

Abstract
The enormous diversity of antibody specificities is generated by random rearrangement of immunoglobulin gene segments and is important for general protection against pathogens. Since random rearrangement harbors the risk of producing self‐destructive antibodies, it is assumed that autoreactive antibody specificities are removed during early B‐cell development leading to a peripheral compartment devoid of autoreactivity. Here, we immunized wild‐type mice with insulin as a common self‐antigen and monitored diabetes symptoms as a measure for autoimmune disease. Our results show that autoreactive anti‐insulin IgM and IgG antibodies associated with autoimmune diabetes can readily be generated in wild‐type animals. Surprisingly, recall immunizations induced increased titers of high‐affinity insulin‐specific IgM, which prevented autoimmune diabetes. We refer to this phenomenon as adaptive tolerance, in which high‐affinity memory IgM prevents autoimmune destruction by competing with self‐destructive antibodies. Together, this study suggests that B‐cell tolerance is not defined by the absolute elimination of autoreactive specificities, as harmful autoantibody responses can be generated in wild‐type animals. In contrast, inducible generation of autoantigen‐specific affinity‐matured IgM acts as a protective mechanism preventing self‐destruction.
Keywords: B‐cell selection, central tolerance, diabetes, autoimmune diseases
Subject Categories: Immunology, Molecular Biology of Disease
Booster immunizations with a typical self‐antigen triggers a phenomenon called adaptive tolerance, in which high‐affinity memory IgM compete with self‐destructive antibodies thereby protecting autoantigens from degradation.

Introduction
Self‐tolerance is crucial for maintaining physiological integrity by avoiding autoimmune reactions. Currently, central and peripheral tolerance mechanisms are believed to control the B‐cell antigen receptor (BCR) repertoire during B‐cell development thereby preventing the generation or survival of self‐reactive B cells (Goodnow et al, 1988; Nemazee & Buerki, 1989; Benschop et al, 2001).
It is assumed that autoreactive B cells are subjected to receptor editing by secondary immunoglobulin (Ig) gene rearrangement that removes the autoreactive specificity and enables the expression of non‐autoreactive BCR (Gay et al, 1993; Tiegs et al, 1993; Zhang et al, 2003). B cells that fail to remove their autoreactivity are subjected to clonal deletion by central tolerance mechanisms during early B‐cell development in the bone marrow (Nossal & Pike, 1978; Nemazee & Buerki, 1989; Hartley et al, 1991; Wardemann et al, 2003). Self‐reactive B cells that circumvent central tolerance and migrate to the periphery are thought to undergo clonal anergy (peripheral tolerance) which is characterized by downmodulation of IgM‐BCR expression and supposed functional unresponsiveness of the respective cells (Nossal & Pike, 1980; Goodnow et al, 1988; Brink et al, 1992; Cyster et al, 1994; O’Neill et al, 2011).
However, the high expression level of IgD‐BCR on the supposedly anergic cells and the differences in responsiveness between IgM and IgD‐BCR suggest that IgD marks maturation and selective B‐cell responsiveness rather than anergy (Übelhart et al, 2015; Setz et al, 2019). In addition, the finding that the vast majority of normal serum IgM is autoreactive is not in agreement with the concept of general elimination of autoreactive B cells by strict tolerance mechanisms (Lobo, 2016). In fact, natural serum IgM plays important roles in homeostasis but is polyreactive and thus possesses a self‐destructive potential (Grönwall et al, 2012). Conventionally, transgenic mouse models expressing a defined autoreactive BCR specificity were used in the presence or absence of cognate antigen to investigate B‐cell tolerance and the prevention of self‐destructive antibody responses (Goodnow et al, 1988, 1989; Nemazee & Buerki, 1989; Nemazee & Bürki, 1989; Hartley et al, 1991). However, replacement of the germline configuration of the Ig genes by expression cassettes for high‐affinity mutated autoreactive BCR creates an abnormal situation for early B‐cell development and results in an unphysiologically monospecific repertoire (Goodnow et al, 1988; Nemazee & Bürki, 1989; Hartley et al, 1991). Moreover, the studied model autoantigens have no relevance to known autoimmune diseases (Cooper & Stroehla, 2003; Suurmond & Diamond, 2015). Therefore, there is urgent need for the development of physiologically relevant experimental models to study the mechanisms of immune tolerance and autoimmune diseases.
Epidemiological studies show that up to 5% of the population in industrialized countries suffers from autoimmune diseases such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), or diabetes type 1 (T1D) (Cooper & Stroehla, 2003). Notably, autoantibodies are present in the vast majority of autoimmune diseases and often are the driving force for pathogenesis (Suurmond & Diamond, 2015).
We recently described that mature B cells are responsive to polyvalent antigen, while monovalent antigen modulates this responsiveness (Übelhart et al, 2015; Setz et al, 2019) suggesting alternative mechanisms for the control of self‐reactive B cells and how autoantibody responses are resolved. To investigate these mechanisms, we selected a disease‐related autoantigen (insulin) and established a novel experimental system in wild‐type mice. The results uncover an unexpected mechanism of adaptive tolerance in which autoantigen‐specific high‐affinity protective memory IgM (PR‐IgM) plays a pivotal role counteracting self‐destructive antibody responses.
