

Key Points
Acquired PNH or 6p CN-LOH clones can distinguish immune-mediated AA from IBMFSs.
Clonal TCR gene rearrangement is not specific for acquired AA and occurs in ∽15% of IBMFS patients.
Visual Abstract

Abstract
Acquired aplastic anemia (AA) is a life-threatening bone marrow aplasia caused by the autoimmune destruction of hematopoietic stem and progenitor cells. There are no existing diagnostic tests that definitively establish AA, and diagnosis is currently made via systematic exclusion of various alternative etiologies, including inherited bone marrow failure syndromes (IBMFSs). The exclusion of IBMFSs, which requires syndrome-specific functional and genetic testing, can substantially delay treatment. AA and IBMFSs can have mimicking clinical presentations, and their distinction has significant implications for treatment and family planning, making accurate and prompt diagnosis imperative to optimal patient outcomes. We hypothesized that AA could be distinguished from IBMFSs using 3 laboratory findings specific to the autoimmune pathogenesis of AA: paroxysmal nocturnal hemoglobinuria (PNH) clones, copy-number–neutral loss of heterozygosity in chromosome arm 6p (6p CN-LOH), and clonal T-cell receptor (TCR) γ gene (TRG) rearrangement. To test our hypothesis, we determined the prevalence of PNH, acquired 6p CN-LOH, and clonal TRG rearrangement in 454 consecutive pediatric and adult patients diagnosed with AA, IBMFSs, and other hematologic diseases. Our results indicated that PNH and acquired 6p CN-LOH clones encompassing HLA genes have ∽100% positive predictive value for AA, and they can facilitate diagnosis in approximately one-half of AA patients. In contrast, clonal TRG rearrangement is not specific for AA. Our analysis demonstrates that PNH and 6p CN-LOH clones effectively distinguish AA from IBMFSs, and both measures should be incorporated early in the diagnostic evaluation of suspected AA using the included Bayesian nomogram to inform clinical application.
Introduction
Acquired aplastic anemia (AA) is a life-threatening bone marrow failure (BMF) disorder caused by T-lymphocyte–mediated autoimmune attack on hematopoietic stem and progenitor cells (HSPCs).1-3 Accurate diagnosis of AA followed by prompt, appropriate therapy is imperative for optimal patient outcomes.4 Currently, no diagnostic tests can definitively establish the autoimmune etiology of marrow aplasia, and diagnosis instead occurs via the systematic exclusion of alternative causes of BMF.2,4 Distinguishing immune-mediated BMF (ie, AA) from inherited BMF syndromes (IBMFSs) is particularly challenging. IBMFSs can mimic AA in children and young adults, and patients may present with few extrahematopoietic manifestations and no family history.5,6 Despite their overlapping clinical presentations, AA and IBMFSs require distinct approaches to treatment and family planning. Specifically, immunosuppressive therapy (IST) that is used to treat immune-mediated AA cannot reverse IBMFS-related marrow aplasia, and misdiagnosis of IBMFSs as AA can delay definitive treatment while exposing patients to serious complications. Patients with undiagnosed IBMFSs may also experience significant bone marrow transplant–related complications with higher-intensity conditioning used for AA or engraftment failure if a related donor carries unrecognized IBMFSs.5,7,8
Exclusion of IBMFSs can take multiple weeks. At a minimum, patients should be evaluated for Fanconi anemia (FA) and dyskeratosis congenita (DC) via chromosome breakage and telomere-length analysis, respectively.2 Genomic testing for IBMFS-associated variants using next-generation sequencing (NGS) may exclude additional IBMFSs but can significantly delay treatment. Thus, there is a pressing need to develop an efficient diagnostic algorithm that accurately discriminates AA from IBMFSs.
We hypothesized that 3 particular laboratory features tied to the autoimmune pathogenesis of AA could hold high predictive power in discriminating AA from other conditions. Previous studies of clonal hematopoiesis in AA have described 2 recurrent acquired changes in the HSPCs of AA patients: expanded clonal populations with paroxysmal nocturnal hemoglobinuria (PNH) immunophenotype and HLA loss through acquired copy number–neutral loss of heterozygosity of chromosome arm 6p (6p CN-LOH).3,9-12 These 2 changes are very rare in age-related clonal hematopoiesis13-15 and are posited to arise specifically in AA by allowing mutant HSPCs to escape autoimmune attack.9-12 Clonally expanded T-cell populations are another characteristic frequently observed in immune-mediated disorders, including AA, and have been proposed as a diagnostic marker for AA.16-18 Here, we evaluated the prevalence of PNH, acquired 6p CN-LOH involving the major histocompatibility complex (MHC) region (6p CN-LOHMHC), and clonal T-cell receptor (TCR) γ gene (TRG) rearrangement, as well as the predictive values of these measures in establishing correct diagnosis, for 454 pediatric and adult patients consecutively enrolled in the retrospective/prospective Children’s Hospital of Philadelphia (CHOP)–University of Pennsylvania (Penn) Bone Marrow Failure Registry. Our results show that PNH and 6p CN-LOHMHC effectively distinguish AA from IBMFSs and mimicking conditions, whereas clonal TRG rearrangement is not specific for AA.
Methods
Subjects
All 454 pediatric and adult patients referred to the CHOP-Penn Comprehensive Bone Marrow Failure Center and consecutively enrolled in the retrospective/prospective institutional review board (IRB)-approved bi-institutional Bone Marrow Failure Research Study and Repository (Penn IRB #811751; CHOP IRB# 10-007569) from 2010 to 2020 were eligible for this study (supplemental Table 1). Race and ethnicity were self-reported. Informed consent was obtained from all participants or their legal guardians before enrollment in accordance with the Declaration of Helsinki.
Diagnostic group assignment
Patients were assigned diagnostic groups using standard diagnostic criteria.19-21 Clinical data were abstracted from the electronic medical record. AA diagnosis was completed by standard criteria, including a bone marrow biopsy examination and metaphase cytogenetic analysis, with fluorescence in situ hybridization performed in case of poor cytogenetic culture growth, followed by systematic exclusion of other disorders and etiologies that mimic AA.6,20 AA severity was classified using the modified Camitta criteria.22,23 Patients with isolated hemolytic PNH, as well as PNH that arose from AA (AA/PNH syndrome) with overt hemolysis and large (>30%) PNH granulocyte (PNHGran) clones, were grouped into the PNH disease category.24 One PNH disease patient has undergone spontaneous remission, as previously described.25
Classical IBMFSs were defined as Shwachman-Diamond syndrome (SDS), DC, Diamond-Blackfan anemia (DBA), FA, and severe congenital neutropenia (SCN). IBMFSs were diagnosed using a combination of clinical diagnoses and syndrome-specific tests, including telomere-length measurement by fluorescence in situ hybridization and flow cytometry for DC, chromosome breakage analysis on lymphocytes or skin fibroblasts for FA, and syndrome-specific genetic testing (supplemental Table 2).21,26 Patients with other inherited disorders, including inherited hematologic diseases associated with mutations in GATA1 (n = 1), GATA2 (n = 2), SAMD9 (n = 2), SAMD9L (n = 1), and MYH9 (n = 1), as well as Noonan syndrome (n = 1), X-linked agammaglobulinemia (n = 1), hereditary xerocytosis (n = 1), sideroblastic anemia (n = 7), congenital dyserythropoietic anemia (n = 2), glycogen storage disease type I (n = 1), pyruvate kinase deficiency (n = 2), and others (n = 16), were classified as other inherited.
