

Abstract
Novel therapies are urgently needed to improve global treatment of SARS-CoV-2 infection. Herein, we briefly provide a concise report on the medicinal chemistry strategies towards the development of effective SARS-CoV-2 inhibitors with representative examples in different strategies from the medicinal chemistry perspective.
KEY WORDS: SARS-CoV-2, COVID-19, Antiviral drugs, Drug design, Medicinal chemistry strategies
Graphical abstract
This review summarizes major medicinal chemistry strategies of developing SARS-CoV-2 inhibitors, including drug repositioning, large-scale screening, rational drug design based on target and utilization of natural compounds.

1. Introduction
Pandemics and epidemics of respiratory viruses continue to be recognized as being among the most common causes of morbidity and mortality around the world. In 2019, some pneumonia cases with indefinite reasons have been reported1. Through the epidemiological investigation, virus isolation and nucleic acid sequencing, it was confirmed that the pathogenic factor was a novel coronavirus named as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) by the International Committee on Taxonomy of Viruses2,3. And this disease is known as COVID-19 which standing for coronavirus disease 2019. With the continuous spread of the epidemic, the WHO announced on March 11, 2020 that COVID-19 is a global pandemic4.
SARS-CoV-2, belongs to the family of Coronavirinae that constitute the largest family of viruses, is an envelope virus composed of a single positive strand RNA genome and a helically symmetric nucleocapsid5. Coronaviruses of Orthocoronavirinae subfamily are divided into four groups: α-, β-, γ-, and δ-CoVs according to phylogenetic relationships and genomic structures6. Coronaviruses in group α such as HCoV-229E can infect people and lead to self-limiting upper respiratory tract diseases7,8. However, some coronaviruses in group β including severe acute respiratory syndrome coronavirus (SARS-CoV), SARS-CoV-2, and Middle East respiratory syndrome coronavirus (MERS-CoV) can cause serious and infectious diseases with high fatality rates9, 10, 11.
Similar to SARS-CoV, respiratory droplet and close contact are the main transmission routes of SARS-CoV-2. In addition, it was reported that there also may be a risk of faecal-oral and aerosol transmission12,13. The clinical symptoms are generally fever, fatigue, dry cough and dyspnea, and the severe ones were accompanied by acute respiratory distress syndrome, septic shock, coagulation dysfunction and multiple organ failure14.
The statistics have considered till the end of July 2021, SARS-CoV-2 has spread to 220 countries or areas, with a total of 198 million confirmed cases and 4.2 million deaths11. Although the mortality rate of SARS-CoV-2 is about 2.0%, lower than that of SARS CoV (8422 confirmed cases, 916 deaths, mortality rate 10.9%) and MERS-CoV (2468 confirmed cases, 851 deaths, case fatality rate 34.5%), SARS-CoV-2 is more contagious and now is spreading very quickly around the globe9,10. The high number of infections has demonstrated the ability of SARS-CoV-2 to spread quickly and sustainably in the community.
Unfortunately, there are no satisfactory anti-SARS-CoV-2 drugs so far. Although the FDA has approved remdesivir for the treatment of mild and moderate patients with COVID-19, WHO suggests no remdesivir for patients with COVID-19 at any severity15. Besides, the clinical trial results of lopinavir-ritonavir and hydroxychloroquine did not show effectiveness with statistical significance16,17. Therefore, there is an unmet medical need to develop novel antiviral drugs and better therapeutic options to combat this deadly disease. Taking these facts into consideration, an in-depth study of the life cycle and pathogenic mechanism of SARS-CoV-2 is urgently needed to pave the way for the development of SARS-CoV-2 inhibitors.
The very first step of SARS-CoV-2 replicative process initiates with the viral spike protein's attachment to host receptor, angiotensin-converting enzyme 2 (ACE2), mediating virion endocytosis or fusion into the host cell18. After the endocytosis, the S protein on the virus surface is activated by cathepsin L, which enables the release of viral RNA. On the other hand, the fusion process of viral envelope with the endosomal membrane follows the activation of the S protein by transmembrane protease serine 2 (TMPRRS2)19. These fusion events promote the release of the single-stranded RNA into cytoplasm, where synthesis and proteolytic cleavage of the replicase polyprotein takes place. Double-stranded RNA is synthesized by the RdRp using the single-stranded RNA as a template. Then, the double-stranded RNA is transcribed to produce genomic and subgenomic RNAs. Translation of genomic RNAs produces the accessory proteins and two polyproteins, autoproteolytically cleaved by papain-like protease (PLpro) and Main protease (Mpro). This cleavage processes render 16 nonstructural proteins including RNA dependent RNA polymerase (RdRp) and helicase. Translation of the subgenomic RNAs renders the CoV structural proteins (E, M, N, S). These proteins are subsequently inserted into the endoplasmic reticulum and trafficking along the secretory pathway into the endoplasmic reticulum-Golgi intermediate compartment (ERGIC)20. Full sequences of virus RNA are packed with the nucleocapsid (N) protein, then assembled with viral structural proteins inside membranes of the ERGIC and form complete viral particles that are released by exocytosis. The replicative cycle of SARS-CoV-2 is shown in Fig. 1. Any steps or any proteins that are essential in the SARS-CoV-2 replication cycle can be targeted to develop antiviral drugs.
Figure 1.

Replicative cycle of SARS-CoV-2.
From the medicinal chemistry point of view, many general approaches might be helpful to identify new therapies to combat SARS-CoV-2 infections. Drug repurposing is an efficient approach where the screening of chemical libraries containing drugs or investigational new drugs (INDs) occurs to find effective SARS-CoV-2 inhibitors. Those drugs or INDs with appropriate pharmaceutical properties possess approved safety profile. They can be massively produced in the pharmaceutical industry which can save a lot of time in this urgent situation. One additional possibility involves the large-scale screening in silico or in vitro. Through the methods of virtual screening and high-throughput screening (HTS), it is possible to identify promising lead compounds in a short time from millions of candidates. Another is the rational drug design based on target structures such as the functional proteins of SARS-CoV-2. However, this process may not respond quickly to such a emergency situation under pandemic, but if we take the great genetic diversity of bat SARS related-CoVs into consideration, it is necessary to enrich our arsenal in case of the emergence of new coronavirus mutants in the future21. To be noted, these three approaches are often used in combination to facilitate drug development. Besides, natural products derived from herbal plants has also been an important source of lead molecules for SARS-CoV-2 treatment.
In this review, we briefly provided a concise report on the medicinal chemistry strategies to develop effective SARS-CoV-2 inhibitors with representative examples from the medicinal chemistry perspective.
2. Medicinal chemistry strategies in seeking effective SARS-CoV-2 inhibitors
The discovery of lead compounds is a crucial step in the research and development of new drugs. Based on the experience of the discovery and development of SARS-CoV and MERS-CoV inhibitors, some general medicinal chemistry strategies can facilitate the discovery of SARS-CoV-2 candidate drugs in this emergent situation. Drug repurposing, large-scale screening in silico or in vitro, target-based rational drug design and exploitation of natural products and traditional Chinese medicine are several representative strategies. Herein, we focus on medicinal chemistry strategies and their successful application cases towards the discovery of safe and efficient SARS-CoV-2 inhibitors.
2.1. Drug repurposing
Repurposing or repositioning the existing drugs is a approach for generating novel clinical usages that beyond the scope of its original medical instruction for known approved or investigational drugs22. New drug development is generally acknowledged as time-consuming and expensive. However, drug repositioning is a shortcut to the discovery of effective coronaviral inhibitors in this pandemic situation. Here we reviewed the strategy of repurposing of existing broad-spectrum antiviral drugs and repositioning of privileged structure based on target similarity. And several representative examples for the discovery of SARS-CoV-2 candidate drugs are also discussed.
2.1.1. Reposition of existing broad-spectrum antiviral drugs
One feasible method for the discovery of effective anti-SARS-CoV-2 drugs with clinical potential would be the reusing of existing broad-spectrum antiviral drugs23. There are generally some classical methods that can be used to develop or screen for novel broad-spectrum coronaviral inhibitors. The first method is the repurposing of nucleoside analogues as the inhibitors of SARS-CoV-2 RdRp which is conserved across different coronavirus family and plays an important part in coronavirus life cycle24. It makes RdRp an ideal target for broad-spectrum coronaviral drugs. Nucleoside analogues are converted into its active triphosphate metabolite by intracellular kinase and incorporated into the growing RNA chain leading to the termination of the replicative cycle, which accounts for their antivirus activity25. However, CoVs are resistant to some nucleoside analogues such as ribavirin (1, Fig. 2) because of the proofreading activity of 3ʹ‒5ʹ exoribonuclease (ExoN)26.
Figure 2.

