. The fractions of 18.5 min to 20 min were collected, kept at −80 °C overnight and then lyophilized into white color powder. Yields: 358 mg (63.4%). 1H NMR (500 MHz, d6-DMSO, 25 °C, TMS): δ 2.57 (t, J = 6.0 Hz, 2H), 2.37 (t, J = 5.9 Hz, 2H), 2.34 (s, 3H), 1.72-1.65 (m, 4H); 13C NMR (125 MHz, d6-DMSO, 25 °C, TMS): δ 193.23, 189.77, 109.60, 32.26, 22.76, 22.71, 21.73, 20.91; 19F NMR (470 MHz, d6-DMSO, 25 °C, CFCl3): [10B]-BF2: δ −138.361 (19.2%), [11B]-BF2: δ −138.423 (80.8%); HRMS (ESI) calc’d for [M]=[C8H11BF2O2]: 188.0820, calc’d for [M-H]− =[C8H10BF2O2]−: 187.0742, found [M-H]−: 187.0748.
4-(4-(Tert-butyl)phenyl)-2,2-difluoro-6-(4-methoxyphenyl)-2H-1λ3,3,2λ4-dioxaborinine (16).
1-(4-(tert-butyl)phenyl)-3-(4-methoxyphenyl)propane-1,3-dione (630 mg, 3 mmol) was dissolved in 10 mL of dry CH2Cl2. BF3 · (OEt)2 (46.5%, 916 mg, 6 mmol) was added to the solution dropwise, under magnetic stirring at room temperature. The reaction was monitored with UPLC. After two hours of reaction, 5 mL of deionized water was added to quench the reaction. The mixture was dried by rotary evaporation. Dried material was re-dissolved in DMF and purified by preparative C18 Reversed Phase LC chromatography. The gradient was water/acetonitrile at a flow rate of 12 mL/min (90/10 to 10/90 linear gradient, 20 min, and then isocratic water/acetonitrile 10/90 for another 25 min). Ultrapure water and HPLC acetonitrile (Sigma, St. Louis, MO, USA) were used. The fractions of 26 min to 28 min were collected, kept at −80 °C overnight and then lyophilized into white color powder. Yields: 691 mg (64.3%). 1H NMR (500 MHz, d6-DMSO, 25 °C, TMS): δ 8.39 (d, J = 9.1 Hz, 2H), 8.29 (d, J = 8.7 Hz, 2H), 7.81 (s, 1H), 7.67 (d, J = 8.7 Hz, 2H), 7.21 (d, J = 9.1 Hz, 2H), 3.94 (s, 3H), 1.34 (s, 9H); 13C NMR (125 MHz, d6-DMSO, 25 °C, TMS): δ 181.78, 181.03, 166.18, 159.44, 132.62, 129.49, 129.44, 126.69, 123.95, 115.48, 93.68, 56.51, 35.68, 31.13; 19F NMR (470 MHz, d6-DMSO, 25 °C, CFCl3): [10B]-BF2: δ −140.117 (20.3%), [11B]-BF2: δ −140.179 (79.7%). HRMS (ESI) calc’d for [M]=[C20H21BF2O3]: 358.1552, found [M]−: 358.1549.
2,2-Difluoro-4,6-bis(4-methoxyphenyl)-2H-1λ3,3,2λ4-dioxaborinine (17).
1,3-Bis(4-methoxyphenyl)propane-1,3-dione (852 mg, 3 mmol) was dissolved in 10 mL of dry CH2Cl2. BF3 · (OEt)2 (46.5%, 916 mg, 6 mmol) was added to the solution dropwise, under magnetic stirring at room temperature. The reaction was monitored with UPLC. After two hours of reaction, 5 mL of deionized water was added to quench the reaction. The mixture was dried by rotary evaporation. Dried material was re-dissolved in DMF and purified by preparative C18 Reversed Phase LC chromatography. The gradient was water/acetonitrile at a flow rate of 12 mL/min (90/10 to 10/90 linear gradient, 20 min, and then isocratic water/acetonitrile 10/90 for another 25 min). Ultrapure water and HPLC acetonitrile (Sigma, St. Louis, MO, USA) were used. The fractions of 23 min to 30 min were collected, kept at −80 °C overnight and then lyophilized into yellow color powder. Yields: 687 mg (69.0%). 1H NMR (500 MHz, d6-DMSO, 25 °C, TMS): δ 8.37 (d, J = 9 Hz, 4H), 7.76 (s, 1H), 7.20 (d, J = 9 Hz, 4H), 3.94 (s, 6H); 13C NMR (125 MHz, d6-DMSO, 25 °C, TMS): δ 180.56, 165.81, 132.25, 124.17, 115.35, 92.87, 56.44; 19F NMR (470 MHz, d6-DMSO, 25 °C, CFCl3): [10B]-BF2: δ −140.493 (20.0%), [11B]-BF2: δ −140.556 (80.0%); HRMS (ESI) calc’d for [M]=[C17H15BF2O4]: 332.1031, found [M]−: 332.1031.