Patients lacking an established, specific IBMFS diagnosis, but clinically diagnosed with a presumed congenital BMF syndrome due to lifelong cytopenias and associated syndromic features, were classified as BMF not otherwise specified (NOS). Isolated neutropenias without an established genetic cause were grouped under neutropenia NOS. Myelodysplastic syndrome (MDS) was diagnosed according to World Health Organization classification criteria.27,28 Finally, patients evaluated due to a suspicion of BMF, but subsequently diagnosed with other hematologic diseases, were categorized as other; such diagnoses included acute myeloid leukemia, acute lymphoid leukemia, juvenile myelomonocytic leukemia, iron-refractory iron-deficiency anemia, and cytopenias associated with autoimmune conditions.
PNH flow cytometry
The presence of PNH clones was established by detection of glycosylphosphatidylinositol (GPI)-anchored protein–deficient granulocytes, monocytes, and erythrocytes using multicolor flow cytometry, as previously described.29,30 PNH testing was performed as part of the patients’ clinical evaluation by Clinical Laboratory Improvement Amendments (CLIA)-certified flow cytometry laboratories at CHOP, the Hospital of the University of Pennsylvania (HUP), or sendout reference facilities in the course of routine clinical care. PNH clone size was determined as the percentage of GPI-anchor–deficient granulocytes.30 PNH testing performed early in the study enrollment period had a lower limit of quantitation of 1% PNH clone size and evaluated PNHGran and PNH erythrocytes. Testing performed after June 2018 used high-resolution PNH flow cytometry with a sensitivity threshold of 0.05% for PNHGran, 0.3% for PNH monocytes, and 0.01% for PNH erythrocytes. PNHGran clone sizes were used for all comparisons because they were available for all patients and were unaffected by hemolysis or transfusions. Patients with GPI-deficient PNHGran were classified into 4 categories based on PNH clone size: subclinical (<1%), small (1% to <10%), moderate (10% to 30%), and large (>30%) (supplemental Table 3). We used a cutoff of PNHGran that corresponded to our assay sensitivity (>0.05%) because the presence of even minor PNH populations has been previously shown to be predictive of immunosuppressive therapy response in AA.31-33 In patients with detectable PNHGran clones, the median interval from presentation to first available PNH testing was 0 months (ie, at diagnosis), ranging from initial presentation to 74 months after original diagnosis.
Acquired 6p CN-LOH detection
The presence of acquired 6p CN-LOH was ascertained by single-nucleotide polymorphism array (SNP-A) analysis of DNA extracted from bone marrow or peripheral blood samples.34 SNP-A was performed as part of the standard clinical evaluation by CLIA-certified cytogenomic testing facilities at the CHOP Division of Genomic Diagnostics or ARUP Laboratories. Per institutional practice, clinical SNP-A testing was performed at the time of initial evaluation and serially upon diagnostic reevaluation for refractory disease or altered hematologic parameters.34 An additional 34 AA patients and 14 PNH patients were tested on a research basis at the CHOP Center for Applied Genomics using Illumina Global Screening Array genotyping, as previously described.34 The presence of 6p CN-LOH was detected by direct visualization of the B-allele frequency (BAF) and log R ratio in GenomeStudio (Illumina, San Diego, CA). Clonality was determined by examination of BAF, in which patients with large regions of mosaic homozygosity involving the terminus of the short arm of chromosome 6 were classified as having acquired 6p CN-LOH, as previously described (supplemental Table 4).34 Internal regions of homozygosity, with BAF of 1 or 0, were assumed to be constitutional, and they were excluded from the determination of acquired 6p CN-LOH.35 For patients with acquired 6p CN-LOH, the median interval from initial cytopenia to available SNP-A genotyping was 5 months, ranging from initial presentation to 117 months after original diagnosis. Of 16 patients with 6p CN-LOH, 15 had 6p CN-LOH detected on the first available SNP-A, while 1 patient developed 6p CN-LOH upon serial testing.
TRG rearrangement assay
Clonal TRG rearrangement testing was performed by the HUP CLIA-certified Molecular Diagnostics Laboratory as previously described.36 TRG rearrangement was used to evaluate T-cell clonality because TCR genes rearrange in a stepwise fashion starting with the TRG locus, such that even TCR-β–expressing cells have TRG rearrangements, which can be used as a biomarker of clonality. Compared with the TCR β gene (TRB) polymerase chain reaction (PCR) assay, the TRG PCR assay has less technical complexity and a comparable or superior sensitivity for detecting clonal expansions in patients with T-cell lymphoproliferative neoplasms.36-38 DNA extracted from peripheral blood or bone marrow specimens was used for PCR detection of clonal variable (V) and joining (J) fragments by amplification across unique VJ junctions with fluorescent primers targeting well-conserved framework regions of TRG V and downstream J segments. After electrophoretic separation, peak sizes and heights were determined. Polyclonal T-cell populations produced Gaussian distributions of peak sizes whereas clonal populations produced distinct peaks. When a prominent peak was observed in an otherwise polyclonal distribution, clonality was interpreted using the ratio of the suspected clonal peak height to the average of the 2 flanking peak heights. All clinical testing was interpreted in a blinded manner, as this study was performed after completion of clinical analysis. Twenty additional samples, selected among IBMFS patients using a random number generator, were tested on a research basis. The interpretation of research samples was performed by a molecular pathologist blinded to the patient diagnosis.
For this study, we used both positive and indeterminate results from the PCR assay as evidence of likely T-cell clonal expansion. Three patients had only TRB PCR results available. Patients with a prior history of solid organ transplant were excluded from TRG analysis.
Statistical analysis
Clinical test results were compared between diagnostic groups using a 2-tailed Fisher exact test with a significant P value < .05. Odds ratios (ORs) were estimated using the Haldane-Anscombe correction, and the Mantel-Haenszel test was used to compute common ORs across several data sets. Sensitivity, specificity, and positive and negative predictive values were calculated. Likelihood ratios were computed using an adjusted specificity of 0.995 instead of 1.0 to prevent division by zero, and Bayesian nomograms were plotted as previously described.39,40
Results
The cohort of 454 pediatric and adult patients included 170 patients with AA, 42 with hemolytic PNH, 109 with classical IBMFSs (DBA, DC, FA, SCN, and SDS), and 38 with other inherited diseases (Table 1; supplemental Table 1). The majority of IBMFS patients had established genetic diagnoses (supplemental Table 2). PNH, 6p CN-LOH, and TRG rearrangement testing results were available for 190, 356, and 160 patients, respectively (Table 1). Metaphase cytogenetics data were available for 377 patients (supplemental Table 5), with 98.0% of AA patients and 91.1% of classical IBMFS patients lacking acquired cytogenetic abnormalities.
Table 1.