The structures of ribavirin, remdesivir, favipiravir, FNC, NHC and EIDD-2801.
Remdesivir (2, Fig. 2) is the representative example of RdRp inhibitors with broad-spectrum coronaviral inhibitory activity. Remdesivir showed inhibitory activity of blocking the replication of SARS-CoV, MERS-CoV and SARS-CoV-2 in HAE cells (EC50s = 69, 74 and 0.77 μmol/L, respectively)27,28. It was demonstrated that the proofreading activity could be covered by an increased and nontoxic concentration of remdesivir29.
A number of clinical trials concerning remdesivir's therapeutic effect of COVID-19 patients with respiratory tract infection have been conducted worldwide. In May 2020, the Gilead Sciences published their final report, claiming remdesivir leads to the better clinical outcome in hospitalized COVID-19 patients, including shortened recovery time and depressed respiratory system infection (NCT04280705)30. However, WHO Solidarity Trial Consortium (NCT04315948) acquired an opposite conclusion. The tested factors including overall mortality, initiation of ventilation, and duration in hospital were not significantly improved by remdesivir administration31. Meanwhile, Spinner CD studied remdesivir treatment on moderate COVID-19 patients in 11 days (NCT04292730). After the injection of remdesivir, patients presented some difference in clinical status, yet these differences could not mean any clinical improvement32. A recent clinical research, however, proved remdesivir could alleviate neuroaxonal injury of COVID-19 patients33. In view of the above contradictory clinical trials results, more clinical data are needed to make a definite conclusion.
Favipiravir (3, T-705, Fig. 2), a novel RdRp inhibitor with broad-spectrum anti-RNA virus activity, was approved in March 2014 to treat new and recurrent influenza. In addition, various studies have shown that favipiravir has a good antiviral effect on various RNA viruses, such as Ebola, norovirus, arenavirus and bunyavirus34. Within a few months of the COVID-19's outbreak, Chinese researchers reported that favipiravir had antiviral activity against SARS-CoV-2 in vitro (EC50 = 61.88 μmol/L, in Vero E6 cell)28. Soon afterwards, several favipiravir phase III clinical trials on COVID-19 were conducted, and the completed trials were reported to be effective for COVID-19. It can alleviate the clinical symptoms and shorten the clinical cure time, particularly for mild to moderate patients35, 36, 37. In view of the above positive clinical trials, the orally administered favipiravir has been authorized the emergency use for COVID-19 in India, Russia, Pakistan and other countries38. However, in December 2020, the Japanese government suspended the approval of favipiravir on the ground that it was difficult to judge its effectiveness. However, this did not negate its effectiveness, and Toyama Chemical Industry Co., Ltd. had decided to restart the clinical trial in Japan in May 2021, which will provide further evidence on its clinical efficacy and safety for the treatment of COVID-19.
Azvudine (4, FNC, Fig. 2), initially developed for HIV, is a safe and effective nucleoside-based SARS-CoV-2 RdRp inhibitor (clinical trial: ChiCTR2000029853), showing a prospect of treating COVID-19. Previous studies have shown that C4ʹ azide is the guarantee for azvudine to inhibit HIV and many other viruses39,40. However, the efficacy and safety in large sample-sized clinical trials still need to be further confirmed41.
β-d-N4-Hydroxycytidine (5, NHC, Fig. 2) is a N-hydrolyzed cytidine analog, which was initially applied to induce genetic mutation in bacteria42. Studies has proved its broad-spectrum activity towards a variety of RNA viruses, including HCoV-NL63 and SARS-CoV43,44. Most importantly, NHC has been proved to effectively inhibit SARS-CoV-2 in cell culture and animal models, with an EC50 of 0.3 μmol/L in Vero E6 and 0.08 μmol/L in Calu-345. EIDD-2801 (6, Fig. 2) is NHC's esterified prodrug, endowed with better pharmacokinetic properties and almost equal activity (EC50 = 0.22 μmol/L) compared with NHC. When NHC molecules are integrated into the growing RNA chain, single mutations are formed and accumulated, leading to lethal mutagenesis in viral genome46. Moreover, NHC can effectively evade from proofreading mechanism and is hardly recognized or excised by ExoN. It has showed prominent activity against remdesivir-resistant SARS-CoV-2 strain45.
The second method involves the development of SARS-CoV-2 inhibitors targeting the essential metabolic steps in host cells. The viral replication depends on the associated substrates, enzymes and organelles provided by the host cells. The classic example of this method is the inhibitors of dihydroorotate dehydrogenase (DHODH), a rate-limiting enzyme in the de novo biosynthesis pathway of pyrimidines nucleotides which are indispensable components of viral genomic RNA or DNA47. Thus, it makes DHODH a suitable target to develop inhibitors of coronaviral replication.
Teriflunomide is the active metabolite of leflunomide (7, 8, Fig. 3), and both are DHODH inhibitors used clinically to treat immunological disease48. According to the latest research, Xiong et al.49 found that leflunomide and teriflunomide inhibit the SARS-CoV-2 with the EC50s of 63.56 and 26.06 μmol/L in Vero E6 cells (MOI = 0.05), respectively. They designed a series of novel DHODH inhibitors based on the target structure and found that S312 and S416 (9, 10, Fig. 3) potently block SARS-CoV-2 replication with the EC50 values of 1.55 and 17 nmol/L in Vero E6 cells. Their work throws light on the application of DHODH inhibitors, including teriflunomide and S416 to combat COVID-19.
Figure 3.

The structures of teriflunomide, leflunomide, S312 and S416.
Ivermectin (11, Fig. 4), an anti-parasitic drug on the market, was previously reported as a broad-spectrum antiviral agent and it demonstrated strong inhibitory effect on SARS-CoV-2 in vitro. Treatment of SARS-CoV-2 the infected Vero-hSLAM cells with ivermectin reduced the viral RNA by about 5000 times after 48 h50. Recently, Yuan et al.51 tested a series of antimicrobial metallodrugs and found that ranitidine bismuth citrate (12, Fig. 4) can block SARS-CoV-2 replication with low cytotoxicity (CC50 = 2243 ± 43 μmol/L) and high selectivity (SI = 975). They observed the viral loads of infected Vero E6 cells decreased by 1000-fold in vitro and the action mechanism study revealed that ranitidine bismuth citrate could potently inhibit the ATPase (IC50 = 0.69 μmol/L) and DNA-unwinding of SARS-CoV-2 helicase (IC50 = 0.70 μmol/L).
Figure 4.

The structures of ivermectin and ranitidine bismuth citrate.
2.1.2. Target similarity-inspired reposition of privileged structure
The conservation or similarity of the target in different viruses makes it a suitable option for repositioning of privileged structures. It means that the privileged structures proved to be effective on the target of a known virus, and these structures potentially be effective on a similar target of a novel virus.
Camostat (13, Fig. 5), targeting TMPRSS2 and related serine proteases, has been clinically used in the treatment of chronic pancreatitis. Shirato et al.52 found that camostat could effectively block the MERS-CoV entry process. MERS-CoV S protein plays an important part in inducing cell fusion and camostat can moderately inhibit syncytium formation at single-digit micromolar concentrations, and the complete inhibition occurs at 100 μmol/L53. In 2020, Hoffmann et al.54 discovered that TMPRSS2 is involved in S protein priming of SARS-CoV-2 and 13 can suppress SARS-CoV-2 replication in lung cells.
Figure 5.

The structure of camostat mesylate.
Based on the similarity among different protease, Ma et al.55 used the FRET-based enzymatic assay to evaluate the biological activity of varying protease inhibitors against SARS-CoV-2 Mpro and reported their discovery of Mpro inhibitors. The enzymatic assay of representative inhibitors from HIV protease (aspartic protease), HCV protease (serine protease), cathepsin protease (cysteine protease), DPP4 (serine protease), and proteasome inhibitors were screened firstly. Based on the results of the first screen, they subsequently performed a re-screening of a focused library of cathepsin/calpain inhibitors and related viral Mpro inhibitors. Furthermore, they evaluated the anti-SARS-CoV-2 activity in vitro, and screened that boceprevir, calpain inhibitors II/XII, and GC-376 (14–17, Fig. 6) were effective in inhibiting the virus replication with EC50 values of 0.49–3.37 μmol/L at cellular level. The eutectic structure of SARS-CoV-2 Mpro and GC-376, which possesses the highest enzyme inhibitory activity with an IC50 of 0.03 μmol/L, revealed two specific binding configurations. These co-crystal results elucidated the molecular interactions and protein conformational flexibility in the binding process of Mpro protein with substrate and inhibitor55.
Figure 6.

Structures of boceprevir, calpain inhibitors II, XII and GC-376.
Recently, researchers at University of Chicago used the strategy of “drug repurposing” to seek anti-SARS-CoV-2 agents from 1900 approved drugs56. Masitinib, an oral tyrosine kinase inhibitor, was found to competitively inhibit the activity of the SARS-CoV-2 main protease, thereby inhibiting the replication of SARS-CoV-2 (18, Fig. 7). The antiviral activity of masitinib in transgenic mice model was evaluated, which showed that the treatment of masitinib reduced the titer of SARS-CoV-2 in lungs and nose to less than 1% only in 6 days. At the same time, the lung inflammation was also decreased. Furthermore, masatinib was also effective on various mutant strains of SARS-CoV-2 (alpha, beta and gamma).
Figure 7.

The structure of masitinib and its position in the Mpro (PDB code: 7JU7).
2.2. Large-scale screening in silico or in vitro
Virtual screening, as a complementary strategy to high-throughput screening57, has been a powerful approach in seeking effective anti-coronavirus drugs owing to the development of bioscience and computer-assisted drug design (CADD)58,59.
2.2.1. Ligand-based virtual screening
Dipyridamole (19, Fig. 8) is an effective drug in anti-coagulant therapy with favourable and broad pharmacological properties60. Dipyridamole inhibits vasodilatation and platelet aggregation through multiple mechanisms, including inhibit phosphodiesterase and block nucleoside uptake61,62. Such mechanism may contribute to its antiviral activity, because viral genome replication always relies on nucleosides from host cells. Dipyridamole has been reported to restrain the reactivation of herpes simplex virus63.
Figure 8.

The structure of dipyridamole and omipalisib.
Recently, Liu et al.64 conducted virtual screening on a library of marketed drugs and confirmed dipyridamole's blocking effect of SARS-CoV-2 replication (EC50 = 100 nmol/L, Vero E6). Therefore, they concluded that COVID-19 patients could benefit from dipyridamole through limited viral replication, suppressed hypercoagulability, and enhanced immune recovery.
Specific compound libraries are applied in screening to identify alternatives of existing drugs. As mentioned, RdRp is a druggable target to SARS-CoV-2, but researchers have proved that certain mutations of RdRp, including P323L, could render SARS-CoV-2 with drug resistance65. To identify novel molecules with potent RdRp inhibition activity, Jang66 screened 6218 drugs either on market or in clinical trial. The research is reinforced with advanced screening method and excluded any false-positive compounds. Omipalisib (20, Fig. 8), as their selected molecule with strongest activity, is about 20 folds more potent than remdesivir (IC50 = 0.49 μmol/L). Unlike traditional nucleoside RdRp inhibitors, Omipalisib has a novel scaffold and more interaction with nearby amnio acid residues, thus it is a valuable hit targeting RdRp for further optimization.
2.2.2. Target-based virtual screening
Zhai screened about 500 thousand bioactive molecules by docking method, and built a quantitative structure–activity relationship (QSAR) model to filter the identified 288 molecules. Among them, 71 compounds were selected to determine their enzyme inhibitory activities. Finally, two compounds 21 and 22 (Fig. 9) with micromolar activity (IC50 = 19 and 38 μmol/L, respectively) were chosen as top hits67.
Figure 9.