4,4′-((1E,1′E)-(2,2-difluoro-2H-1λ3,3,2λ4-dioxaborinine-4,6-diyl)bis(ethene-2,1-diyl))bis(2-methoxyphenol) (5).
Curcumin (1106 mg, 3 mmol) was dissolved in 10 mL of dry CH2Cl2. BF3 · (OEt)2 (46.5%, 916 mg, 6 mmol) was added to the solution dropwise, under magnetic stirring at room temperature. The reaction was monitored with UPLC. After two hours of reaction, 5 mL of deionized water was added to quench the reaction. Dried material was re-dissolved in DMF and purified by preparative C18 Reversed Phase LC chromatography. The gradient was water/acetonitrile at a flow rate of 12 mL/min (90/10 to 10/90 linear gradient, 20 min, and then isocratic water/acetonitrile 10/90 for another 25 min). Ultrapure water and HPLC acetonitrile (Sigma, St. Louis, MO, USA) were used. The fractions of 19 min to 20 min were collected, kept at −80 °C overnight and then lyophilized into red color powder. Yields: 717 mg (57.4%). 1H NMR (500 MHz, d6-DMSO, 25 °C, TMS): δ 10.09 (s, 2H), 7.93 (d, J = 15.6 Hz, H), 7.47 (d, J = 1.8 Hz, 2H), 7.34 (dd, J = 8.3 Hz, 2H), 7.02 (d, J = 15.6 Hz, 2H), 6.88 (d, J = 8.2 Hz, 2H), 6.45 (s, 1H), 3.85 (s, 6H); 13C NMR (125 MHz, d6-DMSO, 25 °C, TMS): δ 179.19, 151.81, 148.64, 147.42, 126.45, 125.70, 118.33, 116.42, 112.89, 101.55, 56.23; 19F NMR (470 MHz, d6-DMSO, 25 °C, CFCl3): δ −140.230 −140.295, −140.361, −140.423; HRMS (ESI) calc’d for [M]=[C21H19BF2O6]: 416.1243, calc’d for [M-H]− =[C21H18BF2O6]−: 415.1165, found [M-H]−: 415.1164.
((1E,1′E)-(2,2-difluoro-2H-1λ3,3,2λ4-dioxaborinine-4,6-diyl)bis(ethene-2,1-diyl))bis(2-methoxy-4,1-phenylene) bis(2,4,6-trifluorobenzoate) (9).
5 (12.5 mg, 0.03 mmol), 2,4,6-Trifluorobenzoic acid (15.8 mg, 0.09 mmol), HOBt hydrate (12.2 mg, 0.09 mmol), EDC hydrochloride (57.6 mg, 0.3 mmol) and 20 μL pyridine were dissolved in 1 mL DMF (HPLC, Sigma) in a 1.5 mL eppendorf tube. The reaction, shielded from light, was left at room temperature overnight. A color change from weak orange-red to bright green is visible under 365 nm UV lamp illumination indicating reaction. The product was isolated by preparative C18 Reversed Phase LC chromatography. The gradient was water/acetonitrile at a flow rate of 12 mL/min (50/50 to 0/100 linear gradient, 20 min, and then isocratic water/acetonitrile 0/100 for another 25 min). Ultrapure water and HPLC acetonitrile (Sigma, St. Louis, MO, USA) were used. The fractions of 21 min to 22 min were collected, kept at −80 °C overnight and then lyophilized into yellow color powder. Yields: 17.2 mg (78.3%). 1H NMR (500 MHz, d6-DMSO, 25 °C, TMS): δ 8.11 (d, J = 15.8 Hz, 2H), 7.76 (s, 2H), 7.57 (d, J = 8.15 Hz, 2H), 7.48 (t, J = 9.25 Hz, 4H), 7.39 (d, J = 8.15 Hz, 2H), 7.34 (d, J = 15.75 Hz, 2H), 6.68 (s, 1H), 3.90 (s, 6H); 13C NMR (125 MHz, d6-DMSO, 25 °C, TMS): δ 180.62, 164.15, 162.98, 162.91, 162.85, 162.78, 160.93, 160.86, 160.80, 160.73, 158.25, 151.52, 146.67, 141.83, 134.24, 131.66, 123.85, 123.38, 122.58, 114.13, 106.25, 106.22, 102.98, 102.95, 102.77, 102.74, 102.56, 102.53, 56.80; 19F NMR (470 MHz, d6-DMSO, 25 °C, CFCl3): δ −102.030 (t, J = 10.25 Hz, 2F), −107.581 (t, J = 10 Hz, 4F), δ −139.296, −139.369, −139.445, −139.508; HRMS (ESI) calc’d for [M]=[C35H21BF8O8]: 732.1202, found [M]−: 732.1185.