The prevalence of 6p CN-LOH, PNH clones, and clonal TRG rearrangement in BMF disorders
| Total cohort, n = 454 |
6p CN-LOH clone | PNH clone | Clonal TRG rearrangement | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diagnostic group | SNP-A, n = 356 | 6p CN-LOH (+), n (%) | Comparison, OR | P | PNH testing, n = 190 |
PNH (+), n (%) | OR | P | TCR testing, n = 160 | n (%) | Comparison, OR | P | |
| AA | 170 | 141 | 16 (11) | 126 | 58 (46) | 89 | 26 (29) | ||||||
| AA vs. PNH, 10.39 | .025 | N/A | N/A | AA vs. PNH, 0.28 | .018 | ||||||||
| PNH disease | 42 | 39 | 0 (0) | 42 | 42 (100) | 20 | 12 (60) | ||||||
| AA vs. IBMFSs, 19.59 | .002 | 11.10 | .035 | AA vs. IBMFSs, 2.27 | .208 | ||||||||
| Classical IBMFSs | |||||||||||||
| SDS | 11 | 10 | 0 (0) | 0 | N/A | 5 | 2 (40) | ||||||
| DC | 34 | 20 | 0 (0) | 3 | 0 (0) | 6 | 0 (0) | ||||||
| DBA | 35 | 22 | 0 (0) | 1 | 0 (0) | 8 | 1 (13) | ||||||
| FA | 19 | 14 | 0 (0) | 2 | 0 (0) | 6 | 1 (17) | ||||||
| SCN | 10 | 8 | 0 (0) | 0 | N/A | 1 | 0 (0) | ||||||
| AA vs. all inherited disorders including IBMFSs, 26.43 | .000 | AA vs. all inherited disorders including IBMFSs, 16.23 | .010 | AA vs. all inherited disorders including IBMFSs, 2.23 | .161 | ||||||||
| Other inherited | 38 | 26 | 0 (0) | 3 | 0 (0) | 6 | 1 (17) | ||||||
| AA vs. all non-AA, 56.67 | .000 | AA vs. all non-AA, Non-PNH, 38.43 | .000 | AA vs. all non-AA, non-PNH, 1.09 | .849 | ||||||||
| BMF NOS | 14 | 13 | 0 (0) | 0 | N/A | 2 | 0 (0) | ||||||
| Neutropenia NOS | 42 | 33 | 0 (0) | 1 | 0 (0) | 9 | 4 (44) | ||||||
| MDS | 14 | 10 | 0 (0) | 5 | 0 (0) | 3 | 2 (67) | ||||||
| Other | 25 | 20 | 0 (0) | 7 | 0 (0) | 5 | 3 (60) | ||||||
N/A, not applicable; OR, odds ratio.
Evaluation of predictive value of PNHGran in AA diagnosis
To evaluate the predictive value of PNH clones for AA diagnosis, we compared the prevalence of PNHGran between AA and other hematologic diseases. PNHGran were detected in 58 of 126 AA patients (46%). In contrast, PNHGran were not identified in 22 patients with other disorders (OR 38.43; P = .000). Crucially, when compared with classical IBMFS patients, AA patients were significantly more likely to have PNHGran (OR 11.10; P = .035). Overall, PNHGran provided a positive predictive value (PPV) of 100% and a negative predictive value (NPV) of 48.5%, along with a specificity of 100% and sensitivity of 46.0%, for AA diagnosis and IBMFS exclusion (Table 2). A prior single-institution report showed no PNHGran in 20 IBMFS patients, with a similarly high prevalence of PNHGran in patients with AA.41 Combined with our data set, PNHGran provided a 100% PPV for AA diagnosis and exclusion of IBMFSs (common OR 25.39; P = .000).
Table 2.
The diagnostic value of PNH, acquired 6p CN-LOH, and clonal TRG rearrangement for the diagnosis of acquired AA
| Laboratory test | Sensitivity, % | Specificity, % | PPV, % | NPV, % |
|---|---|---|---|---|
| PNHGran | 46.0 | 100.0 | 100.0 | 48.5 |
| Acquired 6p CN-LOHMHC | 11.4 | 100.0 | 100.0 | 63.2 |
| Clonal TRG rearrangement PCR | 29.2 | 63.4 | 50.0 | 41.7 |
The prevalence of PNHGran was similar in pediatric-onset and adult-onset AA patients, and was not associated with disease duration or age at testing. Similarly, there was no correlation of PNHGran positivity with AA severity, although PNHGran-positive patients had higher marrow cellularity at AA diagnosis (supplemental Table 6).
Evaluation of predictive value of 6p CN-LOHMHC in AA diagnosis
To ascertain the predictive value of acquired 6p CN-LOH in AA diagnosis, we evaluated SNP-A for 356 patients (Table 1). Sixteen of 141 AA patients (11.3%) had 6p CN-LOH clones, always encompassing the MHC region and HLA genes (supplemental Figure 1; supplemental Table 4). In contrast, none of 215 non-AA patients had acquired 6p CN-LOH (OR 56.67; P = .000). Compared with 74 classical IBMFS patients, none of whom had 6p CN-LOHMHC, 6p CN-LOHMHC (regardless of clone size) was significantly more frequent in AA (OR 19.59; P = .002), with a 100% PPV, 63.2% NPV, 100% specificity, and 11.4% sensitivity for AA diagnosis (Table 2).
6p CN-LOHMHC was detected at similar frequencies in patients across the age spectrum, including 10.3% of pediatric-onset and 12.7% of adult-onset AA patients (P = .786), and was not associated with duration of AA, severity of AA, or marrow cellularity (supplemental Table 6).
Evaluation of predictive value of clonal TRG rearrangement in AA diagnosis
In contrast to PNH and 6p CN-LOH, a clinical test of T-lymphocyte clonality, as measured by positive or indeterminate results on the TRG PCR assay, was less specific for AA. We evaluated the presence of clonal TRG rearrangement in 160 patients (Table 3), and in agreement with prior studies showing increased T-cell clonality with age,42 we found a higher frequency of clonal TRG rearrangement in older patients (41.5% in adults vs 18.8% in children; OR 3.07; P = .021) (supplemental Table 6).
Table 3.
The prevalence of clonal TRG rearrangement in BMF disorders
| Diagnostic group | Total count, n = 160 | Positive, n (%) | Indeterminate, n (%) | Total clonal TRG rearrangement (+), n (%) |
|---|---|---|---|---|
| AA | 89 | 14 (16) | 12 (13) | 26 (29) |
| PNH disease | 20 | 11 (55) | 1 (5) | 12 (60) |
| Classical IBMFSs | ||||
| SDS | 5 | 1 (20) | 1 (20) | 2 (40) |
| DC | 6 | 0 (0) | 0 (0) | 0 (0) |
| DBA | 8 | 1 (13) | 0 (0) | 1 (13) |
| FA | 6 | 1 (17) | 0 (0) | 1 (17) |
| SCN | 1 | 0 (0) | 0 (0) | 0 (0) |
| Other inherited | 6 | 1 (17) | 0 (0) | 1 (17) |
| BMF NOS | 2 | 0 (0) | 0 (0) | 0 (0) |
| Neutropenia NOS | 9 | 2 (22) | 2 (22) | 4 (44) |
| MDS | 3 | 0 (0) | 2 (67) | 2 (67) |
| Other | 5 | 1 (20) | 2 (40) | 3 (60) |
In comparison with 26 classical IBMFS patients, AA patients were ∽2 times more likely to have clonal TRG rearrangement (29% [26 of 89] vs 15% [4 of 26]; OR 2.27), although this difference was not statistically robust (P = .208). This remained true when adjusting for age (common OR 2.41; Padj = .231). Overall, clonal TRG rearrangement provided a 50% PPV, 41.7% NPV, 63.4% specificity, and 29.2% sensitivity for the diagnosis of AA and exclusion of IBMFSs (Table 2). In aggregate, clonal TRG rearrangement was ∽3 times more likely in patients with acquired disorders (AA, PNH, neutropenia NOS, and MDS) than in patients with inherited conditions (36.4% [44 of 121] vs 15.6% [5 of 32]; OR 3.09; P = .032); this difference remained significant after age adjustment (common OR 3.16; Padj = .003).