Results
Polyvalent autoantigen induces autoreactive IgG
To avoid the usage of transgenic mice that artificially harbor monospecific antigen‐reactive B cells, we used wild‐type mice with a physiological BCR repertoire. Here, we selected native insulin and insulin‐related autoantigens as a physiologically relevant system for autoimmune diseases. During biosynthesis in the pancreas, proinsulin is cleaved into the well‐studied hormone insulin and the so‐called C‐peptide (CP), which are both secreted into the bloodstream (Fig 1A). In contrast to insulin, CP is less conserved between mouse and man (Fig 1B). In addition, insulin is found in nanomolar concentrations in the blood and plays pivotal roles in regulating blood glucose levels and diabetes. In contrast, CP is barely detectable in the blood (picomolar concentrations) and has no clearly reported physiological function (Zhu et al, 2016). Using full‐length CP or native insulin, we aimed at investigating autoreactive antibody responses toward an abundant, functionally important, and highly regulated autoantigen (i.e., insulin) compared to a barely detectable autoantigen lacking obvious physiological function (CP).
Figure 1. Polyvalent native self‐molecules induce robust autoreactive IgG responses in vivo .
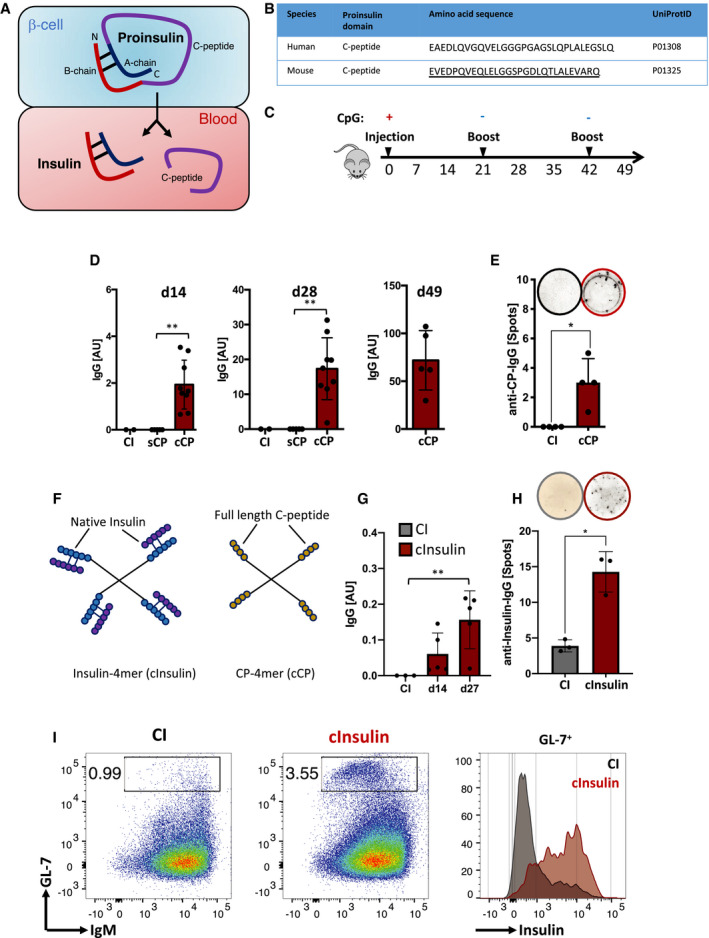
-
ASchematic illustration of proinsulin‐derived full‐length C‐peptide and insulin.
-
BTable comparing human to murine C‐peptide amino acid sequences. Underlined: sequence used as peptide for immunization.
-
CSchematic immunization schedule.
-
D–H(D) Serum anti‐C‐peptide‐immunoglobulins (coating: C‐peptide) titers of mice immunized with cCP (n = 9 for d14 and d28; n = 5 for d49) and (G) anti‐insulin‐immunoglobulins (coating: insulin) titers of mice immunized with complex native insulin (cInsulin, n = 5) and control‐immunized (CI: CpG only, n = 3) measured by ELISA at indicated days. Dots represent individual mice. Mean ± SD, statistical significance was calculated by using Mann–Whitney U‐test. (E, H) ELISpot assays showing CP‐specific IgG (n = 4 mice/group) (E) and insulin‐specific IgG (n = 3 mice/group) (H) producing splenocytes at day 14. Top lane showing representative images of wells. Mean ± SD, statistical significance was calculated by using Mann–Whitney U‐test, *P < 0.05, **P < 0.01. (F) Schematic illustration of native insulin (cInsulin) and full‐length C‐peptide (cCP) tetrameric polyvalent complexes.
-
IFlow cytometric analysis of splenocytes of mice immunized with cInsulin or control immunization on day 26. Left and middle panel showing germinal center (GC) B cells (GL‐7+) pre‐gated on B cells (CD19+ B220+). Right panel showing insulin‐reactive GC B cells pre‐gated on GL‐7+ cells in a histogram. Data are representative of two independent experiments with n = 4/group.