The structures of compounds 21 and 22.
Based on nine structures of Mpro with different conformations, Yang screened a protein mimetic library containing 8960 compounds. The work process mainly contains ensemble docking with the nine Mpro molecules, assisted with GlideScore, binding pose, and scaffold diversity analysis. Further evaluation of enzymatic inhibition and cellular antivirus activity confirmed remarkable activity of two molecules 23 and 24 (Fig. 10)68.
Figure 10.

The structures of compounds 23 and 24.
2.2.3. Target-based high throughput screening in vitro
The method of high throughput screening (HTS) in vitro can screen massive compounds and determine their biological activity in a short time. There are mainly two types of HTS: target-based HTS and fragment-based HTS and these two methods are often combined to discover new hit compounds.
Tripathi et al.69 tested the in vitro Mpro inhibitory of various classes drugs, which could potently cease SARS-CoV-2 replication. They combined the results of different methods including protease activity assay, surface plasmon resonance (SPR), size exclusion chromatography and in silico docking studies. And they discovered that Teicoplanin (25, Fig. 11) is a promising drug against Mpro with an EC50 value of 1.5 μmol/L and works effectively through interacting with the active site on Mpro. Besides, Coelho et al.70 used recombined Mpro as target and performed biochemical screening in a library of established drugs, and rely on fluorescent assay to identify their inhibition activity. The screening process identified thirteen drug candidates with their IC50 values were between 0.2 and 23 μmol/L. Among those 13 inhibitors, Evans blue (26, Fig. 11) exhibited the most distinguished antiviral activity and could be developed as an Mpro inhibitor.
Figure 11.

The structures of teicoplanin and Evans blue.
Nonstructural protein 14 (nsp14) is a bifunctional protein with both N7 methyltransferase and 3ʹ−5ʹ exonuclease which is highly conserved in coronavirus and is responsible for N7 methylation of guanosine in virus RNA and proofreading in the replication of RNA71,72. For nsp14 target, Pearson, LA described the high-throughput screening of 1771 FDA-approved drug library using RapidFire technology at a single concentration of 10 μmol/L. Twelve compounds with statistical significance were sorted out efficiently and accurately, among which nitazoxanide (27, Fig. 12) present the best activity with IC50 of 9.7 μmol/L. Therefore, nitazoxanide can be further optimized and modified as an nsp14 inhibitor73.
Figure 12.

The structures of nitazoxanide and PF-03882845.
Basu S conducted a HTS in vitro for nsp 14 inhibitors in a customized compound library containing more than 5000 drugs74. The compounds in the library have been clinically or fundamentally studied previously. Four potential nsp14 inhibitors were screened out which showing the varying degrees of anti-SARS-CoV-2 activity in Vero E6 cells. The most potential compound is PF-03882845 with the EC50 = 10.97 mmol/L (28, Fig. 12). Moreover, the EC50 value was significantly decreased when combined with remdesivir, indicating synergistic inhibition on the SARS-CoV-2 replication (EC50 + remdesivir = 4.79 mmol/L). This also proves that PF-03882845 is a potential nsp14 inhibitor.
2.3. Target-based rational drug design
High-resolution cocrystal structures confirmed the mechanism of action and illuminated the structural determinants involved in binding. In drug discovery, target-based rational drug design is applied for the identification of potential drug candidates of therapeutic interest according to ligand-target interactions. Molecular docking provides a powerful tool to probe fundamental biological questions at atomistic level. In the SARS-CoV-2 replication cycle, the major drug targets are Mpro, PLpro, RdRp, Spike protein and ACE2.
2.3.1. Mpro or PLpro as the target
2.3.1.1. Mpro as the target
Viral proteases have been important targets to develop antiviral drugs. In CoVs, The Mpro and PLpro would cleave polyproteins PP1a and PP1b to obtain vital nonstructural proteins, such as the viral polymerase (RdRp) and helicase75. Mpro is responsible for 11 cuts and has a protein fold similar to serine proteases like trypsin. PLpro, papain-like protease, uses a Cys residue in the catalytic process, and is responsible for three additional cuts in viral polyproteins. Because of their critical role in CoV propagation and their relatively conserved catalytical active sites, they are good targets for developing new CoV inhibitors through structure-based rational drug design76. Interestingly, HIV protease inhibitors containing a co-formulation ritonavir-boosted lopinavir are being used to treat COVID-19 patients77. However, HIV protease is an aspartyl-protease and mechanistically different from the CoV proteases78. The mechanism of action of ritonavir/lopinavir is not clear. Recent data suggest that therapies based on lopinavir have no clinical efficacy against SARS-CoV-216.
In 2020, Jin et al.79 described a program that focus on rapidly discovering lead compounds with clinical potential rapidly. This program involves different methods including structure-assisted drug design, virtual drug screening and high-throughput screening. They identified the mode of action of N3 (29, Fig. 13A) through CADD and subsequently elucidated the co-crystal structure of Mpro in complex with N3 (Fig. 13B). Then they combine target structure-based virtual screening with HTS to evaluate the Mpro inhibitory activity of more than ten thousand chemical entities including marketed drugs, drug candidates in clinical phase stage and other molecules with evident biological activity. Six of these compounds inhibited Mpro with IC50s ranging from 0.67 to 21.39 μmol/L (30–35, Fig. 13C), and ebselen showed the best antiviral activity with an EC50 value of 4.67 μmol/L in cell-level assays.
Figure 13.

(A) Structure of N3; (B) the co-crystal structure with ligand N3; (C) the structures and IC50 values of six compounds.
On the basis of the crystal structure of SARS-CoV-2 Mpro, compounds 11a and 11b (36, 37, Fig. 14A) were designed and synthetized, which exhibited excellent inhibitory activity and potently blocked SARS-CoV-2 replication. Subsequently, the co-crystal structures of the Mpro in complex with 11a or 11b were determined at 1.5 Å resolution, which revealed that the aldehyde warhead in 11a and 11b can covalently bind with Cys145 of Mpro (Fig. 14B). In addition, the two compounds exhibited good pharmacokinetic properties in vivo, and 11a also possessed low toxicity, indicating that it could act as promising drug candidates for further development of SARS-CoV-2 Mpro inhibitors80.
Figure 14.

(A) Structures of 11a and 11b; (B) the interactions between 11a and SARS-CoV-2 Mpro (PDB code: 6LZE).
Recently, using telaprevir and boceprevir as the starting point, Yang group of Sichuan University obtained 32 new and highly active small molecule inhibitors of Mpro, in which MI-23 has an IC50 value of 7.6 nmol/L (38, Fig. 15). Cocrystal structure proved that MI-23 could effectively bind to Cys145–His41 catalystic site of Mpro and the rest of the fragments occupies the S1, S2 and S4 cavities, meanwhile the aldehyde group warhead attaching to Cys145 with covalent interreaction. In addition, MI-23 exhibits good pharmacokinetic properties in vivo81. Finally, the effect of MI-23 on SARS-CoV-2 infection in transgenic mice (hACE2) was evaluated, and the result showed the significant antiviral and anti-inflammatory effects. This study laid a good foundation for the development of oral anti-SARS-CoV-2 inhibitors.
Figure 15.

The location of MI-23 (PDB code: 7D3I) in the cavity of active site.
In addition, seeking non-covalent Mpro inhibitors is also of great significance. Perampanel (39, Fig. 16) was identified as a weak SARS-CoV-2 Mpro inhibitor through kinetic assay82. According to Zhang's work83, in docking model of perampanel and Mpro, three aromatic rings stretched from its central pyridinone ring and occupied S1, S2 and S1′ pockets of the catalytic center. The initial modification of Perampanel produced compound 40, with IC50 value in single-digit micromolar level (Fig. 16). Subsequently, the researcher extended substituent groups of phenyl ring in S2 pocket to occupy S3 and S4 pocket, leading to the discovery of 41 with an enhanced activity (Fig. 16)83. Compound 42 (Fig. 16) was also synthesized in their following work, and showed favorable activity and solubility84.
Figure 16.

Structures of perampanel and compounds 40–42.
2.3.1.2. PLpro as the target
PLpro, also known as nsp3, is responsible for three additional cuts in viral polyproteins. Recent studies shed light on PLpro's mechanism of suppressing immune response of host cell and facilitate coronavirus infection85, making it a valuable antiviral target. Rut analyzed amnio acid sequence of PLpro cleavage site and used library-derived substrates to further study its substrate specificity86. Based on sequence of fluorescence substrates, they designed two tetrapeptide molecules VIR250 and VIR251 (43 and 44, Fig. 17) with covalent warhead and inhibit PLpro deubiquitinating activity both at an EC50 of 0.1 μmol/L. Furthermore, the two molecules showed relatively high selectivity, which inhibition effect is about 100 folds lower when targeted to human deubiquitinase UCH-L3.
Figure 17.

The designation and structures of VIR250 and VIR251.
Previously, in seeking SARS-CoV PLpro inhibitors, Báez-Santos et al.87 modified small-molecule lead compound 45 (Fig. 18) and designed a series of inhibitors with flexible conformation and hydrophobic binding sites, represented by compound 46 (Fig. 18). As several analogs of 45 has been proved to effectively inhibit SARS-CoV-2 PLpro activity, such non-peptide compounds could provide more options for designing novel SARS-CoV-2 PLpro inhibitors in the future.
Figure 18.