((1E,1′E)-(2,2-difluoro-2H-1λ3,3,2λ4-dioxaborinine-4,6-diyl)bis(ethene-2,1-diyl))bis(2-methoxy-4,1-phenylene) bis(3-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)propanoate) (10).
5 (12.5 mg, 0.03 mmol), 3-Maleimidopropionic acid (15.2 mg, 0.09 mmol), HOBt hydrate (12.2 mg, 0.09 mmol), EDC hydrochloride (57.6 mg, 0.3 mmol) and 20 μL pyridine were dissolved in 1 mL DMF (HPLC, Sigma) in a 1.5 mL eppendorf tube. The reaction, shielded from light, was left at room temperature overnight. A color change from weak orange-red to bright green that is visible under 365 nm UV lamp illumination indicates reaction. The product was isolated by preparative chromatography. The gradient was water/acetonitrile at a flow rate of 12 mL/min (90/10 to 0/100 linear gradient, 20 min, and then isocratic water/acetonitrile 0/100 for another 25 min). Ultrapure water and HPLC acetonitrile (Sigma, St. Louis, MO, USA) were used. The fractions of 20 min to 21 min were collected, kept at −80 °C overnight and then lyophilized into yellow color powder. Yields: 7.2 mg (33.4%). 1H NMR (500 MHz, d6-DMSO, 25 °C, TMS): δ 8.06 (d, J = 15.7 Hz, 2H), 7.66 (s, 2H), 7.51 (d, J = 7.55 Hz, 2H), 7.29 (d, J = 15.8 Hz, 2H), 7.23 (d, J = 8.2 Hz, 2H), 7.07 (d, J = 3.0 Hz, 4H), 6.65 (s, 1H), 3.83 (s, 6H), 3.78 (t, J = 7.0 Hz, 4H), 2.91 (t, J = 7.0 Hz, 4H); 13C NMR (125 MHz, d6-DMSO, 25 °C, TMS): δ 180.53, 171.27, 171.21, 168.99, 151.65, 146.79, 142.40, 135.17, 133.67, 123.94, 123.29, 123.13, 122.26, 113.78, 102.67, 56.63, 56.60, 33.66, 32.57; 19F NMR (470 MHz, d6-DMSO, 25 °C, CFCl3): δ −139.418, −139.483, −139.550, −139.614. HRMS (ESI) calc’d for [M]=[C35H29BF2N2O12]: 718.1782, found [M]−: 718.1759.
General Radiolabeling of compounds 1, 2, 3, 5, 6, 9, 10, 13, 14, 15, 16, and 17.
General radiolabeling of compounds were performed in 4 steps (A-D):
Step A (F-18 drying): ~ 10 mCi F-18 aqueous solution received from a cyclotron (NCM, Bronx, NY) was added to a borosilicate Agilent screw top micro sampling v-vial (1 mL volume). The vial was capped and fitted with a 16G needle and a 20G needle. Nitrogen at a pressure of 40psi was flushed through the 20G needle, while the 16G needle was fitted so that outlet nitrogen was bubbled through >1M potassium hydroxide (KOH) solution. No heat was applied.
Step B (Catalyst solution preparation): Fresh SnCl4 catalytic solution was prepared by mixing 20 μL of neat SnCl4 (Sigma, 99.995% trace metals basis, #217913) in 1 mL dry acetonitrile (HPLC, Sigma, product of USA). This catalytic solution should be freshly prepared and used within 1 h.
Step C (Re-suspension of F-18 in catalyst solution): A ~200 μL volume of catalyst solution (Step B) was added into the v-vial containing dried F-18 (Step A). The vial was sealed and sonicated for 3-5 min.
Step D (Radiolabeling): A 100 μL re-suspended F-18/catalyst solution (Step C) was used to dissolve 0.5 mg cold, non-radioactive difluoro-dioxaborinin (DK). This reaction was kept at room temperature (~ 21 °C) in a 1.5 mL Eppendorf tube or PCR tube for up to 30 min.