To this end, we used biotinylated full‐length CP to generate polyvalent antigen complexes (cCP) by incubation with streptavidin (SAV). The non‐complexed, soluble form of the CP (sCP) was used as monovalent antigen. We injected wild‐type mice with sCP, cCP to test their potential to induce autoreactive antibody responses (Fig 1C). Similar to control immunization (CI), sCP induced no detectable IgG immune responses, whereas the polyvalent form cCP induced IgG as measured at d14 and d28, respectively (Fig 1D). In addition to ELISA experiments, a Western blot analysis was performed to determine the specificity of the generated antibody responses. The serum of mice immunized with cCP contained IgG antibodies recognizing pancreatic Proinsulin (Appendix Fig S1). To confirm the memory response against CP, we performed another recall immunization at d42 using cCP again without adjuvant and detected at d49 a robust IgG response against CP (Fig 1D). To confirm these findings, we performed ELISpot analysis to determine the number of anti‐CP‐IgG secreting cells. In agreement with the serum Ig results, the ELISpot experiments showed that mice immunized with cCP possess increased numbers of IgG secreting cells (Fig 1E).
Together, these data show that antibody responses can be directed against an autoantigen, that is, CP, suggesting that the respective autoreactive B cells were neither clonally deleted by central tolerance nor functionally silenced by anergy.
Native insulin induces autoreactive IgG responses
To test our hypothesis that autoreactive anti‐insulin B cells are naturally present in the periphery and are not deleted by central tolerance nor turned unresponsive by anergy, we generated polyvalent insulin complexes (cInsulin) by incubating biotinylated native insulin with streptavidin (Fig 1F). Importantly, the biotinylated insulin used for the reported experiments is biologically active when injected in monovalent form into mice as determined in our own previous experiments.
Wild‐type mice were injected with 10 µg of cInsulin and monitored over time for the presence of anti‐insulin antibodies in serum. In parallel, we tested whether the immunized mice developed a diabetes‐like dysregulation of glucose metabolism by monitoring glucose levels in blood and urine as well as signs of acute pancreatitis. We detected considerable amounts of anti‐insulin IgG at d14 and after booster immunization at d27 (Fig 1G). In order to exclude any drastic effects of CpG that we used in initial injections, we performed immunization experiments by injecting InsA‐KLH on day 0 without CpG. The mice immunized without CpG showed a robust anti‐insulin IgM response (Appendix Fig S2A and B). To confirm the ELISA results, we performed ELISpot analysis and detected increased numbers of anti‐insulin IgG secreting B cells in the spleen of cInsulin‐immunized mice (Fig 1H). Furthermore, we analyzed spleens of mice immunized with cInsulin on day 5 after boost (d26). We observed a drastic increase in the germinal center (GC) B‐cell population (GL‐7+) in cInsulin‐immunized mice as compared to the control mice (Fig 1I). Interestingly, GC B cells of cInsulin‐immunized mice showed clear binding to native insulin (Fig 1I). In contrast to polyvalent streptavidin, immunization with insulin coupled to a monovalent streptavidin failed to induce antibody responses suggesting that the foreign carrier is not required to induce autoantibody responses and that the antigen valency is essential for eliciting the autoimmune response (Appendix Fig S3A and B).
Mice immunized with cInsulin showed substantial signs of diabetes as measured by increased concentrations of blood glucose, starting at d7 and continuing through d14 to d26 after booster immunization (see below).
Together, these data show that regardless of concentration, antibody responses can easily be directed against autoantigens suggesting that the respective autoreactive B cells are present in the periphery and capable of generating autoimmune responses.
B cells are required for autoimmune diabetes induced by polyvalent insulin
To test whether antibody production and thus B cells and autoantibodies induce the elevated blood glucose levels, we injected 10 µg cInsulin into B‐cell‐deficient, mb1‐knockout mice (Hobeika et al, 2006) lacking the BCR component Igα, also known as CD79A (Fig 2A). In contrast to wild‐type animals, no increase in blood glucose levels was observed in the B‐cell‐deficient mice suggesting that the presence of B cells and autoantibody secretion are crucial for the development of diabetes symptoms observed in wild‐type mice (Fig 2B). Moreover, the increase in blood glucose was accompanied by detectable glucose in the urine of wild‐type mice injected with cInsulin (Fig 2C). In agreement with diabetes development, water consumption of wild‐type mice injected with cInsulin dramatically increased (Fig 2D). Due to the severity of diabetes symptoms, we sacrificed the mice at day 26 and analyzed the pancreas and spleen.
Figure 2. Polyvalent native insulin induces autoreactive IgG responses and autoimmune diabetes in wild‐type mice.