Co-occurrence of PNHGran, 6p CN-LOHMHC, and clonal TRG rearrangement in AA and PNH disease
Of 72 AA patients evaluated for all 3 findings, 46 patients (64%) tested positive for at least 1 (Figure 1A). Forty-one patients (57%) tested positive for either PNHGran or 6p CN-LOHMHC. Most patients with clonal TRG rearrangement also had PNHGran (17 patients; 23.6%) or 6p CN-LOHMHC clones (4 patients; 5.6%).
Figure 1.

Comparison of the prevalence of 3 AA-associated laboratory findings (6p CN-LOH, PNH clones, and clonal TRG rearrangement) in AA and PNH patients. (A) The prevalence of 3 AA-associated laboratory findings (PNH clones, acquired 6p CN-LOHMHC, and clonal TRG rearrangement) in the 72 AA patients within the CHOP-Penn cohort who had all 3 tests performed. The bar graph on the left depicts a combined prevalence of 64% for PNH clones, acquired 6p CN-LOHMHC, and clonal TRG rearrangement, with the frequency and co-occurrence of each finding shown in the Venn diagram on the right. (B) A paired bar plot presenting the prevalence of the indicated laboratory findings in patients with AA and PNH disease. PNH patients were defined as those with clinical hemolysis and >30% PNH granulocytes on flow cytometry, and, by definition, all PNH disease patients had PNH clones. In contrast to AA patients, who had 11% prevalence of 6p CN-LOH, none of the 39 PNH disease patients had detectable 6p CN-LOH clones (OR 10.39; P = .025) at the level of sensitivity of the SNP-A assay (∽5% clone size). Conversely, among 13 AA patients who had acquired 6p CN-LOH, PNH clones were found in 4 patients (31%), ranging from 0.01% to 1.30% granulocyte clone size. Compared with AA (29%, 26 of 89 patients), PNH disease patients had a higher prevalence of clonal TRG rearrangement (60%, 12 of 20 patients; OR 3.63; P = .018); this trend endured following age adjustment, although it was no longer statistically significant (common OR 3.07; Padj = .068).
Because of the shared autoimmune pathogenesis of AA and hemolytic PNH,43,44 we also evaluated the prevalence and co-occurrence of 6p CN-LOHMHC and clonal TRG rearrangement in PNH patients. In contrast to AA patients, who had 11% prevalence of 6p CN-LOHMHC, none of 39 PNH patients had detectable 6p CN-LOH clones (OR 10.39; P = .025) at the level of sensitivity of the SNP-A (∽3% to 5% clone size) (Figure 1B). Conversely, among 13 AA patients who had acquired 6p CN-LOHMHC, minute PNHGran clones were identified in 4 patients, ranging from 0.01% to 1.30% PNHGran clone size. None of the AA patients with 6p CN-LOHMHC developed clinically significant PNHGran clones. These data suggest that PNH and 6p CN-LOHMHC represent alternative means of immune escape in AA, such that having both in the same cell confers no further advantage to the HSPCs, leading to the clonal dominance of 1 or the other immune escape event but not both together.
In agreement with previous reports showing frequent skewing of TCR repertoires in hemolytic PNH disease,24,45,46 we found a 60% prevalence (12 of 20) of clonal TRG rearrangement in PNH patients. Clonal TRG rearrangement was more prevalent in patients with PNH disease than AA (OR 3.63; P = .018), although this difference was not statistically significant after age adjustment (common OR 3.07; Padj = .068).
Discussion
Our study provides a comprehensive analysis of the predictive power of 3 laboratory findings associated with the autoimmune pathogenesis of AA for the diagnosis of immune-mediated AA: PNH clones, acquired 6p CN-LOHMHC, and clonal TRG rearrangement. Our results demonstrate that PNH and acquired 6p CN-LOHMHC clones are highly specific for immune-mediated AA, hold a ∽100% PPV for distinguishing immune-mediated AA from IBMFSs, and can facilitate AA diagnosis in up to 57% of patients. Importantly, the absence of PNH and 6p CN-LOHMHC should not be used to exclude AA, as their NPVs are low (Table 2). In contrast to PNH and acquired 6p CN-LOHMHC, general T-cell clonality as measured by TRG PCR assay is not specific to AA, is influenced by other clinical factors including age, and can be found in up to 15% of IBMFS patients. The Bayes nomogram for positive PNHGran and acquired 6p CN-LOHMHC demonstrates the tests’ clinical utility in greatly increasing the posttest probability of immune-mediated AA (Figure 2). Our findings support the routine clinical use of PNH and 6p CN-LOH testing early in the diagnostic algorithm to facilitate more efficient diagnosis of immune-mediated AA and to exclude IBMFSs (Figure 3).
Figure 2.