The structure of compounds 45 and 46.
2.3.2. RdRp as the target
Viral RdRp is conserved in many virus families88. Hence, it is an idealistic target to develop inhibitors active against many different coronaviruses (Fig. 19A). Coronaviruses have large genomes and relatively lower mutation rates compared to other RNA viruses, mainly because the ExoN activity provides some proofreading capability that increases the fidelity of viral genome replication.
Figure 19.

(A) X-ray crystal structure of SARS-CoV-2 RdRp (B) Interactions of remdesivir at the nucleotide binding site.
Remdesivir (Fig. 2) is a representative example of SARS-CoV-2 RdRp inhibitors (Fig. 19B). Although there are some disputes on the clinical results, one COVID-19 patient who is cured with the treatment of remdesivir has been reported89. In Jul 2020, Pruijssers et al.90 discovered that remdesivir effectively blocked the replication of SARS-CoV-2 in human lung cells with an EC50 of 0.01 μmol/L. The infected mice were treated with remdesivir, resulting in reducing viral loads and improving clinical outcomes. These experimental results in cellular level support the application of remdesivir to COVID-19 patient. Unfortunately, the subsequent Phase III trials have shown that remdesivir is a specific drug for relieving the symptom of COVID-19.
Kokic et al.91 present a widely accepted explanation of remdesivir's inhibition mechanism. remdesivir is first converted to its triphosphate metabolite (referred to as remdesivir triphosphate, RTP) form, which acts as a substrate of RdRp (Fig. 20). Then remdesivir monophosphate (RMP) was incorporated into the growing RNA chain, with three more normal nucleotides followed. At this moment, C1ʹ-cyano group on the RMP ribose ring immediately clashes with Ser861 residue in nsp12 sidechain, thus halts nsp12's continuous moving on RNA template and limits the following process of RNA replication (Fig. 20). Under such a state, the 3ʹ-nucleotide is still buried in active center and can escape exonuclease proofreading to some extent, but not completely92. Proofreading activity towards RMP-incorporated RNA chain may account for remdesivir's inadequate clinical efficiency29. Thus, it's necessary to develop inhibitors based on unique property and structure of SARS-CoV-2 RdRp. Also, specially designed exoribonuclease inhibitors are in urgent need to combine with RdRp inhibitors in therapy, which will create a coordination effect and improve clinical outcome significantly.
Figure 20.

Inhibition mechanism of remdesivir (spheres: Atom Van Deer Waals surface; purple: RMP ribose C1ʹ-cyano; red: Ser861 residue on RdRp side chain).
2.3.3. Nsp14 as the target
Nsp14 of SARS-CoV-2 is bifunctional enzyme, as mentioned in Section 2.2.3. In life cycle of SARS-CoV-2, N-cap structure formed by MTase is vital for mRNA stability and translation, while exoribonuclease leads to drug resistance of RdRp inhibitors71. Both functions of nsp14 are promising targets in anti-SARS-CoV-2 drug discovery. The cryo-EM structure of the SARS-CoV-2 Nsp10 and Nsp14 are show in Fig. 21.
Figure 21.

(A) cryo-EM structure of the SARS-CoV-2 Nsp10 and Nsp14 (PDB code: 7DIY); (B) The primary structure of SARS-CoV-2 Nsp10 and Nsp14.
MTase domain of nsp14 depends on S-adenosyl-l-methionine (SAM, 47, Fig. 22), a common methyl group donor to convert 5ʹ-guanosine triphosphate to m7GPPPN, also known as “cap” structure. MTase inhibitors are mostly pseudosubstrates mimicking SAM structure, thus can interrupt the methyl transferring of MTase. Bobilȩva93 chose non-selective MTase inhibitor sinefungin as lead (48, Fig. 22), and modified methionine chain with bulkier and more rigid structures. Of all the compounds synthesized, 49 shows the optimum activity towards nsp14 with the IC50 value of 0.16 μmol/L, lower than that of sinefungin (Fig. 22). Meanwhile, based on cavity near SAM N7 site of nsp14, Otava et al.94 replaced N7 by carbon atom attached to an aromatic moiety. Some of the designed compounds, including 50 (Fig. 22), have reached nanomolar level activity, along with excellent selectivity of human-source RNMT (RNA guanine-7-methyltransferase).
Figure 22.

The structures of SAM, sinefugin, compounds 49 and 50.
Targeting ExoN, several existing drugs were identified as its inhibitors, but it is still far from specific drug design and application95. For SARS-CoV-2, ExoN deficiency mutant is unable to replicate, thus highlights importance of ExoN inhibitors applied in either monotherapy or combined with RdRp inhibitors96.
2.3.4. Spike protein as the target
The combination of S glycoprotein and ACE2 initiates the SARS-CoV-2 replication cycle18. Researchers have depicted the structure of the SARS-CoV-2 trimeric spike protein through cryo-electron microscopy (Fig. 23A) and reveal that it contains two subunits, S1 and S2, which facilitate receptor-binding and membrane fusion process, respectively (Fig. 23B)97. The RBD which can bind ACE2 exists in S1 subunit. The process that S protein binding to the ACE2 induces complicated conformational changes, mediating the S protein switching from a prefusion conformation to a postfusion conformation98. The S1 and S2 subunits in viral S are the key components for virus binding, fusion and entry99. RBD located at the middle region of S1 subunit, and S2 subunit includes a fusion peptide (FP) and two heptapeptide repeats (HR1 and HR2) which can fuse with the target cell membrane (Fig. 23C)100. Therefore, the entry or fusion process is of great importance for viral replication and blocking the binding of ACE2 and the S1 RBD region is a proper way to develop novel SARS-CoV-2 inhibitors101, 102, 103.
Figure 23.

(A) cryo-EM structure of the SARS-CoV-2 S trimer (PDB code: 6VYB); (B) Crystal structure of S monomer; (C) The primary structure of S protein.
In 2019, Xia et al.101 found that the optimized form of polypeptide OC43-HR2P, the compound EK1 (Table 1), has potent fusion inhibitory activity against many kinds of coronaviruses (IC50 values were at 0.19–0.62 μmol/L) and pseudoviral infection activity. To improve the antiviral activity, the reachers redesigned EK1 peptide and acquired EK1C4 with cholesterol molecular attached, which gained potent inhibitory activity against SARS-CoV-2 invasion and pseudovirus infection with IC50 values of 1.3 and 15.8 nmol/L, respectively. Futhermore, EK1C4 also exhibit a broad-spectrum inhibitory activity of human coronaviruses. Intranasal injection of EK1C4 before or after the HCoV-OC43 infection can protect mice from infection, indicating it is capable to prevent and treat the current epidemic of SARS-CoV-2 and emerging SARS-related coronavirus pandemic in the future103.
Table 1.
The amino acid sequences of EK1 and EK1C4103.
| Compd. | Amino acid sequence |
|---|---|
| EK1 | SLDQINVTFLDLEYEMKLEEAIKLEESYIDLKEL |
| EK1C4 | SLDQINVTFLDLEYEMKLEEAIKLEESYIDLKEL-GSGSG-PEG4-Chol |
In 2020, Cao et al.104 combined the de novo design methods of computer-generated scaffolds and molecular docking followed by rational design. They designed some small, stable proteins with high affinity to the SARS-CoV-2 S protein and block it from intereacting with ACE2. Ten obtained miniproteins efficiently combined with the RBD with high binding affinities between 0.1 and 10 nmol/L. The miniproteins prevented Vero E6 cells from being infected by SARS-CoV-2 with median IC50 values ranging from 0.024 to 35 nmol/L, and 56- and 64-residue proteins present the most potentiality (IC50 = 0.16 nmol/L approximately). These minibinders provide a potential option to discover and develop the effective SARS-CoV-2 therapeutics.
Also, Cheng found that furin substrate cleavage in S protein is essential for viral replication and cytopathic effects. They obtained two inhibitors targeting furin, decanoyl-RVKR-chloromethylketone (CMK, 51, Fig. 24) and naphthofluorescein (52, Fig. 24), which can be developed as anti-SARS-CoV-2 agents105. The compound 51 not only blocked the entry of virus, but also further inhibited the cleavage of S protein and syncytium. However, small molecules directly targeting S protein binding site is still hard to discover and develop19.
Figure 24.

Structures of CMK and naphthofluorescein.
Glasgow et al.106 adopted “ACE2 receptor trap” strategy and developed a series of soluble variants of the ACE2 extracellular domain. The redesigned peptide molecules prevent the com-bination of S protein and host cell ACE2, including SARS-CoV, SARS-CoV-2 and any coronaviruses sharing similar cell entry pathway. Furthermore, the receptor trap molecules were modified by computational designing and fused with extra functional domains, enabling the most effective molecules among them reached an IC50 of 28 ng/mL. Such molecules provide a promising option of peptidic SARS-CoV-2 S protein inhibitor, as well as a strategy to redesign other entry receptors of future novel coronaviruses.
2.3.5. Host ACE2 as antiviral drug target
SARS-CoV-2 replication requires the host cell to provide necessary elements, including organelles, proteins and enzymes to support viral structure and function. When SARS-CoV-2 infection occurs, host cell receptors and associated enzymes is an essential component in the process of attachment and fusion.
The receptor ACE2 for SARS-CoV can recognize the RBD situated in the S1 subunit of the virus spike glycoprotein107. Researchers have demonstrated that SARS-CoV-2 uses exactly the ACE2 receptor and entry pathway as well as SARS-CoV18,108. Furthermore, it has been proven that the affinity between ACE2 and SARS-CoV-2 S glycoprotein is higher than that of SARS-CoV spike glycoprotein98. In conclusion, the facts above suggest that ACE2 plays a significant role in the life cycle of SARS-CoV-2 and is a druggable target to develop SARS-CoV-2 entry inhibitors109. Rao et al.110 reported the cryo-electron microscopy structures of full-length human ACE2 binded or unbinded with RBD of the surface S glycoprotein of SARS-CoV-2, which provided valuable insights into seeking SARS-CoV-2 entry inhibitors.
Recently, Lei et al.111 discovered that recombined human ACE2 extra-cellular domain fused with the Fc region of the human immunoglobulin IgG1 (termed as ACE2-Ig, Fig. 25) presents high-affinity binding to the RBD of SARS-CoV-2 and exhibited expected pharmacological activities. Furthermore, ACE2-Ig displays strong inhibitory activity on multiple coronaviruses in vitro. Therefore, these findings support the deeper studies of ACE2-Ig to exploit the entry inhibitor of SARS-CoV-2.
Figure 25.