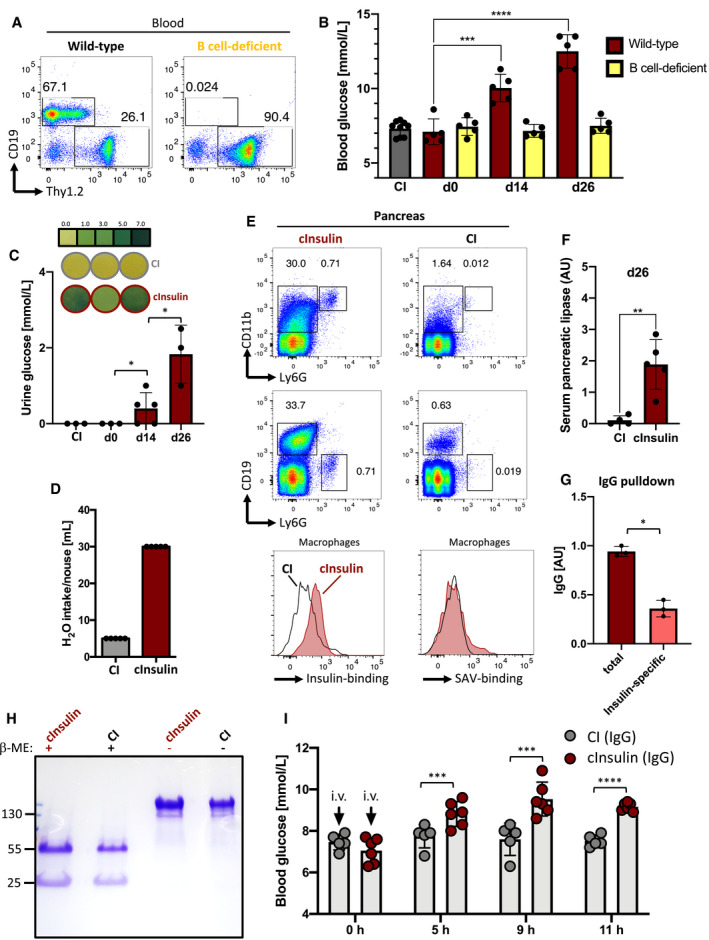
- Flow cytometric analysis of peripheral blood showing B cells (CD19+ Thy1.2‐) and T cells (Thy1.2+ CD19‐) of wild‐type (left) and B‐cell‐deficient (right) mice. Cells were pre‐gated on lymphocytes. Representative of three independent experiments (n = 5/group).
- Blood glucose levels of cInsulin‐immunized (red: WT, n = 5; yellow: B‐cell‐deficient, n = 5) and CI mice (gray, n = 8) were assessed at indicated days post‐immunization. Dots represent individual mice, mean ± SD. Statistical significance was calculated by using repeated measure ANOVA test, ***P < 0.001, ****P < 0.0001.
- Urine glucose levels of cInsulin‐immunized (red, n = 3 for d0, d26; n = 5 for d14) and CI mice (gray, n = 3) were monitored at indicated days post‐immunization. Upper panel showing visualization of glucose standard (top lane) and representative images of tested animals (middle and bottom lanes), respectively. Dots represent individual mice, mean ± SD. Statistical significance was calculated by using repeated measure ANOVA test, *P < 0.05.
- Water intake of cInsulin‐immunized (n = 5) and CI mice (gray, n = 5) monitored from d21 to d26, mean ± SD.
- Flow cytometric analysis of the pancreas of cInsulin‐immunized (red) and CI mice (gray) at day 26. Upper panel showing pancreatic macrophages (CD11b+ Ly6G‐), neutrophils (Ly6G+ CD11b+), and B cells (CD19+) pre‐gated on living cells (FVD‐). Lower panel showing histograms for insulin binding (left) and streptavidin (SAV)‐binding (right). Representative of two independent experiments with n = 5/group. F: Serum pancreatic lipase titers of mice immunized with cInsulin (n = 5) or control immunization (n = 4) measured by ELISA. Dots represent individual mice. Mean ± SD, statistical significance was calculated by using Mann–Whitney U‐test, **P < 0.01.
- Serum pancreatic lipase titers of mice immunized with cInsulin (n = 5) or control immunization (n = 4) measured by ELISA. Dots represent individual mice. Mean ± SD, statistical significance was calculated by using Mann–Whitney U‐test, **P < 0.01.
- Quantification of total (red) and insulin‐specific (salmon) IgG after serum IgG purification of cInsulin‐immunized mice via ELISA with n = 3/group (coating: anti‐IgG). Mean ± SD, statistical significance was calculated by using Mann–Whitney U‐test, *P < 0.05.
- Coomassie‐stained SDS–PAGE showing purified serum IgG of cInsulin‐immunized (red) and CI mice (gray) under reducing (+β‐ME), and non‐reducing conditions. Representative of two independent experiments.
- Blood glucose levels of intravenously (i.v.) injected wild‐type mice at indicated hours post‐injection. 20 µg of purified serum IgG from cInsulin‐immunized mice (red, n = 6) or CI mice (gray, n = 5) was used for injection. Dots represent individual mice. Mean ± SD, statistical significance was calculated by using repeated measure ANOVA test, ***P < 0.001, ****P < 0.0001.
Source data are available online for this figure.