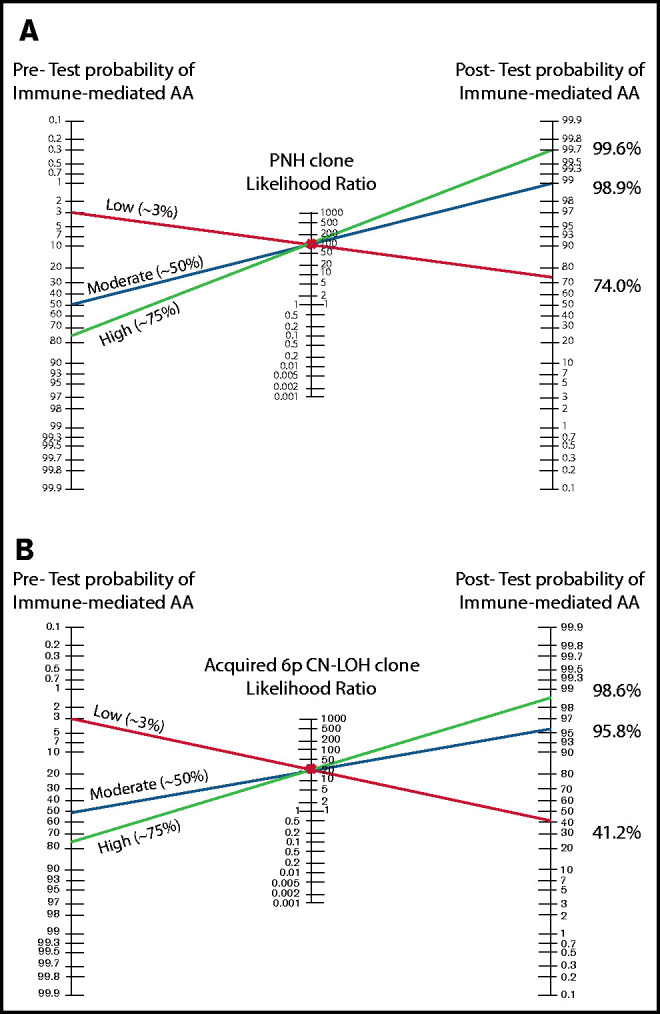
The Bayes theorem/Fagan nomograms demonstrating the pretest and posttest probabilities of immune-mediated AA diagnosis with positive PNH or 6p CN-LOHMHC test results. (A) The Bayes likelihood ratio (LR) nomogram illustrating the predictive value of PNH clones for the diagnosis of AA. PNH clones are ∽100% specific for the diagnosis of immune-mediated AA, and have a positive likelihood ratio of ∽92 for the diagnosis of immune-mediated AA. The patient’s clinical presentation and various clinical factors including age, congenital defects, medications, and comorbidities can influence the estimated pretest probability of immune-mediated AA diagnosis. Shown are 3 examples of patients with a high (75%), moderate (50%), and low (3%) pretest clinical suspicion of immune-mediated AA. Identifying a PNH clone raises the posttest probability of immune-mediated AA to >98% for moderate and high probability patients, allowing clinicians to confidently and efficiently establish a diagnosis of immune-mediated AA. In the patient with a very low suspicion of AA based on other factors, a PNH clone strongly suggests AA diagnosis, with a 74% posttest probability of immune-mediated AA. (B) The Bayes LR nomogram illustrating the predictive value of acquired 6p CN-LOHMHC for AA. Acquired 6p CN-LOHMHC is ∽100% specific for the diagnosis of immune-mediated AA, and has a positive LR of ∽23 for the diagnosis of immune-mediated AA. Shown are 3 examples of patients with high (75%), moderate (50%), and low (3%) pretest clinical suspicion of immune-mediated AA. Identifying an acquired 6p CN-LOHMHC clone raises the posttest probability of immune-mediated AA to >98% for the high probability and to >95% for the moderate probability patients, allowing clinicians to confidently and efficiently establish AA diagnosis. The patient who was previously not suspected of having AA due to various clinical characteristics now has a posttest probability of ∽41% of an immune-mediated AA diagnosis, which should be further considered in the clinical evaluation of this patient.
Figure 3.

The diagnostic utility of PNHGran clones, acquired 6p CN-LOHMHC, and clonal TRG rearrangement for acquired AA. Following initial hematologic evaluation, which includes a complete blood count (CBC) with differential, a bone marrow aspirate and biopsy, and cytogenetics analysis, the diagnostic evaluation of patients with suspected AA should include upfront testing for AA-specific laboratory findings using peripheral blood flow cytometry to detect PNH and single-nucleotide polymorphism array testing of bone marrow or peripheral blood DNA to detect acquired 6p CN-LOHMHC in addition to exclusion of alternative etiologies of pancytopenia. The presence of PNH clones or acquired 6p CN-LOHMHC is predictive of AA, which may allow clinicians to bypass extensive IBMFS testing. In the absence of these findings, AA diagnosis is established by excluding alternative etiologies of marrow aplasia, including exclusion of telomere biology disorders via telomere-length measurements and FA via chromosome breakage testing. Select patients suspected of having other IBMFSs may require comprehensive genetic testing. As PNH and acquired 6p CN-LOHMHC are clonal changes that emerge in response to autoimmune attack on the bone marrow, their prevalence may rise over the disease course. Thus, serial testing for these markers (along with repeating a bone marrow biopsy with cytogenetic analysis and genetic testing for unrecognized germline mutations) may increase the diagnostic yield later in disease. This may be particularly helpful for patients who are refractory to IST, for whom there may be lingering concerns that failure to respond to IST could indicate a mistaken diagnosis of immune-mediated AA, and that a patient’s marrow failure is caused by other, non–immune-mediated etiologies such as IBMFSs. LDH, lactate dehydrogenase.
Although the pathogenesis of PNH has long been linked to immune-mediated AA,3,43,44,47,48 studies examining the predictive value of PNH clones for the diagnosis of AA and exclusion of IBMFSs are sparse. Our data show that PNHGran clones are highly specific for immune-mediated AA, with ∽38-fold higher frequency of PNHGran in immune-mediated AA compared with other disorders. Our study is the first validation of an earlier single-institutional study that reported no PNHGran clones in 20 patients with classical IBMFSs. Combining both studies to increase statistical power, we found that AA patients have ∽25-fold higher odds of expanded PNHGran populations compared with IBMFS patients. Although testing in these studies was driven by clinical suspicion of PNH, our results are supported by a previous flow cytometry–based analysis of an unselected cohort of 28 children with SDS and an NGS-based study of 110 SDS patients, which found no PNH clones or PIGA mutations, respectively.49,50 Two historical cases of patients with presumed FA and PNH have been reported; however, neither used modern testing for FA. In 1944, Dacie and Gilpin described 2 brothers with pancytopenia and hemolysis including features reminiscent of contemporaneous descriptions of FA and PNH; however, neither FA nor PNH were established by modern laboratory testing, and the apparent familial nature of hemolysis challenges PNH diagnosis.51 In 2003, Wainwright et al reported a patient with severe pancytopenia and PNH clone expansion confirmed by flow cytometry; however, FA diagnosis was rendered despite 2 normal chromosome breakage studies, raising the strong possibility of a false-positive third chromosome breakage test.52 Taken together, all available data provide compelling evidence that clonal expansions of PNHGran are highly specific to immune-mediated AA and can be used to distinguish AA from IBMFSs.
We and others previously identified somatic HLA loss as a recurrent finding in AA, present in ∽17% of patients.10,12,19,53,54 Somatic HLA loss occurs by 2 distinct genetic mechanisms, acquired 6p CN-LOHMHC or inactivating mutations in HLA class I genes12,19,54; however, only 6p CN-LOHMHC is currently amenable to clinical testing by SNP-A genotyping. In contrast, somatic HLA mutation identification requires targeted NGS of HLA genes and specialized bioinformatic analysis,12 which, at the time of this publication, is not available clinically. Notably, acquired 6p CN-LOHMHC is the more common mechanism of somatic HLA loss, whereas mutational inactivation of HLA alleles without accompanying 6p CN-LOH occurs in ∽5% of AA patients.12 We expect that somatic-inactivating mutations in HLA alleles are also specific for AA, and their clinical testing would facilitate AA diagnosis in another ∽5% of AA patients.