The structure of ACE2-Ig.
2.4. Natural products
Natural products constitute essential resources to develop new drugs. Therefore, it is worthy of exploring to find SARS-CoV-2 inhibitors from natural products and TCM.
2.4.1. Natural products
Natural products have also provided rich resources for the development of novel antiviral agents112, 113, 114. From January 1981 to September 2019, there are 1881 new drugs have been developed. And 49.2% among these new drugs are natural products or natural product derivatives115. To speed up the discovery and development of anti-coronavirus drugs, researchers have explored the inhibitory effect of natural products of various structures on human coronaviruses. This part summarizes several types of structures that have been widely studied.
Recently, Su et al.116 identified a flavonoid natural product, baicalein (53, Fig. 26), as a non-covalent small-molecule inhibitors of SARS-CoV-2 Mpro confirmed the co-crystal structure of the Mpro in complex with 53 (EC50 = 1.69 μmol/L in Vero E6 cells). Remarkably, the co-crystal structure showed that baicalein can be perfectly fixed in the core of the substrate-binding pocket by interacting with several residues of Mpro (L141−S144), and acting as a “shield” to protect the active site from any substrate approaching (Fig. 26). This unique binding mode, combined with its high ligand binding efficiency (Kd = 4.03 nmol/L) and low molecular weight (270.24 Da), emphasizes that 53 provides an efficient starting point to develop anti-coronavirus drugs.
Figure 26.

The structure of baicalein and the X-ray costructure of baicalein in SARS-CoV-2 Mpro (PDB code: 6M2N).
In addition, Clementi et al.117 found that the activity of human endo-lysosomal Two-Pore Channels (TPC) can be inhibited by the natural flavonoid compound Naringenin (54, Fig. 27). It can protect cells from SARS-CoV-2 infection in a time- and concentration-dependent manner. And they observed a substantial decrease of cytopathic effect (>90%) at 48 h post-infection when Vero E6 cells were treated with both 250 and 62.5 μmol/L naringenin. Besides, they found the replication of HCoV-OC43 and HCoV-229E (MOI = 0.01) can be totally inhibited at the concentration of 62.5 μmol/L within no relevant toxicity. In conclusion, they proved Naringenin as a safe anti-SARS-CoV-2 agent with pan-coronavirus inhibitory activity.
Figure 27.
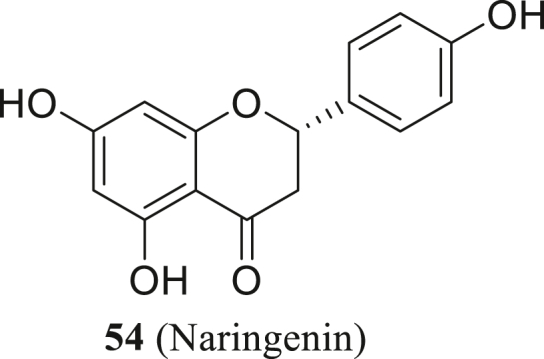
The structure of naringenin.
Colchicine (55, Fig. 28) is a widely applied phytochemical compound, now mainly used to treat gout and Familial Mediterranean Fever (FMF). It had been confirmed to inhibit inflammation response and cytokine storm through many pathways118,119. In the clinical treatment of COVID-19 and previous coronavirus infections, stopping the hyperinflammatory state is vital to reduce complications and improve patient health. Thus, colchicine is considered to have positive effects on SARS-CoV-2 cases, meanwhile, some clinical trials on the effectiveness of colchicine in SARS-CoV-2 patients are in progress120. Recently, Al-Kuraishy121 hypothesizes that a combination of colchicine-doxycycline therapy would have more contribution to COVID-19 treatment, which calls for further clinical evaluation.
Figure 28.

The structure of colchine, emetine, lycorine and glycyrrhizin.
Wang et al.122 reported the emetine (56, Fig. 28) can significantly influence SARS-CoV-2 entry and replication, meanwhile reducing inflammatory factor level in vitro. Lycorine (57, Fig. 28), a natural non-nucleoside RdRp inhibitor, was reported that it could limit SARS-CoV-2 replication with IC50 value of 0.88 ± 0.022 μmol/L, comparable to remdesivir in the same condition123. Van et al.124 predicted glycyrrhizin's (58, Fig. 28) inhibitory activity of SARS-CoV-2 main protease via computational simulation. Development and modification of these phytochemical compounds may provide better treatment options in the future.
Myricetin (59, Fig. 29) is a common flavonoid derived from a wide range of botanical sources. Recent research shows myricetin has high inhibition activity to SARS-CoV-2 Mpro by covalently binding to Cys145 residue (can't show in PyMOL)125. Under oxidative conditions, such a covalent bond could be formed with high specify towards the catalytic center. This discovery encourages further exploration of phytochemicals containing polyphenol group to identify novel covalent SARS-CoV-2 Mpro inhibitors.
Figure 29.