In contrast to control immunization, cInsulin‐immunized mice showed highly increased recruitment of macrophages (CD11b+), neutrophils (Ly6G+), and B cells (CD19+) to the pancreas (Fig 2E). Interestingly, increased recruitment of neutrophils has previously been shown to be associated with severe pancreatitis (Yang et al, 2015). Moreover, IgG+ macrophages of mice immunized with cInsulin showed augmented binding to native insulin (Fig 2E) which supports autoantibody‐mediated acute inflammatory processes at the pancreas (Yang et al, 2015). In addition, characterization of inflammatory cytokines by bead arrays using pancreas supernatants showed elevated levels of TNF‐α and IL‐12 in mice immunized with cInsulin supporting the notion of acute pancreatitis (Appendix Fig S4A and B). Consequently, the observed acute pancreatitis triggered organ damage indicated by highly elevated serum pancreatic lipase levels (Smith et al, 2005; Gomez et al, 2012) (Fig 2F). FACS analysis of the spleen showed no difference of B cells between control immunization mice and those immunized with cInsulin (Appendix Fig S5).
To show that the secreted insulin‐specific IgG was responsible for the diabetes symptoms, we performed IgG pulldown experiments using serum from mice immunized with cInsulin or control immunization (Fig 2G and H). Since the IgG purification may result in dissociation of endogenous insulin from insulin‐specific serum IgG, we determined the anti‐insulin binding capacity. Here, we compared insulin binding of total IgG before and after purification by pulldown. We found that a considerable amount of the IgG isolated from cInsulin mice was reactive to insulin, which may suggest that direct serum IgG measurements may not detect the entire insulin‐specific IgG due to masking of the antibody binding sites by endogenous insulin (Fig 2G). To test the pathogenicity of purified anti‐insulin IgG, we injected equal amounts of IgG from control immunization or mice immunized with cInsulin intravenously into wild‐type animals and monitored blood glucose. We found that injecting total IgG containing 2 µg anti‐insulin IgG was sufficient to induce hyperglycemia in recipient mice suggesting that anti‐insulin‐IgG neutralizes insulin thereby causing diabetes symptoms (Fig 2I).
These data demonstrate that autoreactive B cells recognizing a metabolism‐regulating hormone are neither deleted nor functionally silenced and can induce severe autoimmunity when the autoantigen valency is shifted toward polyvalency.
Presence of monovalent autoantigen suppresses IgG antibody responses
It is conceivable that the immune response toward polyvalent CP or polyvalent cInsulin used in this study was affected by the presence of endogenous monovalent counterparts. Therefore, we tested the effect of combining the monovalent and polyvalent antigen forms in immunization experiments.
To this end, we injected wild‐type mice with sCP alone, cCP alone, or combinations of both and monitored the generation of autoreactive antibody responses over time. While the monovalent sCP alone induced no detectable IgM or IgG responses, it modulated the immune responses elicited by the polyvalent CP (cCP). In fact, the IgG response at d14 was significantly reduced in mice immunized with sCP:cCP ratio of 20:1 as compared to mice immunized with only cCP (0:1) (Fig 3A). In contrast to IgG, CP‐specific IgM was slightly increased in mice immunized with sCP:cCP combination at 20:1 ratio (Fig 3A). Importantly, no difference was detected in the IgG response against streptavidin (SAV) which was used to generate polyvalent CP complexes (Appendix Fig S6).
Figure 3. Ratios of monovalent to polyvalent autoantigen control the isotype composition of secreted antibodies.
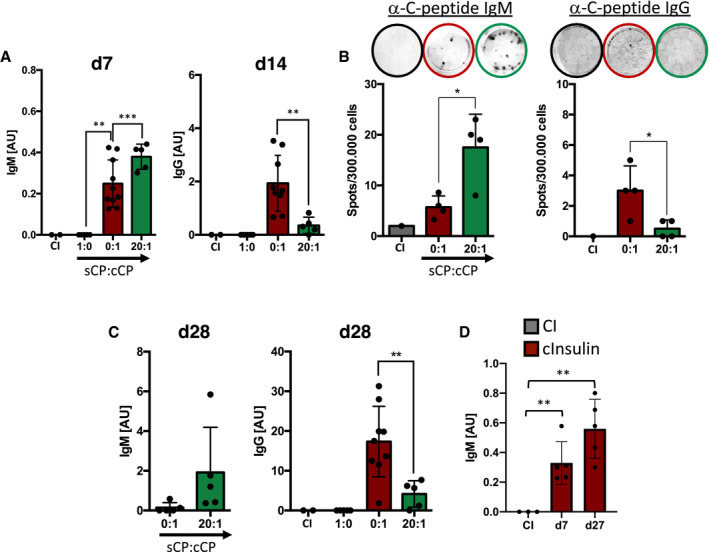
-
A–D(A, C) Serum anti‐C‐peptide‐immunoglobulins titers of mice immunized with CP (0:1 n = 10; 20:1 n = 5) (coating: C‐peptide) and (D) anti‐insulin‐IgM titers of mice immunized with complex insulin (cInsulin, n = 5) (coating: insulin) and control immunization (CI: CpG only, n = 3) measured by ELISA at indicated days. Dots represent individual mice. Mean ± SD, statistical significance was calculated by using Kruskal–Wallis test. (B) ELISpot assay showing CP‐specific immunoglobulin producing splenocytes at day 7 (left panel) and day 14 (right panel). Top lane showing representative images of wells. Dots represent individual mice (n = 4/group) Mean ± SD, statistical significance was calculated by using Kruskal–Wallis test, *P < 0.05, **P < 0.01, ***P < 0.001.