General Radio-HPLC analysis methods of compounds 1, 2, 3, 5, 6, 9, 10, 13, 14, 15, 16, and 17.
Radiolabeled solutions (~30 min) were analyzed on a Varian HPLC equipped with a radioactivity detector to confirm radiolabeling. ~10 μL of solutions containing 2% SnCl4, [18F]-fluoride ion and the radiolabeled difluoro-dioxaborinin (DK) were run through a Hamilton PRP-C18 analytical column (5 μm, 4.6 × 50 mm column, #79675, 1 mL/min). UV-Vis absorbance and a radioactivity detector was used to monitor difluoro-dioxaborinin (DK) elution. Absorbance and radioactive traces correlate in all radiolabelings. For all reported compounds, purity is >95% by both radio and UV-absorbance HPLC. HPLC Elution gradients used are reported in supporting information Pages S14–S25.
Calculation of radiochemical yields (RCYs) for compounds 1, 2, 3, 5, 6, 9, 10, 13, 14, 15, 16, and 17 based on Radio-HPLC.
Radiochemical yields (RCYs) were determined by Radio-HPLC: After 30 min radiolabeling, the crude reaction mixture was analyzed by analytical HPLC equipped with a radio detector. A minor peak at ~ 1 min in the radiotrace channel represents the residual fluoride-18 ion. The major peak in the radiotrace channels represent the radiolabeled difluoro-dioxaborinin (DK). The areas of the peaks are integrated, respectively, and the RCY is calculated as follows:
18F-Autoradiographic TLC characterization of difluoro-dioxaborinin (DK) radiolabeling for compounds 1, 2, 3, 5, 6, 13, 14, 15, 16, and 17.
Radiochemical yields (RCYs) were determined by silica TLC: Crude reaction mixtures aliquots (0.2 μL) were dropped at the baseline on silica TLC at defined time points. Methanol was used as the mobile phase to develop TLC (10-15 min). Silica TLC were allowed to dry before exposure to phosphorimaging screens. A 5 min exposure was sufficient for resolving phosphorimaging data. Exposed screens were imaged by phosphorimager.
Calculation of the radiochemical yields (RCYs) based on silica TLC for compounds 1, 2, 3, 5, 6, 13, 14, 15, 16, and 17.
Silica TLC phosphorimages were analyzed with ImageJ software: Unreacted [18F]-fluoride ion is retained at the baseline (drop point) and [18F]-difluoro-dioxaborinins resolve close to the solvent front. In ImageJ, areas of interest are drawn over [18F]-fluoride ion at baseline, [18F]-difluoro-dioxaborinins near the solvent front, and over a blank control (background). ImageJ was used to calculate respective mean area intensity values. RCY was calculated from the following equation:
Isotopic exchange rate determination of difluoro-dioxaborinin (1) by 18F-autoradiographic TLC quantitation for compounds 1, 2, 3, 5, 6, 13, 14, 15, 16, and 17.
A 0.5 mg quantity of 1 was dissolved with a SnCl4/acetonitrile solution containing 0.6 mCi [18F]-fluoride ion for radiolabeling. At different time points, 0.2 μL aliquots of the crude reaction mixture were placed at the baseline of silica TLC plates with 100% methanol as the mobile phase. A phosphorimaging film was used to expose silica TLC plates for 5 min. Exposed film was scanned by phosphorimager. The RCYs at different time points were calculated according to SI section 6 method.
Determination of RCY as a function of starting [18F]-fluoride ion activity for compounds 1, 2, 3, 5, 6, 13, 14, 15, 16, and 17.
1 (0.5 mg) was dissolved in 500 μL of a 2% SnCl4/acetonitrile solution (prepared by the general radiolabeling steps in SI section 2). Dry [18F]-fluoride ion was resuspended with 800 μL of a 2% SnCl4/acetonitrile solution (prepared by the general radiolabeling steps in SI section 2) and diluted to different specific concentrations using 2% SnCl4/acetonitrile solution. 50 μL 1/SnCl4/acetonitrile solution aliquots were mixed with 50 μL F-18/SnCl4/acetonitrile solution at different specific concentrations. The activity of resulting mixtures were measured immediately after mixing. They were 2.5, 5, 10, 20, 40, 80, 160, 320 and 640 μCi, respectively. After 30 min, 0.2 μL aliquots of the crude reaction mixture were placed at the baseline of silica TLC plates that were developed with 100% methanol. RCY were calculated from phosphorimages (method in SI section 6).