The structure of myricetin and the X-ray costructure of myricetin in SARS-CoV-2 Mpro (PDB code: 7DPP).
3. Conclusions and prospects
The current COVID-19 pandemic have brought major challenge to public health. Since then, significant efforts have been made by scientists around the world and impressive progress has been gained in the research of structural biology, epidemiology and antiviral interventions of the SARS-CoV-2. Unfortunately, there are no effective drugs to combat SARS-CoV-2 until now. It is an enormous challenge for the development of effective drugs, especially for halting the suddenly outbroken SARS-CoV-2.
In face of this urgent situation, some strategies could facilitate the development of effective SARS-CoV-2 inhibitors. Firstly, drug repurposing involves the reuse of existing drugs for a new applications. The advantage of drug repurposing is that the drug security has been tested, so this method can bypass the pharmacokinetic, pharmacodynamic and toxic studies, resulting in entering phase Ⅱ or Ⅲ clinical trials directly, which will significantly promote the drug development process. The FDA-approved drug library, clinical and preclinical drug library and broad-spectrum antivirals library are the common libraries used for drug repurposing. Secondly, large-scale screening in silico and in vitro can evaluate tens of millions of compounds efficiently, which will increase the amount and quality of novel inhibitors. To be noted, large-scale screening can facilitate new drug development only with the exception of false-positive results, along with further evaluation of antiviral activity126. Thirdly, de novo drug design based on the target is a rational process to design a new structure on a molecular level. This is a time-consuming process, but it is necessary to expand our drug reservoir to deal with the potential outbreak of potential coronavirus variants.
It's worth noting that SARS-CoV-2 has a highly mutative genetic sequence, resulting in the generation of drug-resistant mutant strains (alpha, beta, gamma, delta), Among them, delta mutant has stronger transmission ability and greater virus load. The pandemic changed the strategies and methods of researchers, and it showed them what works and what doesn't during a global emergency. The method of target-based drug design is a proper way to overcome the drug resistance of coronavirus. The specific chemical interactions can be introduced into the molecular structure resulting in specifically targeting the regions which are most unlikely for the emergence of drug resistance. Besides, the strategy of the degradation of viral components is another new way to overcome drug resistance. Proteolysis-targeting chimeras (PROTAC)127. and ribonuclease targeting chimeras (RIBOTAC)128. are the representative approaches to degrade viral genomes or proteins which are pivotal in the viral life cycle.
In summary, the development of novel and effective SARS-CoV-2 inhibitors is urgently-needed to respond to the situation what we are facing now. Novel compounds are expected take a significant part in the combination against circulating and emerging coronavirus epidemic. We need to increase the investment in the basic research of antiviral drugs, to build the systematic platform for de novo drug design, lead compound discovery and optimization, drug-likeness evaluation and PK/PD study. In this way, enough drug reservoir could be developed to respond any possible coronavirus out break in the current crisis and future.
Acknowledgments
We gratefully acknowledge financial support from the Shandong Provincial Key Research and Development Project (No. 2019JZZY021011, China), Foreign Cultural and Educational Experts Project (GXL20200015001, China), Outstanding Youth Fund of Shandong Province (ZR2020JQ31, China), Qilu Young Scholars Program of Shandong University and the Taishan Scholar Program at Shandong Province.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Contributor Information
Peng Zhan, Email: zhanpeng1982@sdu.edu.cn.
Xinyong Liu, Email: xinyongl@sdu.edu.cn.
Author contributions
Shenghua Gao and Tianguang Huang drafted the manuscript. Shenghua Gao and Letian Song constructed the Figures. Shujing Xu, Yusen Cheng, Lin Sun and Jian Zhang participated in the collation and classification of literature. Peng Zhan and Dongwei Kang provided constructive suggestions. Srinivasulu Cherukupalli and Tong Zhao contributed to the editing. Peng Zhan and Xinyong Liu conceived the project and revised the manuscript.
Conflicts of interest
The authors declare no conflict of interest. All binding mode illustrations were produced with PyMol (www.pymol.org).
References
- 1.Wu F., Zhao S., Yu B., Chen Y., Wang W., Song Z., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu Y., Ho W., Huang Y., Jin D.Y., Li S., Liu S.L., et al. SARS-CoV-2 is an appropriate name for the new coronavirus. Lancet. 2020;395:949–950. doi: 10.1016/S0140-6736(20)30557-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coronaviridae Study Group of the International Committee on Taxonomy of Viruses The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5:536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.World Health Organization . 11 March 2020. Coronavirus disease 2019 (COVID-19) situation report-51.https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports/ Available from: [Google Scholar]
- 5.Woo P.C.Y., Lau S.K.P., Huang Y., Yuen K.Y. Coronavirus diversity, phylogeny and interspecies jumping. Exp Biol Med. 2009;234:1117–1127. doi: 10.3181/0903-MR-94. [DOI] [PubMed] [Google Scholar]
- 6.Woo P.C., Lau S.K., Lam C.S., Lau C.C., Tsang A.K., Lau J.H., et al. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J Virol. 2012;86:3995–4008. doi: 10.1128/JVI.06540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simon A., Völz S., Höfling K., Kehl A., Tillman R., Müller A., et al. Acute life threatening event (ALTE) in an infant with human coronavirus HCoV-229E infection. Pediatr Pulmonol. 2007;42:393–396. doi: 10.1002/ppul.20595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mayer K., Nellessen C., Hahn-Ast C., Schumacher M., Pietzonka S., Eis-Hübinger A.M., et al. Fatal outcome of human coronavirus NL63 infection despite successful viral elimination by IFN-alpha in a patient with newly diagnosed ALL. Eur J Haematol. 2016;97:208–210. doi: 10.1111/ejh.12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.World Health Organization . 2003. Summary table of SARS cases by country, 1st November 2002–7th August 2003.https://www.who.int/csr/sars/country/2003_08_15/en/ Avaliable from: [Google Scholar]
- 10.World Health Organization . 30 January, 2020. MERS situation update in January 2020.http://www.emro.who.int/health-topics/mers-cov/mers-outbreaks.html Available from: [Google Scholar]
- 11.World Health Organization . 2020. Coronavirus disease (COVID-2019) situation reports.https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports/ Available from: [Google Scholar]
- 12.Wang D., Hu B., Hu C., Zhu F., Liu X., Zhang J., et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. J Am Med Assoc. 2020;323:1061–1069. doi: 10.1001/jama.2020.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen H., Guo J., Wang C., Luo F., Yu X., Zhang W., et al. Clinical characteristics and intrauterine vertical transmission potential of COVID-19 infection in nine pregnant women: a retrospective review of medical records. Lancet. 2020;395:809–815. doi: 10.1016/S0140-6736(20)30360-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.World Health Organization . 20 November 2020. WHO recommends against the use of remdesivir in COVID-19 patients.https://www.who.int/news-room/feature-stories/detail/who-recommends-against-the-use-of-remdesivir-in-covid-19-patients Available from: [Google Scholar]
- 16.Cao B., Wang Y., Wen D., Liu W., Wang J., Fan G., et al. A Trial of lopinavir-ritonavir in adults hospitalized with severe COVID-19. N Engl J Med. 2020;382:1787–1799. doi: 10.1056/NEJMoa2001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boulware D.R., Pullen M.F., Bangdiwala A.S., Pastick K.A., Lofgren S.M., Okafor E.C., et al. A randomized trial of hydroxychloroquine as postexposure prophylaxis for COVID-19. N Engl J Med. 2020;383:517–525. doi: 10.1056/NEJMoa2016638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 19.Chen Z., Du R., Galvan Achi J.M., Rong L., Cui Q. SARS-CoV-2 cell entry and targeted antiviral development. Acta Pharm Sin B. 2021;12:3879–3888. doi: 10.1016/j.apsb.2021.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krijnse-Locker J., Ericsson M., Rottier P.J., Griffiths G. Characterization of the budding compartment of mouse hepatitis virus: evidence that transport from the RER to the Golgi complex requires only one vesicular transport step. J Cell Biol. 1994;124:55–70. doi: 10.1083/jcb.124.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cui J., Li F., Shi Z.L. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17:181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ashburn T.T., Thor K.B. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. 2004;3:673–683. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- 23.Li C.C., Wang X.J., Wang H.C.R. Repurposing host-based therapeutics to control coronavirus and influenza virus. Drug Discov Today. 2019;24:726–736. doi: 10.1016/j.drudis.2019.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ng K.K.S., Arnold J.J., Cameron C.E. Structure-function relationships among RNA-dependent RNA polymerases. Curr Top Microbiol Immunol. 2008;320:137–156. doi: 10.1007/978-3-540-75157-1_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sofia M.J., Chang W., Furman P.A., Mosley R.T., Ross B.S. Nucleoside, nucleotide, and non-nucleoside inhibitors of hepatitis C virus NS5B RNA-dependent RNA-polymerase. J Med Chem. 2012;55:2481–2531. doi: 10.1021/jm201384j. [DOI] [PubMed] [Google Scholar]