These data suggest that soluble monovalent antigen modulates the immune response and determines the IgG:IgM ratio of antibody‐secreting cells during immune responses. To confirm this conclusion, we performed ELISpot analysis to determine the number of IgG or IgM secreting cells in the spleens of the different groups of mice. In agreement with the serum Ig results, the ELISpot experiments showed that mice immunized with a ratio of 20:1 of sCP:cCP possess increased numbers of IgM secreting cells while the numbers of IgG secreting cells were decreased as compared to mice immunized with cCP (stated as sCP:cCP = 0:1) (Fig 3B).
Later challenge with the same antigen at d21 resulted in further increase of CP‐specific IgM at d28 in mice immunized with sCP:cCP ratio of 20:1 while the IgG response was reduced in these mice as compared with mice immunized with only with cCP, sCP:cCP ratio of 0:1 (Fig 3C).
To further support the notion that the presence of endogenous monovalent antigen augments antigen‐specific IgM antibody responses, we measured the IgM responses against polyvalent cInsulin and observed an increased anti‐insulin IgM response at d7 and d27 (Fig 3D).
In summary, the results show that B‐cell responses are induced by polyvalent antigen and modulated by monovalent counterparts thereby regulating B‐cell responsiveness and the isotype composition of generated antibody. This leads to a dynamic and pivotal B‐cell function that differs from the conventional view.
Insulin‐derived epitope induces harmful IgG response
Since monovalent native insulin has a drastic effect on blood glucose levels, we were not able to test combinations of polyvalent and monovalent native insulin in immunization experiments. However, to further confirm the presented findings we performed immunization experiments using an insulin‐A‐chain‐derived peptide, referred to as InsA, which is a frequently reported epitope targeted by human autoantibody responses (Padoa et al, 2005). We coupled the selected peptide to the protein‐carrier KLH to generate a polyvalent antigen complex (cInsA) which was used in further experiments either alone or in combination with the monovalent peptide (sInsA). We found that InsA induced IgM and IgG autoantibody responses binding native insulin (Fig 4A). However, the polyvalent cInsA alone (sInsA:cInsA ratio of 0:1) readily induced the production of anti‐insulin‐IgG, whereas addition of soluble peptide (sInsA:cInsA ratio of 100:1) resulted in significant reduction of autoreactive IgG at d14 and, one week after boost, at d28 (Fig 4A). Most likely, the amount of autoreactive anti‐insulin IgG is higher than detected in direct serum ELISA as anti‐insulin‐IgG bound to endogenous insulin escapes detection as described above.
Figure 4. Soluble insulin‐A peptides modulate autoimmune responses.
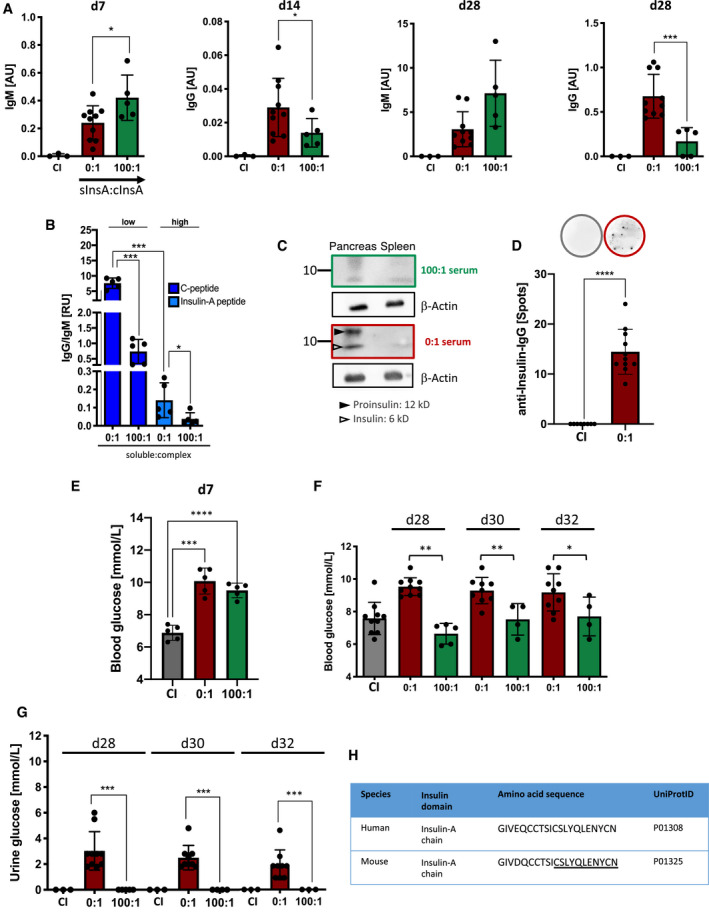
-
ASerum anti‐insulin‐immunoglobulin titers of mice immunized with InsA peptide (red: 0:1, n = 10 and green: 100:1, n = 5) and control immunization (CI: CpG only, gray, n = 3) measured by ELISA at indicated days (coating: insulin). Dots represent individual mice. Mean ± SD, statistical significance was calculated by using Kruskal–Wallis test, *P < 0.05, ***P < 0.001.