Determination of RCYs as a function of quantity for compound 1.
A 10 mg quantity of 1 was dissolved in 2% SnCl4/acetonitrile solution (see method in SI section 2) and was further diluted into solutions of different concentrations with 2% SnCl4/acetonitrile solution. Acetonitrile solutions containing 50 μL of 1 at different concentrations were placed into 8 PCR tubes at room temperature (21 °C). Quantities of 1 were 5, 10, 20, 40, 60, 80, 100 and 200 nmol, respectively. A 50 μL volume of F-18/SnCl4/acetonitrile solution (SI section 2) was added into each PCR tube containing different quantity 1. The F-18 activity in each PCR tube was 320 μCi, measured immediately after mixing. After 30 min, 0.2 μL aliquots of the crude reaction mixture were placed at the baseline of silica TLC plates that were developed with 100% methanol. RCY were calculated from phosphorimages (see method in SI section 6).
General method of 19F NMR quantification of difluoro-dioxaborinins (DKs) solvolysis for compounds for compounds 1, 2, 3, 5, 6, 9, 10, 13, 14, 15, 16, and 17.
A ~ 10 mg quantity of lyophilized difluoro-dioxaborinin (DK) was dissolved in a 0.6 mL solution composed of 90% d6-DMSO/10% water or 10% d6-DMSO/90% water and then submitted for dynamic 19F NMR analysis (25 °C). 19F NMR spectrum were integrated. The fraction of 19F on difluoro-dioxaborinin (P) at different time points (t) was calculated as follows:
Where Ss is the integrations of only 19F on difluoro-dioxaborinin and Su is the sum of all fluoride integrations (including Ss, TFA reference standards added to certain spectra were not integrated). All aqueous solutions (10%, 90% water) in SI section 13 contain 1X PBS buffer (pH 7.4, 10 mM) unless noted otherwise. “Water” or “H2O” are used to differentiate from DMSO.
Calculated values of P were plot vs. time (t) in the first order exponential decay regression analysis equation P = exp(−k*t) in sigmaplot 10. The half-life of solvolysis (t1/2) = ln(2/k).
Animal experiments conducted with compounds 5 and 9.
All studies conducted were approved by the Institutional Animal Care and Use Committee (Istanbul University and Weill Cornell Medicine no. 2014-0030) and are consistent with the recommendations of the American Veterinary Medical Association and the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Human Trials conducted with compound 9.
Images shown are part of a dosing study. All studies were conducted following IRB approval from Istanbul University Cerrahpasa Medical Faculty, Ethical Committee (IRB number: 83045809-604.01.02, Cerrahpasa University School of Medicine, Department of Nuclear Medicine). IUC IRB approval is in compliance with European Union standards. Informed consent was obtained from all individual participants included in the study. All procedures performed involving human participants were performed in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Supplementary Material
ACKNOWLEDGMENTS
The project was supported in part by NIH grants (EB013904, P30CA008748, S10OD016320).
Non-standard Abbreviations Used
- Bal b/c
Bagg and Albino common strain of laboratory mice
- BaSO4
Barium Sulfate
- C18
18-chain hydrocarbon
- CT
Computed tomography (X-ray)
- d(6)
deuterium (6) e.g. deuterated (6) DMSO
- F-18
Fluoride-18 (radioactive)
- F-19
Fluoride-19 (stable)
- ICG
indocyanine green (FDA approved fluorescent contrast)
- IEX
isotopic exchange (radiolabeling)
- LC
liquid chromatography
- M
Exact mass
- MBq
Megabecquerel
- MIP
maximum intensity projection
- MR(I)
Magnetic resonance imaging
- NIH
National Institutes of Health
- mCi
Millicurie
- PET
positron emission tomography
- RCY
Radiochemical yield
- SnCl4
Tin (IV) Chloride
- μCi
microcurie
Footnotes
Supporting Information
The following supporting information is available free of charge on the ACS Publications website. Supporting Information Availability:
Molecular Formula Strings CSV file.
CT-only maximum intensity projection video of mouse in Figure 6
PET/CT maximum intensity projection video of mouse in Figure 6
The authors declare no competing financial interest.