Importantly, unlike acquired 6p CN-LOHMHC, which we found to be highly specific for immune-mediated AA, constitutional homozygosity of 6p and acquired 6p CN-LOH excluding the MHC region are unrelated to AA pathogenesis and should not be used in the diagnostic workup. For instance, a SNP-A analysis of 130 FA patients reported 2 cases of 6p CN-LOH.55 However, unlike acquired 6p CN-LOHMHC in AA, which includes the MHC region and leads to somatic HLA loss, these 2 cases involved the distal end of chromosome 6p with the breakpoints outside of the MHC region (supplemental Figure 1). The same study uncovered no cases of acquired 6p CN-LOH among 15 743 population controls.55 Similarly, 2 independent SNP-A analyses found no instances of 6p CN-LOH in 73 FA and 68 DBA patients, respectively.56,57 To the best of our knowledge, there have been no published reports of acquired 6p CN-LOHMHC in IBMFSs, reinforcing our findings that it is highly specific to immune-mediated AA.
Although our study focused on distinguishing AA from IBMFSs, our results support the use of PNH and acquired 6p CN-LOHMHC to identify patients with AA-type immune-mediated marrow failure more broadly. Patients diagnosed with hypocellular MDS or refractory cytopenia of childhood (RCC) may have varying etiologies of marrow dysfunction, including immune-mediated marrow failure, occult IBMFSs, sporadic MDS, and other inherited MDS predisposition syndromes (eg, GATA2 deficiency).58-62 In fact, a subset of MDS patients was previously reported to have durable objective responses to AA-type IST with horse antithymocyte globulin and cyclosporine.63 Although available studies of immunosuppression in MDS have largely been underpowered, factors predictive of IST response included marrow hypocellularity, young age, HLA-DR15, and PNH clones.63-65 Similarly, pediatric RCC patients who have PNH clones were previously reported to have improved responses to IST, suggesting that their marrow dysfunction is likely immune-mediated.33,66 The close link of PNHGran and acquired 6p CN-LOHMHC to the autoimmune pathogenesis of AA can facilitate efficient identification of patients with AA-type autoimmune marrow failure among those diagnosed with RCC or hypoplastic MDS.
This study has limitations. Particularly, this is a retrospective analysis of testing performed as part of routine clinical care. Although not all patients had all 3 laboratory tests completed, each of these findings has been independently evaluated, and our statistical analysis shows significant differences in the frequencies of PNHGran and acquired 6p CN-LOHMHC in AA compared with IBMFSs and other hematologic diseases. Although not all patients had testing performed at initial presentation, the median time from presentation to testing for PNH and 6p CN-LOH was 0 and 5 months, respectively, with all but 1 patient having a positive finding on the first available testing. Notably, the time of testing would not impact the 100% PPV for AA diagnosis, although it may affect the diagnostic sensitivity, as the prevalence of PNH and 6p CN-LOH may rise with disease progression and ongoing treatment. Thus, we recommend the use of serial testing to potentially increase diagnostic yield by affirming AA later in the disease course, a strategy that, alongside repeating a bone marrow biopsy that includes cytogenetic analysis and considering genetic testing for occult IBMFS germline mutations, may be especially helpful in poorly responding patients with enduring diagnostic uncertainty (Figure 3). Furthermore, our study does not include testing for somatic mutations in myeloid malignancy–associated genes. Notably, multiple authoritative analyses have previously shown that somatic mutations occur frequently in healthy aged individuals, as well as in patients with AA, PNH, MDS, and various inherited disorders.9,11-14,19,67-71 Thus, with the noteworthy exception of mutations that lead to PNH clones or acquired HLA loss and are specific to AA, which we describe in this study, MDS-associated mutations have not yet been shown to discriminate AA from IBMFSs effectively, and their utility in the diagnostic evaluation of AA requires further study.
In sum, our study provides a comprehensive analysis of the diagnostic value of PNH, acquired 6p CN-LOHMHC, and clonal TRG rearrangement for AA in a large cohort of patients with BMF disorders and other hematologic diseases. Our results establish 100% specificity and PPV of both PNH clones and acquired 6p CN-LOHMHC for the diagnosis of immune-mediated AA. In a clinical setting, discovering either of these 2 markers can significantly increase the posttest probability of immune-mediated AA. In contrast, a positive TRG PCR-based assay of general T-cell clonality is not specific to immune-mediated BMF. Future studies using more sophisticated assays of autoreactive T-cell expansion, perhaps including TRB sequencing or functional assays of T-cell autoreactivity, may help to define the role of T-cell analyses in AA diagnosis. In conclusion, our results support the routine early use of PNH and 6p CN-LOHMHC testing to effectively distinguish immune-mediated marrow failure from inherited conditions. The next frontier in BMF diagnostics will include combining these 2 current assays of immune-mediated BMF with more sophisticated T-cell analyses and faster, more comprehensive somatic and germline genetic studies to improve the accuracy and efficiency of diagnosis of acquired and inherited BMF disorders.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the patients and their families for participation in the Bone Marrow Failure Repository. The authors also thank the clinical care and clinical research administration teams for supporting their work.
This work was supported in part by National Institutes of Health National Heart, Lung, and Blood Institute grant K08 HL132101 (D.V.B.), Department of Defense grant BM170031 (T.S.O.), the Institute for Translational Medicine and Therapeutics of the Perelman School of Medicine at the University of Pennsylvania, and National Institutes of Health National Center for Advancing Translational Sciences grant UL1TR001878 (D.V.B. and T.S.O.).
Authorship
Contribution: Y.B.S. and D.V.B. collected and analyzed the data, performed literature review, and wrote and revised the manuscript; S.F.P. and C.N.T. performed TRG rearrangement analysis; Y.L. performed statistical review; P.N., T.S.O., and D.V.B. enrolled patients, processed and provided critical patient samples, and oversaw the human subjects aspects of the research; P.K. and T.S.O. provided pediatric BMF expertise; D.V.B. conceived and oversaw the study; and all authors edited and approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Daria V. Babushok, Division of Hematology-Oncology, Department of Medicine, Hospital of the University of Pennsylvania, Room 808 BRB II/III, 421 Curie Blvd, Philadelphia, PA 19104; e-mail: daria.babushok@pennmedicine.upenn.edu.