- 26.Smith E.C., Blanc H., Vignuzzi M., Denison M.R. Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: evidence for proofreading and potential therapeutics. PLoS Pathog. 2013;9 doi: 10.1371/journal.ppat.1003565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sheahan T.P., Sims A.C., Graham R.L., Menachery V.D., Gralinski L.E., Case J.B., et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aal3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agostini M.L., Andres E.L., Sims A.C., Graham R.L., Sheahan T.P., Lu X., et al. Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio. 2018;9 doi: 10.1128/mBio.00221-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beigel J.H., Tomashek K.M., Dodd L.E., Mehta A.K., Zingman B.S., Kalil A.C., et al. Remdesivir for the treatment of COVID-19-final report. N Engl J Med. 2020;383:1813–1826. doi: 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.WHO Solidarity Trial Consortium. Pan H., Peto R., Henao-Restrepo A.M., Preziosi M.P., Sathiyamoorthy V., et al. Repurposed antiviral drugs for COVID-19—interim WHO solidarity trial results. N Engl J Med. 2021;384:497–511. doi: 10.1056/NEJMoa2023184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spinner C.D., Gottlieb R.L., Criner G.J., Arribas López J.R., Cattelan A.M., Viladomiu A.S., et al. Effect of remdesivir vs standard care on clinical status at 11 days in patients with moderate COVID-19: a randomized clinical trial. J Am Med Assoc. 2020;324:1048–1057. doi: 10.1001/jama.2020.16349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prudencio M., Erben Y., Marquez C.P., Jansen-West K.R., Franco-Mesa C., Heckman M.G., et al. Serum neurofilament light protein correlates with unfavorable clinical outcomes in hospitalized patients with COVID-19. Sci Transl Med. 2021;13 doi: 10.1126/scitranslmed.abi7643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Furuta Y., Komeno T., Nakamura T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc Jpn Acad Ser B Phys Biol Sci. 2017;93:449–463. doi: 10.2183/pjab.93.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cai Q., Yang M., Liu D., Chen J., Shu D., Xia J., et al. Experimental treatment with favipiravir for COVID-19: an open-label control study. Engineering. 2020;6:1192–1198. doi: 10.1016/j.eng.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Udwadia Z.F., Singh P., Barkate H., Patil S., Rangwala S., Pendse A., et al. Efficacy and safety of favipiravir, an oral RNA-dependent RNA polymerase inhibitor, in mild-to-moderate COVID-19: a randomized, comparative, open-label, multicenter, phase 3 clinical trial. Int J Infect Dis. 2021;103:62–71. doi: 10.1016/j.ijid.2020.11.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ivashchenko A.A., Dmitriev K.A., Vostokova N.V., Azarova V.N., Blinow A.A., Egorova A.N., et al. AVIFAVIR for treatment of patients with moderate COVID-19: interim results of a phase II/III multicenter randomized clinical trial. Clin Infect Dis. 2020;9 doi: 10.1093/cid/ciaa1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joshi S., Parkar J., Ansari A., Vora A., Talwar D., Tiwaskar M., et al. Role of favipiravir in the treatment of COVID-19. Int J Infect Dis. 2021;102:501–508. doi: 10.1016/j.ijid.2020.10.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu N., Yang J., Zheng B., Zhang Y., Cao Y., Huan C., et al. The pyrimidine analog FNC potently inhibits the replication of multiple enteroviruses. J Virol. 2020;94:e00204–e00220. doi: 10.1128/JVI.00204-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun L., Peng Y., Yu W., Zhang Y., Liang L., Song C., et al. Mechanistic insight into antiretroviral potency of 2ʹ-eeoxy-2ʹ-β-fluoro-4ʹ-azidocytidine (FNC) with a long-lasting effect on HIV-1 prevention. J Med Chem. 2020;63:8554–8566. doi: 10.1021/acs.jmedchem.0c00940. [DOI] [PubMed] [Google Scholar]
- 41.Ren Z.G., Luo H., Yu Z.J., Song J., Liang L., Wang L., et al. A randomized, open-label, controlled clinical trial of azvudine tablets in the treatment of mild and common COVID-19, a pilot study. Adv Sci. 2020;7:2001435. doi: 10.1002/advs.202001435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salganik R.I., Vasjunina E.A., Poslovina A.S., Andreeva I.S. Mutagenic action of N4-hydroxycytidine on Escherichia coli B cyt- Mutat Res. 1973;20:1–5. doi: 10.1016/0027-5107(73)90091-2. [DOI] [PubMed] [Google Scholar]
- 43.Barnard D.L., Hubbard V.D., Burton J., Smee D.F., Morrey J.D., Otto M.J., et al. Inhibition of severe acute respiratory syndrome-associated coronavirus (SARSCoV) by calpain inhibitors and beta-d-N4-hydroxycytidine. Antivir Chem Chemother. 2004;15:15–22. doi: 10.1177/095632020401500102. [DOI] [PubMed] [Google Scholar]
- 44.Pyrc K., Bosch B.J., Berkhout B., Jebbink M.F., Dijkman R., Rottier P., et al. Inhibition of human coronavirus NL63 infection at early stages of the replication cycle. Antimicrob Agents Chemother. 2006;50:2000–2008. doi: 10.1128/AAC.01598-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sheahan T.P., Sims A.C., Zhou S., Graham R.L., Pruijssers A.J., Agostini M.L., et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci Transl Med. 2020;12 doi: 10.1126/scitranslmed.abb5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pruijssers A.J., Denison M.R. Nucleoside analogues for the treatment of coronavirus infections. Curr Opin Virol. 2019;35:57–62. doi: 10.1016/j.coviro.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Evans D.R., Guy H.I. Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway. J Biol Chem. 2004;279:33035–33038. doi: 10.1074/jbc.R400007200. [DOI] [PubMed] [Google Scholar]
- 48.Donahue K.E., Gartlehner G., Jonas D.E., Lux L.J., Thieda P., Jonas B.L., et al. Systematic review: comparative effectiveness and harms of disease-modifying medications for rheumatoid arthritis. Ann Intern Med. 2008;148:124–134. doi: 10.7326/0003-4819-148-2-200801150-00192. [DOI] [PubMed] [Google Scholar]
- 49.Xiong R., Zhang L., Li S., Sun Y., Ding M., Wang Y., et al. Novel and potent inhibitors targeting DHODH are broad-spectrum antivirals against RNA viruses including newly-emerged coronavirus SARS-CoV-2. Protein Cell. 2020;11:723–739. doi: 10.1007/s13238-020-00768-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caly L., Druce J.D., Catton M.G., Jans D.A., Wagstaff K.M. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir Res. 2020;178:104787. doi: 10.1016/j.antiviral.2020.104787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yuan S., Wang R., Chan J., Zhang A., Cheng T., Chik K., et al. Metallodrug ranitidine bismuth citrate suppresses SARS-CoV-2 replication and relieves virus-associated pneumonia in Syrian hamsters. Nat Microbiol. 2020;5:1439–1448. doi: 10.1038/s41564-020-00802-x. [DOI] [PubMed] [Google Scholar]
- 52.Zhou Y., Vedantham P., Lu K., Agudelo J., Carrion R., Jr., Nunneley J.W., et al. Protease inhibitors targeting coronavirus and filovirus entry. Antivir Res. 2015;116:76–84. doi: 10.1016/j.antiviral.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shirato K., Kawase M., Matsuyama S. Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J Virol. 2013;87:12552–12561. doi: 10.1128/JVI.01890-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma C., Sacco M.D., Hurst B., Townsend J.A., Hu Y., Szeto T., et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30:678–692. doi: 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Drayman N., DeMarco J.K., Jones K.A., Azizi S.A., Froggatt H.M., Tan K., et al. Masitinib is a broad coronavirus 3CL inhibitor that blocks replication of SARS-CoV-2. Science. 2021;373:931–936. doi: 10.1126/science.abg5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bajorath J. Integration of virtual and high-throughput screening. Nat Rev Drug Discov. 2002;1:882–894. doi: 10.1038/nrd941. [DOI] [PubMed] [Google Scholar]
- 58.Kitchen D.B., Decornez H., Furr J.R., Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3:935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 59.Wingert B.M., Camacho C.J. Improving small molecule virtual screening strategies for the next generation of therapeutics. Curr Opin Chem Biol. 2018;44:87–92. doi: 10.1016/j.cbpa.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thomé M.P., Borde C., Larsen A.K., Henriques J.A., Lenz G., Escargueil A.E., et al. Dipyridamole as a new drug to prevent Epstein-Barr virus reactivation. Antivir Res. 2019;172:104615. doi: 10.1016/j.antiviral.2019.104615. [DOI] [PubMed] [Google Scholar]
- 61.Gresele P., Momi S., Falcinelli E. Anti-platelet therapy: phosphodiesterase inhibitors. Br J Clin Pharmacol. 2011;72:634–646. doi: 10.1111/j.1365-2125.2011.04034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harker L.A., Kadatz R.A. Mechanism of action of dipyridamole. Thromb Res. 1983;29:39–46. doi: 10.1016/0049-3848(83)90356-0. [DOI] [PubMed] [Google Scholar]
- 63.Tenser R.B., Gaydos A., Hay K.A. Inhibition of herpes simplex virus reactivation by dipyridamole. Antimicrob Agents Chemother. 2001;45:3657–3659. doi: 10.1128/AAC.45.12.3657-3659.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu X., Li Z., Liu S., Chen Z., Zhao Z., Huang Y.Y., et al. Therapeutic effects of dipyridamole on COVID-19 patients with coagulation dysfunction. Acta Pharm Sin B. 2020;10:1205–1215. doi: 10.1016/j.apsb.2020.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pachetti M., Marini B., Benedetti F., Giudici F., Mauro E., Storici P., et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J Transl Med. 2020;18:179. doi: 10.1186/s12967-020-02344-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jang W.D., Jeon S., Kim S., Lee S.Y. Drugs repurposed for COVID-19 by virtual screening of 6,218 drugs and cell-based assay. Proc Natl Acad Sci U S A. 2021;118 doi: 10.1073/pnas.2024302118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhai T., Zhang F., Haider S., Kraut D., Huang Z. An integrated computational and experimental approach to identifying inhibitors for SARS-CoV-2 3CL protease. Front Mol Biosci. 2021;8:661424. doi: 10.3389/fmolb.2021.661424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang J., Lin X., Xing N., Zhang Z., Zhang H., Wu H., et al. Structure-based discovery of novel nonpeptide inhibitors targeting SARS-CoV-2 Mpro. J Chem Inf Model. 2021;61:3917–3926. doi: 10.1021/acs.jcim.1c00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tripathi P.K., Upadhyay S., Singh M., Raghavendhar S., Bhardwaj M., Sharma P., et al. Screening and evaluation of approved drugs as inhibitors of main protease of SARS-CoV-2. Int J Biol Macromol. 2020;164:2622–2631. doi: 10.1016/j.ijbiomac.2020.08.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Coelho C., Gallo G., Campos C.B., Hardy L., Würtele M. Biochemical screening for SARS-CoV-2 main protease inhibitors. PLoS One. 2020;15 doi: 10.1371/journal.pone.0240079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu C., Shi W., Becker S.T., Schatz D.G., Liu B., Yang Y. Structural basis of mismatch recognition by a SARS-CoV-2 proofreading enzyme. Science. 2021;373:1142–1146. doi: 10.1126/science.abi9310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yan L., Yang Y., Li M., Zhang Y., Zheng L., Ge J., et al. Coupling of N7-methyltransferase and 3′−5′ exoribonuclease with SARS-CoV-2 polymerase reveals mechanisms for capping and proofreading. Cell. 2021;184:3474–3485. doi: 10.1016/j.cell.2021.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pearson L.A., Green C.J., Lin D., Petit A.P., Gray D.W., Cowling V.H., et al. Development of a high-throughput screening assay to identify inhibitors of the SARS-CoV-2 guanine-N7-methyltransferase using RapidFire mass spectrometry. SLAS Discov. 2021;26:749–756. doi: 10.1177/24725552211000652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Basu S., Mak T., Ulferts R., Wu M., Deegan T., Fujisawa R., et al. Identifying SARS-CoV-2 antiviral compounds by screening for small molecule inhibitors of Nsp14 RNA cap methyltransferase. Biochem J. 2021;478:2481–2497. doi: 10.1042/BCJ20210219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zumla A., Chan J.F., Azhar E.I., Hui D.S., Yuen K.Y. Coronaviruses-drug discovery and therapeutic options. Nat Rev Drug Discov. 2016;15:327–347. doi: 10.1038/nrd.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chuck C.P., Chen C., Ke Z., Wan D.C.C., Chow H.F., Wong K.B. Design, synthesis and crystallographic analysis of nitrile-based broad-spectrum peptidomimetic inhibitors for coronavirus 3C-like proteases. Eur J Med Chem. 2013;59:1–6. doi: 10.1016/j.ejmech.2012.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.National Health Commission of China . 2020. Notice on issuing the new coronavirus pneumonia diagnosis and treatment plan (7th ed. in trial)http://www.nhc.gov.cn/yzygj/s7653p/202003/46c9294a7dfe4cef80dc7f5912eb1989.shtml Available from: [Google Scholar]