REFERENCES:
- (1).(a) Yuan Z; Cheng R; Chen P; Liu G; Liang SH Efficient Pathway for the Preparation of Aryl(isoquinoline)iodonium(III) Salts and Synthesis of Radiofluorinated Isoquinolines. Angew. Chem. Int. Ed 2016, 55, 11882–11886. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chansaenpak K; Vabre B; Gabbai FP [18F]-Group 13 Fluoride Derivatives as Radiotracers for Positron Emission Tomography. Chem. Soc. Rev 2016, 45, 954–971. [DOI] [PubMed] [Google Scholar]; (c) van der Born D; Pees A; Poot AJ; Orru RVA; Windhorst AD; Vugts DJ Fluorine-18 Labelled Building Blocks for PET Tracer Synthesis. Chem. Soc. Rev 2017, 46, 4709–4773. [DOI] [PubMed] [Google Scholar]; (d) Deng X; Rong J; Wang L; Vasdev N; Zhang L; Josephson L; Liang SH Chemistry for Positron Emission Tomography: Recent Advances in 11C-, 18F-, 13N-, and 15O-Labeling Reactions. Angew. Chem. Int. Ed 2019, 58, 2580–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Zheng J; Wang L; Lin JH; Xiao JC; Liang SH Difluorocarbene-Derived Trifluoromethylthiolation and [18F]Trifluoromethylthiolation of Aliphatic Electrophiles. Angew. Chem. Int. Ed 2015, 54, 13236–13240. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jacobson O; Kiesewetter DO; Chen X Fluorine-18 Radiochemistry, Labeling Strategies and Synthetic Routes. Bioconjug. Chem 2015, 26, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nodwell MB; Yang H; Čolović M; Yuan Z; Merkens H; Martin RE; Bénard F; Schaffer P; Britton R 18F-Fluorination of Unactivated C–H Bonds in Branched Aliphatic Amino Acids: Direct Synthesis of Oncological Positron Emission Tomography Imaging Agents. J. Am. Chem. Soc 2017, 139, 3595–3598. [DOI] [PubMed] [Google Scholar]; (d) Gendron T; Sander K; Cybulska K; Benhamou L; Sin PKB; Khan A; Wood M; Porter MJ; Arstad E Ring-Closing Synthesis of Dibenzothiophene Sulfonium Salts and Their Use as Leaving Groups for Aromatic 18F-Fluorination. J. Am. Chem. Soc 2018, 140, 11125–11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Fier PS; Hartwig JF Selective C-H Fluorination of Pyridines and Diazines Inspired by a Classic Amination Reaction. Science 2013, 342, 956–960. [DOI] [PubMed] [Google Scholar]; (b) Thompson S; Zhang Q; Onega M; McMahon S; Fleming I; Ashworth S; Naismith JH; Passchier J; O’Hagan D A Localized Tolerance in the Substrate Specificity of the Fluorinase Enzyme enables “Last-Step” 18F Fluorination of a RGD Peptide under Ambient Aqueous Conditions. Angew. Chem. Int. Ed 2014, 53, 8913–8918. [DOI] [PubMed] [Google Scholar]; (c) Buckingham F; Kirjavainen AK; Forsback S; Krzyczmonik A; Keller T; Newington IM; Glaser M; Luthra SK; Solin O; Gouverneur V Organomediated Enantioselective 18F Fluorination for PET Applications. Angew. Chem. Int. Ed 2015, 54, 13366–13369. [DOI] [PubMed] [Google Scholar]; (d) Neumann CN; Hooker JM; Ritter T Concerted Nucleophilic Aromatic Substitution with 19F− and 18F−. Nature 2016, 534, 369–373. [DOI] [PubMed] [Google Scholar]; (e) Levin MD; Chen TQ; Neubig ME; Hong CM; Theulier CA; Kobylianskii IJ; Janabi M; O’Neil JP; Toste FD A Catalytic Fluoride-rebound Mechanism for C(sp3)-CF3 Bond Formation. Science 2017, 356, 1272–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zeng X; Li J; Ng CK; Hammond GB; Xu B (Radio)fluoroclick Reaction Enabled by a Hydrogen-Bonding Cluster. Angew. Chem. Int. Ed 2018, 57, 2924–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Verhoog S; Kee CW; Wang Y; Khotavivattana T; Wilson TC; Kersemans V; Smart S; Tredwell M; Davis BG; Gouverneur V 18F-Trifluoromethylation of Unmodified Peptides with 5-18F-(Trifluoromethyl)dibenzothiophenium