References
- 1.Young NS.Current concepts in the pathophysiology and treatment of aplastic anemia. Hematology Am Soc Hematol Educ Program. 2013;2013:76-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peslak SA, Olson T, Babushok DV.. Diagnosis and treatment of aplastic anemia. Curr Treat Options Oncol. 2017;18(12):70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luzzatto L, Risitano AM.. Advances in understanding the pathogenesis of acquired aplastic anaemia. Br J Haematol. 2018;182(6):758-776. [DOI] [PubMed] [Google Scholar]
- 4.Scheinberg P, Young NS.. How I treat acquired aplastic anemia. Blood. 2012;120(6):1185-1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fogarty PF, Yamaguchi H, Wiestner A, et al. Late presentation of dyskeratosis congenita as apparently acquired aplastic anaemia due to mutations in telomerase RNA. Lancet. 2003;362(9396):1628-1630. [DOI] [PubMed] [Google Scholar]
- 6.Blombery P, Fox L, Ryland GL, et al. Utility of clinical comprehensive genomic characterisation for diagnostic categorisation in patients presenting with hypocellular bone marrow failure syndromes. Haematologica. 2020;106(1):64-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orfali RF, Wynn RF, Stevens RF, Chopra R, Ball SE. Failure of red cell production following allogenic BMT for Diamond Blackfan anaemia (DBA) illustrates functional significance of high erythrocyte adenosine deaminase (eADA) activity in the donor Blood 1999 94: (Suppl. 1) 414 [Abstract 1832] [Google Scholar]
- 8.Kahn JM, Brazauskas R, Tecca HR, et al. Subsequent neoplasms and late mortality in children undergoing allogeneic transplantation for nonmalignant diseases. Blood Adv. 2020;4(9):2084-2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kulasekararaj AG, Jiang J, Smith AE, et al. Somatic mutations identify a subgroup of aplastic anemia patients who progress to myelodysplastic syndrome. Blood. 2014;124(17):2698-2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katagiri T, Sato-Otsubo A, Kashiwase K, et al. ; Japan Marrow Donor Program . Frequent loss of HLA alleles associated with copy number-neutral 6pLOH in acquired aplastic anemia. Blood. 2011;118(25):6601-6609. [DOI] [PubMed] [Google Scholar]
- 11.Yoshizato T, Dumitriu B, Hosokawa K, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. 2015;373(1):35-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Babushok DV, Duke JL, Xie HM, et al. Somatic HLA mutations expose the role of class I-mediated autoimmunity in aplastic anemia and its clonal complications. Blood Adv. 2017;1(22):1900-1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Genovese G, Kähler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477-2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488-2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laurie CC, Laurie CA, Rice K, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet. 2012;44(6):642-650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Risitano AM, Maciejewski JP, Green S, Plasilova M, Zeng W, Young NS.. In-vivo dominant immune responses in aplastic anaemia: molecular tracking of putatively pathogenetic T-cell clones by TCR beta-CDR3 sequencing. Lancet. 2004;364(9431):355-364. [DOI] [PubMed] [Google Scholar]
- 17.Plasilova M, Risitano A, Maciejewski JP.. Application of the molecular analysis of the T-cell receptor repertoire in the study of immune-mediated hematologic diseases. Hematology. 2003;8(3):173-181. [DOI] [PubMed] [Google Scholar]
- 18.Giudice V, Feng X, Lin Z, et al. Deep sequencing and flow cytometric characterization of expanded effector memory CD8+CD57+ T cells frequently reveals T-cell receptor Vβ oligoclonality and CDR3 homology in acquired aplastic anemia. Haematologica. 2018;103(5):759-769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Babushok DV, Perdigones N, Perin JC, et al. Emergence of clonal hematopoiesis in the majority of patients with acquired aplastic anemia. Cancer Genet. 2015;208(4):115-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Incidence of aplastic anemia: the relevance of diagnostic criteria. By the International Agranulocytosis and Aplastic Anemia Study. Blood. 1987;70(6):1718-1721. [PubMed] [Google Scholar]
- 21.Wilson DB, Link DC, Mason PJ, Bessler M.. Inherited bone marrow failure syndromes in adolescents and young adults. Ann Med. 2014;46(6):353-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Camitta BM.Pathogenesis and treatment of aplastic anemia. Rinsho Ketsueki. 1984;25(4):459-469. [PubMed] [Google Scholar]
- 23.Rovó A, Tichelli A, Dufour C; SAA-WP EBMT . Diagnosis of acquired aplastic anemia. Bone Marrow Transplant. 2013;48(2):162-167. [DOI] [PubMed] [Google Scholar]
- 24.Karadimitris A, Manavalan JS, Thaler HT, et al. Abnormal T-cell repertoire is consistent with immune process underlying the pathogenesis of paroxysmal nocturnal hemoglobinuria. Blood. 2000;96(7):2613-2620. [PubMed] [Google Scholar]
- 25.Babushok DV, Stanley N, Xie HM, et al. Clonal replacement underlies spontaneous remission in paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2017;176(3):487-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Armanios M, Blackburn EH.. The telomere syndromes [published correction appears in Nat Rev Genet. 2013;14(3):235]. Nat Rev Genet. 2012;13(10):693-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937-951. [DOI] [PubMed] [Google Scholar]
- 28.Hong M, He G.. The 2016 Revision to the World Health Organization Classification of Myelodysplastic Syndromes. J Transl Int Med. 2017;5(3):139-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parker CJ.Management of paroxysmal nocturnal hemoglobinuria in the era of complement inhibitory therapy. Hematology Am Soc Hematol Educ Program. 2011;2011:21-29. [DOI] [PubMed] [Google Scholar]
- 30.Illingworth AJ, Marinov I, Sutherland DR.. Immunophenotyping of paroxysmal nocturnal hemoglobinuria (PNH). Methods Mol Biol. 2019;2032:323-354. [DOI] [PubMed] [Google Scholar]
- 31.Nakao S, Sugimori C, Yamazaki H.. Clinical significance of a small population of paroxysmal nocturnal hemoglobinuria-type cells in the management of bone marrow failure. Int J Hematol. 2006;84(2):118-122. [DOI] [PubMed] [Google Scholar]
- 32.Sugimori C, Chuhjo T, Feng X, et al. Minor population of CD55-CD59- blood cells predicts response to immunosuppressive therapy and prognosis in patients with aplastic anemia. Blood. 2006;107(4):1308-1314. [DOI] [PubMed] [Google Scholar]
- 33.Fattizzo B, Ireland R, Dunlop A, et al. Clinical and prognostic significance of small paroxysmal nocturnal hemoglobinuria clones in myelodysplastic syndrome and aplastic anemia [published online ahead of print 4 March 2021]. Leukemia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Babushok DV, Xie HM, Roth JJ, et al. Single nucleotide polymorphism array analysis of bone marrow failure patients reveals characteristic patterns of genetic changes. Br J Haematol. 2014;164(1):73-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Afable MG II, Tiu RV, Maciejewski JP.. Clonal evolution in aplastic anemia. Hematology Am Soc Hematol Educ Program. 2011;2011:90-95. [DOI] [PubMed] [Google Scholar]
- 36.Hodges E, Krishna MT, Pickard C, Smith JL.. Diagnostic role of tests for T cell receptor (TCR) genes. J Clin Pathol. 2003;56(1):1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu H, Bench AJ, Bacon CM, et al. A practical strategy for the routine use of BIOMED-2 PCR assays for detection of B- and T-cell clonality in diagnostic haematopathology. Br J Haematol. 2007;138(1):31-43. [DOI] [PubMed] [Google Scholar]
- 38.van Dongen JJ, Langerak AW, Brüggemann M, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003;17(12):2257-2317. [DOI] [PubMed] [Google Scholar]
- 39.Fagan TJ.Letter: nomogram for Bayes theorem. N Engl J Med. 1975;293(5):257. [DOI] [PubMed] [Google Scholar]
- 40.Landsheer H. UncertainInterval: Uncertain Interval Methods for Three-Way Cut-Point Determination in Test Results. R package version 0.6.0; 2020.