- 78.Menéndez-Arias L., Tözsér J. HIV-1 protease inhibitors: effects on HIV-2 replication and resistance. Trends Pharmacol Sci. 2008;29:42–49. doi: 10.1016/j.tips.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 79.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 80.Dai W., Zhang B., Jiang X.M., Su H., Li J., Zhao Y., et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Qiao J., Li Y., Zeng R., Liu F., Luo R., Huang C., et al. SARS-CoV-2 M(pro) inhibitors with antiviral activity in a transgenic mouse model. Science. 2021;371:1374–1378. doi: 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ghahremanpour M.M., Tirado-Rives J., Deshmukh M., Ippolito J.A., Zhang C.H., Cabeza de Vaca I., et al. Identification of 14 known drugs as inhibitors of the main protease of SARS-CoV-2. ACS Med Chem Lett. 2020;11:2526–2533. doi: 10.1021/acsmedchemlett.0c00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang C.H., Stone E.A., Deshmukh M., Ippolito J.A., Ghahremanpour M.M., Tirado-Rives J., et al. Potent noncovalent inhibitors of the main protease of SARS-CoV-2 from molecular sculpting of the drug perampanel guided by free energy perturbation calculations. ACS Cent Sci. 2021;7:467–475. doi: 10.1021/acscentsci.1c00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang C.H., Spasov K.A., Reilly R.A., Hollander K., Stone E.A., Ippolito G.A., et al. Optimization of triarylpyridinone inhibitors of the main protease of SARS-CoV-2 to low-nanomolar antiviral potency. ACS Med Chem Lett. 2021;12:1325–1332. doi: 10.1021/acsmedchemlett.1c00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shin D., Mukherjee R., Grewe D., Bojkova D., Baek K., Bhattacharya A., et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587:657–662. doi: 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rut W., Lv Z., Zmudzinski M., Patchett S., Nayak D., Snipas S.J., et al. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: a framework for anti-COVID-19 drug design. Sci Adv. 2020;6 doi: 10.1126/sciadv.abd4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Báez-Santos Y.M., Barraza S.J., Wilson M.W., Agius M.P., Mielech A.M., Davis N.M., et al. X-ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain-like proteases. J Med Chem. 2014;57:2393–2412. doi: 10.1021/jm401712t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ng K.K.-S., Arnold J.J., Cameron C.E. Structure-function relationships among RNA-dependent RNA polymerases, RNA interference. Curr Top Microbiol Immunol. 2008;320:137–156. doi: 10.1007/978-3-540-75157-1_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Holshue M.L., DeBolt C., Lindquist S., Lofy K.H., Wiesman J., Bruce H., et al. First case of 2019 novel coronavirus in the United States. N Engl J Med. 2020;382:929–936. doi: 10.1056/NEJMoa2001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pruijssers A.J., George A.S., Schäfer A., Leist S.R., Gralinksi L.E., Dinnon K.H., et al. Remdesivir inhibits SARS-CoV-2 in human lung cells and chimeric SARS-CoV expressing the SARS-CoV-2 RNA polymerase in mice. Cell Rep. 2020;32:107940. doi: 10.1016/j.celrep.2020.107940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kokic G., Hillen H., Tegunov D., Dienemann C., Seitz F., Schmitzova J., et al. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat Commun. 2021;12:279. doi: 10.1038/s41467-020-20542-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bouvet M., Imbert I., Subissi L., Gluais L., Canard B., Decroly E. RNA 3ʹ-end mismatch excision by the severe acute respiratory syndrome coronavirus nonstructural protein nsp10/nsp14 exoribonuclease complex. Proc Natl Acad Sci U S A. 2012;109:9372–9377. doi: 10.1073/pnas.1201130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bobiļeva O., Bobrovs R., Kaņepe I., Patetko L., Kalniņš G., Šišovs M., et al. Potent SARS-CoV-2 mRNA cap methyltransferase inhibitors by bioisosteric replacement of methionine in SAM cosubstrate. ACS Med Chem Lett. 2021;12:1102–1107. doi: 10.1021/acsmedchemlett.1c00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Otava T., Šála M., Li F., Fanfrlík J., Devkota K., Perveen S., et al. The structure-based design of SARS-CoV-2 nsp14 methyltransferase ligands yields nanomolar inhibitors. ACS Infect Dis. 2021;7:2214–2220. doi: 10.1021/acsinfecdis.1c00131. [DOI] [PubMed] [Google Scholar]
- 95.Wang X., Sacramento C.Q., Jockusch S., Chaves O.A., Tao C., Fintelman-Rodrigues N., et al. Combination of antiviral drugs to inhibit SARS-CoV-2 polymerase and exonuclease as potential COVID-19 therapeutics. bioRxiv. 2021 doi: 10.1038/s42003-022-03101-9. https://www.biorxiv.org/content/10.1101/2021.07.21.453274v1 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ogando N.S., Zevenhoven-Dobbe J.C., van der Meer Y., Bredenbeek P.J., Posthuma C.C., Snijder E.J. The enzymatic activity of the nsp14 exoribonuclease is critical for replication of MERS-CoV and SARS-CoV-2. J Virol. 2020;94 doi: 10.1128/JVI.01246-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cantuti-Castelvetri L., Ojha R., Pedro L.D., Djannatian M., Franz J., Kuivanen S., et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020;370:856–860. doi: 10.1126/science.abd2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.L., Abiona O., et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lu L., Liu Q., Zhu Y., Chan K.H., Qin L., Li Y., et al. Structure-based discovery of Middle East respiratory syndrome coronavirus fusion inhibitor. Nat Commun. 2014;5:3067. doi: 10.1038/ncomms4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang C., Zhao L., Xia S., Zhang T., Cao R., Liang G., et al. De Novo design of α-helical lipopeptides targeting viral fusion proteins: a promising strategy for relatively broad-spectrum antiviral drug discovery. J Med Chem. 2018;61:8734–8745. doi: 10.1021/acs.jmedchem.8b00890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li J., Zhan P., Liu X. Targeting the entry step of SARS-CoV-2: a promising therapeutic approach. Signal Transduct Target Ther. 2020;17:98. doi: 10.1038/s41392-020-0195-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xiu S., Dick A., Ju H., Mirzaie S., Abdi F., Cocklin S., et al. Inhibitors of SARS-CoV-2 entry: current and future opportunities. J Med Chem. 2020;63:12256–12274. doi: 10.1021/acs.jmedchem.0c00502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xia S., Zhu Y., Liu M., Lan Q., Xu W., Wu Y., et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell Mol Immunol. 2020;17:765–767. doi: 10.1038/s41423-020-0374-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cao L., Goreshnik I., Coventry B., Case J.B., Miller L., Kozodoy L., et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science. 2020;370:426–431. doi: 10.1126/science.abd9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cheng Y.W., Chao T.L., Li C.L., Chiu M.F., Kao H.C., Wang S.H., et al. Furin inhibitors block SARS-CoV-2 spike protein cleavage to suppress virus production and cytopathic effects. Cell Rep. 2020;33:108254. doi: 10.1016/j.celrep.2020.108254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Glasgow A., Glasgow J., Limonta D., Solomon P., Lui I., Zhang Y., et al. Engineered ACE2 receptor traps potently neutralize SARS-CoV-2. Proc Natl Acad Sci U S A. 2020;117:28046–28055. doi: 10.1073/pnas.2016093117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bian J., Li Z. Angiotensin-converting enzyme 2 (ACE2): SARS-CoV-2 receptor and RAS modulator. Acta Pharm Sin B. 2021;1:1–12. doi: 10.1016/j.apsb.2020.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lei C., Qian K., Li T., Zhang S., Fu W., Ding M., et al. Neutralization of SARS-CoV-2 spike pseudotyped virus by recombinant ACE2-Ig. Nat Commun. 2020;11:2070. doi: 10.1038/s41467-020-16048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Koehn F.E. Drug discovery from natural products. Nat Rev Drug Discov. 2009;8:678. doi: 10.1038/nrd2950. [DOI] [PubMed] [Google Scholar]
- 113.Harvey A.L., Edrada-Ebel R., Quinn R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat Rev Drug Discov. 2015;14:111–129. doi: 10.1038/nrd4510. [DOI] [PubMed] [Google Scholar]
- 114.Koehn F.E., Carter G.T. The evolving role of natural products in drug discovery. Nat Rev Drug Discov. 2005;4:206–220. doi: 10.1038/nrd1657. [DOI] [PubMed] [Google Scholar]
- 115.Newman D.J., Cragg G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J Nat Prod. 2020;83:770–803. doi: 10.1021/acs.jnatprod.9b01285. [DOI] [PubMed] [Google Scholar]
- 116.Su H., Yao S., Zhao W., Li M., Liu J., Shang W., et al. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol Sin. 2020;41:1167–1177. doi: 10.1038/s41401-020-0483-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Clementi N., Scagnolari C., D'Amore A., Palombi F., Criscuolo E., Frasca F., et al. Naringenin is a powerful inhibitor of SARS-CoV-2 infection in vitro. Pharmacol Res. 2021;163:105255. doi: 10.1016/j.phrs.2020.105255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Andreu J., Timasheff S. Tubulin bound to colchicine forms polymers different from microtubules. Proc Natl Acad Sci U S A. 1982;79:6753–6756. doi: 10.1073/pnas.79.22.6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Benhamou O., Geva S., Jacobs M., Drew J., Waldman M., Dekel K. The use of colchicine in respiratory diseases or current respiratory medicine. Curr Respir Med Rev. 2013;9:300–304. [Google Scholar]
- 120.Vitiello A., Ferrara F. Colchicine and SARS-CoV-2: management of the hyperinflammatory state. Respir Med. 2021;178:106322. doi: 10.1016/j.rmed.2021.106322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Al-Kuraishy H., Al-Gareeb A., Qusty N., Cruz-Martins N., El-Saber Batiha G. Sequential doxycycline and colchicine combination therapy in COVID-19: the salutary effects. Pulm Pharmacol Therapeut. 2021;67:102008. doi: 10.1016/j.pupt.2021.102008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang A., Sun Y., Liu Q., Wu H., Liu J., He J., et al. Low dose of emetine as potential anti-SARS-CoV-2 virus therapy: preclinical in vitro inhibition and in vivo pharmacokinetic evidences. Mol Biomed. 2020;1:14. doi: 10.1186/s43556-020-00018-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jin Y.H., Min J.S., Jeon S., Lee J., Kim S., Park T., et al. Lycorine, a non-nucleoside RNA dependent RNA polymerase inhibitor, as potential treatment for emerging coronavirus infections. Phytomed. 2021;86:153440. doi: 10.1016/j.phymed.2020.153440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Van de Sand L., Bormann M., Alt M., Schipper L., Heilingloh C., Steinmann E., et al. Glycyrrhizin effectively inhibits SARS-CoV-2 replication by inhibiting the viral main protease. Viruses. 2021;13:609. doi: 10.3390/v13040609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Su H., Yao S., Zhao W., Zhang Y., Liu J., Shao Q., et al. Identification of pyrogallol as a warhead in design of covalent inhibitors for the SARS-CoV-2 3CL protease. Nat Commun. 2021;12:3623. doi: 10.1038/s41467-021-23751-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Xiang R., Yu Z., Wang Y., Wang L., Huo S., Li Y., et al. Recent advances in developing small-molecule inhibitors against SARS-CoV-2. Acta Pharm Sin B. 2021 doi: 10.1016/j.apsb.2021.06.016. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cao C., Yang J., Chen Y., Zhou P., Wang Y., Du W., et al. Discovery of SK-575 as a highly potent and efficacious proteolysis-targeting chimera degrader of PARP1 for treating cancers. J Med Chem. 2020;63:11012–11033. doi: 10.1021/acs.jmedchem.0c00821. [DOI] [PubMed] [Google Scholar]
- 128.Haniff H.S., Tong Y., Liu X., Chen J.L., Suresh B.M., Andrews R.J., et al. Targeting the SARS-CoV-2 RNA genome with small molecule binders and ribonuclease targeting chimera (RIBOTAC) degraders. ACS Cent Sci. 2020;6:1713–1721. doi: 10.1021/acscentsci.0c00984. [DOI] [PMC free article] [PubMed] [Google Scholar]