Trifluoromethanesulfonate. J. Am. Chem. Soc 2018, 140, 1572–1575. [DOI] [PubMed] [Google Scholar]; (h) Chen W; Huang Z; Tay NES; Giglio B; Wang M; Wang H; Wu Z; Nicewicz DA; Li Z Direct Arene C–H Fluorination with 18F− via Organic Photoredox Catalysis. Science 2019, 364, 1170–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Huiban M; Tredwell M; Mizuta S; Wan Z; Zhang X; Collier TL; Gouverneur V; Passchier J A Broadly Applicable [18F]trifluoromethylation of Aryl and Heteroaryl Iodides for PET Imaging. Nat. Chem 2013, 5, 941–944. [DOI] [PubMed] [Google Scholar]; (b) Tredwell M; Preshlock SM; Taylor NJ; Gruber S; Huiban M; Passchier J; Mercier J; Génicot C; Gouverneur V A General Copper-Mediated Nucleophilic 18F Fluorination of Arenes. Angew. Chem. Int. Ed 2014, 53, 7751–7755. [DOI] [PubMed] [Google Scholar]; (c) Born D. v. d.; Sewing C; Herscheid JDM; Windhorst AD; Orru RVA; Vugts DJ A Universal Procedure for the [18F]Trifluoromethylation of Aryl Iodides and Aryl Boronic Acids with Highly Improved Specific Activity. Angew. Chem. Int. Ed 2014, 53, 11046–11050. [DOI] [PubMed] [Google Scholar]; (d) Rotstein BH; Stephenson NA; Vasdev N; Liang SH Spirocyclic Hypervalent Iodine(III)-mediated Radiofluorination of Non-activated and Hindered Aromatics. Nat. Commun 2014, 5, 4365. [DOI] [PubMed] [Google Scholar]; (e) Khotavivattana T; Verhoog S; Tredwell M; Pfeifer L; Calderwood S; Wheelhouse K; Collier TL; Gouverneur V 18F-Labeling of Aryl-SCF3, -OCF3 and -OCHF2 with [18F]Fluoride. Angew. Chem. Int. Ed 2015, 54, 9991–9995. [DOI] [PubMed] [Google Scholar]
- (5).(a) Perrin DM [18F]-Organotrifluoroborates as Radioprosthetic Groups for PET Imaging: From Design Principles to Preclinical Applications. Acc. Chem. Res 2016, 49, 1333–1343. [DOI] [PubMed] [Google Scholar]; (b) Preshlock S; Tredwell M; Gouverneur V 18F-Labeling of Arenes and Heteroarenes for Applications in Positron Emission Tomography. Chem. Rev 2016, 116, 719–766. [DOI] [PubMed] [Google Scholar]
- (6).Chen P-Z; Niu L-Y; Chen Y-Z; Yang Q-Z Difluoroboron β-Diketonate Dyes: Spectroscopic Properties and Applications. Coord. Chem. Rev 2017, 350, 196–216. [Google Scholar]
- (7).Taylor NJ; Emer E; Preshlock S; Schedler M; Tredwell M; Verhoog S; Mercier J; Genicot C; Gouverneur V Derisking the Cu-Mediated 18F-Fluorination of Heterocyclic Positron Emission Tomography Radioligands. J. Am. Chem. Soc 2017, 139, 8267–8276. [DOI] [PubMed] [Google Scholar]
- (8).(a) Schirrmacher R; Bradtmoeller G; Schirrmacher E; Thews O; Tillmanns J; Siessmeier T; Buchholz HG; Bartenstein P; Waengler B; Niemeyer CM; Jurkschat K, 18F-labeling of Peptides by means of an Organosilicon-based Fluoride Acceptor. Angew. Chem. Int. Ed 2006, 45, 6047–6050. [DOI] [PubMed] [Google Scholar]; (b) Liu Z; Pourghiasian M; Radtke MA; Lau J; Pan J; Dias GM; Yapp D; Lin KS; Bénard F; Perrin DM An Organotrifluoroborate for Broadly Applicable One-Step 18F-Labeling. Angew. Chem. Int. Ed 2014, 53, 11876–11880. [DOI] [PubMed] [Google Scholar]
- (9).Liu S; Lin T-P; Li D; Leamer L; Shan H; Li Z; Gabbaï FP; Conti PS Lewis Acid-Assisted Isotopic 18F-19F Exchange in BODIPY Dyes: Facile Generation of Positron Emission Tomography/Fluorescence Dual Modality Agents for Tumor Imaging. Theranostics 2013, 3, 181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ilhan H; Lindner S; Todica A; Cyran CC; Tiling R; Auernhammer CJ; Spitzweg C; Boeck S; Unterrainer M; Gildehaus FJ; Böning G; Jurkschat K; Wängler C; Wängler B; Schirrmacher R; Bartenstein P Biodistribution and First Clinical Results of 18F-SiFAlin-TATE PET: a Novel 18F-labeled Somatostatin Analog for Imaging of Neuroendocrine Tumors. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 870–880. [DOI] [PubMed] [Google Scholar]