-
BRatios of IgG to IgM derived from ELISA values plotted against molar ratios of antigens for n = 5/group. Mean ± SD, statistical significance was calculated by using Kruskal–Wallis test, *P < 0.05, ***P < 0.001.
-
CWestern blot analysis of insulin‐specific serum IgG derived from InsA peptide‐immunized mice. Top panel (green): 100:1 serum, lower panel (red): 0:1 serum (sInsA:cInsA). Serum of mice was used as primary antibody and detection was done with anti‐mouse‐IgG‐HRP. Black filled arrow: Proinsulin (12 kD), Black non‐filled arrow: insulin (6 kD), β‐actin (42 kD, loading control). Representative of two independent experiments.
-
DELISpot of InsA peptide‐immunized (red, n = 11) and CI mice (gray, n = 8) showing insulin‐specific IgG‐producing splenocytes on day 14. Representative wells are shown (top lane). Mean ± SD, statistical significance was calculated by using Mann–Whitney U‐test, ****P < 0.0001.
-
E, FBlood glucose levels of InsA peptide‐immunized (red: 0:1, n = 5 for E and n = 10 for F; green: 100:1, n = 5 and n = 4 for F, d30, d32) and CI (gray, n = 5 for E and n = 10 for F) mice were assessed at indicated days. Dots represent individual mice. Mean ± SD, statistical significance was calculated by using Kruskal–Wallis test (E) and repeated measure ANOVA test (F), *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
-
GUrine glucose levels of InsA‐peptide‐immunized (red: n = 10 for d28, d32 and n = 8 for d30; green: n = 5 for d28, n = 4 for d30, n = 3 for d32) and CI (gray, n = 3) mice were monitored at indicated days post‐immunization. Dots represent individual mice. For 100:1: representative data are shown for two independent experiments with total n = 10. Mean ± SD, statistical significance was calculated by using repeated measure ANOVA test, ***P < 0.001.
-
HTable comparing human to murine insulin‐A‐chain amino acid sequences. Underlined: sequence used as peptide.
Source data are available online for this figure.
Notably, comparable IgM and IgG responses were observed against the carrier protein KLH underlining the autoantigen‐specific effect of the monovalent InsA antigen on the suppression of IgG responses induced by cInsA in the KLH‐InsA complexes (Appendix Fig S7A and B).
Moreover, the presence of soluble InsA resulted in robust insulin‐specific IgM production at d7 and d28, which was slightly higher than in mice immunized with polyvalent cInsA alone (sInsA:cInsA ratio of 0:1) (Fig 4A). Since insulin is present in high amounts in the circulatory system, we conclude that the presence of endogenous monovalent insulin modulates the immune response elicited by cInsA, thereby leading to increased autoreactive IgM responses as compared to IgG. Taken together, our data indicate that the ratio of polyvalent to monovalent antigen is mirrored by the ratio of antigen‐specific IgG to IgM (γ/μ ratio) during memory responses (Fig 4B).
Serum IgG of mice immunized with polyvalent peptide only (sInsA:cInsA ratio of 0:1) readily detected native insulin in Western blot analysis (Fig 4C), while serum IgG of mice immunized in the presence of soluble peptide (sInsA:cInsA ratio of 100:1) was insufficient to clearly detect insulin (Fig 4C). In addition, ELISpot analysis using splenic B cells from mice immunized with cInsA confirmed the increased presence of anti‐insulin IgG secreting cells in respective mice (Fig 4D).
To confirm that the increased anti‐insulin IgG is associated with harmful autoimmune responses, we tested whether mice immunized with cInsA show signs of diabetes. One week after immunization at d7, we found that both groups of mice immunized with cInsA only (sInsA:cInsA ratio of 0:1), or combination of monovalent and polyvalent cInsA (sInsA:cInsA ratio of 100:1) show increased blood glucose as compared to control immunization (Fig 4E).
One week after booster immunization, however, increased blood glucose was observed only in mice immunized with polyvalent InsA (0:1 ratio of sInsA:cInsA) (Fig 4F). In addition, we tested whether the glucose concentration was also increased in the urine. Indeed, the increased autoreactive anti‐insulin IgG led to detectable glucose in the urine (Fig 4G). In contrast to IgG, no detectable signs of autoimmune diabetes were observed in mice immunized with 100:1 ratio and possessing increased amounts of autoreactive anti‐insulin IgM after booster immunization (Fig 4F, for IgM amount compare Fig 4A).
FACS analysis of splenic B cells at d28 after immunization suggests that InsA immunization resulted in an increase of B cells with elevated amount of IgM (Appendix Fig S8A and B). Moreover, mice immunized with cInsA only (sInsA:cInsA = 0:1) showed increased number of macrophages in the pancreas which bound autoreactive IgG as determined by the increased InsA peptide binding (Appendix Fig S8C). Similar results were observed in the spleen (Appendix Fig S9).