- 41.DeZern AE, Symons HJ, Resar LS, Borowitz MJ, Armanios MY, Brodsky RA.. Detection of paroxysmal nocturnal hemoglobinuria clones to exclude inherited bone marrow failure syndromes. Eur J Haematol. 2014;92(6):467-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshida K, Cologne JB, Cordova K, et al. Aging-related changes in human T-cell repertoire over 20 years delineated by deep sequencing of peripheral T-cell receptors. Exp Gerontol. 2017;96:29-37. [DOI] [PubMed] [Google Scholar]
- 43.Rotoli B, Luzzatto L.. Paroxysmal nocturnal haemoglobinuria. Baillieres Clin Haematol. 1989;2(1):113-138. [DOI] [PubMed] [Google Scholar]
- 44.Rotoli B, Luzzatto L.. Paroxysmal nocturnal hemoglobinuria. Semin Hematol. 1989;26(3):201-207. [PubMed] [Google Scholar]
- 45.Karadimitris A, Li K, Notaro R, et al. Association of clonal T-cell large granular lymphocyte disease and paroxysmal nocturnal haemoglobinuria (PNH): further evidence for a pathogenetic link between T cells, aplastic anaemia and PNH. Br J Haematol. 2001;115(4):1010-1014. [DOI] [PubMed] [Google Scholar]
- 46.Risitano AM, Maciejewski JP, Muranski P, et al. Large granular lymphocyte (LGL)-like clonal expansions in paroxysmal nocturnal hemoglobinuria (PNH) patients. Leukemia. 2005;19(2):217-222. [DOI] [PubMed] [Google Scholar]
- 47.Luzzatto L, Bessler M, Rotoli B.. Somatic mutations in paroxysmal nocturnal hemoglobinuria: a blessing in disguise? Cell. 1997;88(1):1-4. [DOI] [PubMed] [Google Scholar]
- 48.Dameshek W.Riddle: what do aplastic anemia, paroxysmal nocturnal hemoglobinuria (PNH) and “hypoplastic” leukemia have in common? Blood. 1967;30(2):251-254. [PubMed] [Google Scholar]
- 49.Keller P, Debaun MR, Rothbaum RJ, Bessler M.. Bone marrow failure in Shwachman-Diamond syndrome does not select for clonal haematopoiesis of the paroxysmal nocturnal haemoglobinuria phenotype. Br J Haematol. 2002;119(3):830-832. [DOI] [PubMed] [Google Scholar]
- 50.Kennedy AL, Myers KC, Bowman J, et al. Distinct genetic pathways define pre-malignant versus compensatory clonal hematopoiesis in Shwachman-Diamond syndrome. Nat Commun. 2021;12(1):1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dacie JV, Gilpin A.. Refractory anaemia (Fanconi type): its incidence in three members of one family, with in one case a relationship to chronic haemolytic anaemia with nocturnal haemo-globinuria (Marchiafava-Micheli disease or ‘nocturnal haemoglobinuria’). Arch Dis Child. 1944; 19(100):155-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wainwright L, Brodsky RA, Erasmus LK, Poyiadjis S, Naidu G, MacKinnon D.. Paroxysmal nocturnal hemoglobinuria arising from Fanconi anemia. J Pediatr Hematol Oncol. 2003;25(2):167-168. [DOI] [PubMed] [Google Scholar]
- 53.Betensky M, Babushok D, Roth JJ, et al. Clonal evolution and clinical significance of copy number neutral loss of heterozygosity of chromosome arm 6p in acquired aplastic anemia. Cancer Genet. 2016;209(1-2):1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zaimoku Y, Takamatsu H, Hosomichi K, et al. Identification of an HLA class I allele closely involved in the autoantigen presentation in acquired aplastic anemia [published correction appears in Blood. 2017;130(8):1072]. Blood. 2017;129(21):2908-2916. [DOI] [PubMed] [Google Scholar]
- 55.Reina-Castillón J, Pujol R, López-Sánchez M, et al. Detectable clonal mosaicism in blood as a biomarker of cancer risk in Fanconi anemia [published correction appears in Blood Adv. 2017;1(18):1368]. Blood Adv. 2017;1(5):319-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y, Zhou W, Alter BP, et al. Chromosomal aberrations and survival after unrelated donor hematopoietic stem cell transplant in patients with fanconi anemia. Biol Blood Marrow Transplant. 2018;24(10):2003-2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Farrar JE, Vlachos A, Atsidaftos E, et al. Ribosomal protein gene deletions in Diamond-Blackfan anemia. Blood. 2011;118(26):6943-6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kallen ME, Dulau-Florea A, Wang W, Calvo KR.. Acquired and germline predisposition to bone marrow failure: diagnostic features and clinical implications. Semin Hematol. 2019;56(1):69-82. [DOI] [PubMed] [Google Scholar]
- 59.Niemeyer CM, Baumann I.. Classification of childhood aplastic anemia and myelodysplastic syndrome. Hematology Am Soc Hematol Educ Program. 2011;2011:84-89. [DOI] [PubMed] [Google Scholar]
- 60.Yang W, Zhang P, Hama A, Ito M, Kojima S, Zhu X.. Diagnosis of acquired bone marrow failure syndrome during childhood using the 2008 World Health Organization classification system. Int J Hematol. 2012;96(1):34-38. [DOI] [PubMed] [Google Scholar]
- 61.Tanaka TN, Bejar R.. MDS overlap disorders and diagnostic boundaries. Blood. 2019;133(10):1086-1095. [DOI] [PubMed] [Google Scholar]
- 62.Aalbers AM, van den Heuvel-Eibrink MM, Baumann I, et al. Bone marrow immunophenotyping by flow cytometry in refractory cytopenia of childhood. Haematologica. 2015;100(3):315-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stahl M, DeVeaux M, de Witte T, et al. The use of immunosuppressive therapy in MDS: clinical outcomes and their predictors in a large international patient cohort. Blood Adv. 2018;2(14):1765-1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sloand EM, Wu CO, Greenberg P, Young N, Barrett J.. Factors affecting response and survival in patients with myelodysplasia treated with immunosuppressive therapy. J Clin Oncol. 2008;26(15):2505-2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saunthararajah Y, Nakamura R, Wesley R, Wang QJ, Barrett AJ.. A simple method to predict response to immunosuppressive therapy in patients with myelodysplastic syndrome. Blood. 2003;102(8):3025-3027. [DOI] [PubMed] [Google Scholar]
- 66.Aalbers AM, van der Velden VH, Yoshimi A, et al. The clinical relevance of minor paroxysmal nocturnal hemoglobinuria clones in refractory cytopenia of childhood: a prospective study by EWOG-MDS. Leukemia. 2014;28(1):189-192. [DOI] [PubMed] [Google Scholar]
- 67.Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126(1):9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shen W, Clemente MJ, Hosono N, et al. Deep sequencing reveals stepwise mutation acquisition in paroxysmal nocturnal hemoglobinuria. J Clin Invest. 2014;124(10):4529-4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Noy-Lotan S, Krasnov T, Dgany O, et al. Incorporation of somatic panels for the detection of haematopoietic transformation in children and young adults with leukaemia predisposition syndromes and with acquired cytopenias. Br J Haematol. 2021;193(3):570-580. [DOI] [PubMed] [Google Scholar]
- 70.Tsai FD, Lindsley RC.. Clonal hematopoiesis in the inherited bone marrow failure syndromes. Blood. 2020;136(14):1615-1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schratz KE, Haley L, Danoff SK, et al. Cancer spectrum and outcomes in the Mendelian short telomere syndromes. Blood. 2020;135(22):1946-1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.