- (11).Saha GB Basics of PET Imaging: Physics, Chemistry, and Regulations, 3rd ed.Springer International Publishing, New York, 2016, pp 161–178. [Google Scholar]
- (12).(a) Ting R; Harwig CW; Lo J; Li Y; Adam MJ; Ruth TJ; Perrin DM Substituent Effects on Aryltrifluoroborate Solvolysis in Water: Implications for Suzuki-Miyaura Coupling and the Design of Stable 18F-Labeled Aryltrifluoroborates for Use in PET Imaging. J. Org. Chem 2008, 73, 4662–4670. [DOI] [PubMed] [Google Scholar]; (b) Liu Z; Chao D; Li Y; Ting R; Oh J; Perrin DM From Minutes to Years: Predicting Organotrifluoroborate Solvolysis Rates. Chem. Eur. J 2015, 21, 3924–3928. [DOI] [PubMed] [Google Scholar]
- (13).Ran C; Xu X; Raymond SB; Ferrara BJ; Neal K; Bacskai BJ; Medarova Z; Moore A Design, Synthesis, and Testing of Difluoroboron-Derivatized Curcumins as Near-Infrared Probes for in vivo Detection of Amyloid-β Deposits. J. Am. Chem. Soc 2009, 131, 15257–15261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Collot M; Fam TK; Ashokkumar P; Faklaris O; Galli T; Danglot L; Klymchenko AS Ultrabright and Fluorogenic Probes for Multicolor Imaging and Tracking of Lipid Droplets in Cells and Tissues. J. Am. Chem. Soc 2018, 140, 5401–5411. [DOI] [PubMed] [Google Scholar]
- (15).(a) Zhang X; Tian Y; Zhang C; Tian X; Ross AW; Moir RD; Sun H; Tanzi RE; Moore A; Ran C Near-infrared Fluorescence Molecular Imaging of Amyloid Beta Species and Monitoring Therapy in Animal Models of Alzheimer’s Disease. Proc. Natl. Acad. Sci. U.S.A 2015, 112, 9734–9739. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang X; Tian Y; Zhang H; Kavishwar A; Lynes M; Brownell A-L; Sun H; Tseng Y-H; Moore A; Ran C Curcumin Analogues as Selective Fluorescence Imaging Probes for Brown Adipose Tissue and Monitoring Browning. Sci. Rep 2015, 5, 13116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Eaton DF International Union of Pure and Applied Chemistry Organic Chemistry Division Commission on Photochemistry: Reference Materials for Fluorescence Measurement. J. Photochem. Photobiol. B: Biol 1988, 2, 523–531. [DOI] [PubMed] [Google Scholar]
- (17).(a) Oh MH; Lee N; Kim H; Park SP; Piao Y; Lee J; Jun SW; Moon WK; Choi SH; Hyeon T Large-Scale Synthesis of Bioinert Tantalum Oxide Nanoparticles for X-ray Computed Tomography Imaging and Bimodal Image-Guided Sentinel Lymph Node Mapping. J. Am. Chem. Soc 2011, 133, 5508–5515. [DOI] [PubMed] [Google Scholar]; (b) Jung Y; Reif R; Zeng Y; Wang RK Three-Dimensional High-Resolution Imaging of Gold Nanorods Uptake in Sentinel Lymph Nodes. Nano Lett. 2011, 11, 2938–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Noh Y-W; Kong S-H; Choi D-Y; Park HS; Yang H-K; Lee H-J; Kim HC; Kang KW; Sung MH; Lim YT Near-Infrared Emitting Polymer Nanogels for Efficient Sentinel Lymph Node Mapping. ACS Nano 2012, 6, 7820–7831. [DOI] [PubMed] [Google Scholar]; (d) Pan D; Cai X; Yalaz C; Senpan A; Omanakuttan K; Wickline SA; Wang LV; Lanza GM Photoacoustic Sentinel Lymph Node Imaging with Self-Assembled Copper Neodecanoate Nanoparticles. ACS Nano 2012, 6, 1260–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kitai T; Inomoto T; Miwa M; Shikayama T Fluorescence Navigation with Indocyanine Green for Detecting Sentinel Lymph Nodes in Breast Cancer. Breast Cancer 2005, 12, 211–215. [DOI] [PubMed] [Google Scholar]
- (19).Luo S; Zhang E; Su Y; Cheng T; Shi C A Review of NIR Dyes in Cancer Targeting and Imaging. Biomaterials 2011, 32, 7127–7138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.