Interestingly, InsA is highly conserved between mouse and man (Fig 4H) and anti‐InsA antibodies bind native insulin and are often involved in human autoimmune diabetes (Padoa et al, 2005). Therefore, this model might be of great use for investigating the molecular mechanisms underlying autoantibody‐driven diabetes development and pancreatitis.
Together, our data suggest that increased ratio of complex polyvalent autoantigen leads to increased amount of autoreactive IgG and subsequent self‐destructive autoimmune responses in wild‐type animals. In contrast, an increase in monovalent antigen results in increased autoreactive memory IgM which seems to prevent self‐destructive immune responses.
Adaptive immune tolerance by autoreactive memory IgM
The data presented above suggest that high titers of memory anti‐insulin‐IgM during secondary immune responses protect from dysregulation of glucose metabolism and autoimmune diabetes in mice immunized with InsA.
To test whether increased ratios of secondary autoreactive anti‐insulin‐IgM counter the pathological effects on glucose metabolism induced by autoreactive anti‐insulin‐IgG, we challenged the mice initially immunized in the presence of monovalent InsA peptide (sInsA:cInsA ratio 100:1) with polyvalent antigen cInsA at d51 (Fig 5A). This treatment usually induces autoimmune diabetes from d7 to 28 (Appendix Fig S10A–C, d7 vs. d14). However, mice pre‐immunized in the presence of monovalent InsA peptide (sInsA:cInsA ratio 100:1) showed no dysregulation of glucose metabolism at d51 to 59 (Fig 5B and C). These mice produced autoreactive anti‐insulin‐IgM but no anti‐insulin IgG (Fig 5D and E). Our data suggest that primary immunization in the presence of monovalent InsA peptide (sInsA:cInsA ratio 100:1) induced adaptive tolerance against the pathogenic immunization with polyvalent cInsA. Moreover, these findings indicate that this unique tolerance mechanism creates a novel class of memory responses by eliciting and maintaining the production of protective regulatory IgM (PR‐IgM).
Figure 5. Monovalent insulin‐A peptides induce immune tolerance via autoreactive memory IgM.
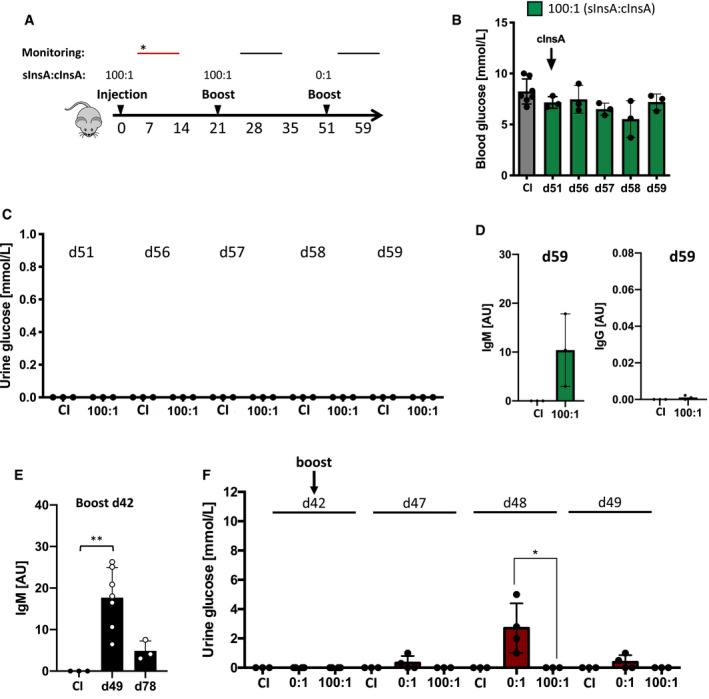
-
AImmunization scheme. *monitoring: diabetes symptoms observed (red bar).
-
BBlood glucose levels of InsA‐peptide‐immunized mice (n = 3) and CI (n = 7) mice were assessed at indicated days post‐immunization. Dots represent individual mice. Mean ± SD, statistical significance was calculated by using repeated measure ANOVA test, all comparisons were not significant.
-
C–F(C, F) Urine glucose levels of InsA‐peptide‐immunized (n = 3) and CI (n = 3) mice were monitored at indicated days post‐immunization. Dots represent individual mice. Mean ± SD, statistical significance was calculated by using repeated measure ANOVA test, *P < 0.05. (D, E) Serum anti‐insulin‐immunoglobulin titers of mice immunized with InsA peptide (day 59, day 78: n = 3, day 49: n = 7) and control immunization (CI: CpG only, n = 3) measured by ELISA at indicated days (coating: insulin). Dots represent individual mice. Mean ± SD, statistical significance was calculated by using Mann–Whitney U‐test (D) and repeated measure ANOVA test (E). *P < 0.05, **P < 0.01.
We tested whether mice immunized with only cInsA (sInsA:cInsA ratio 0:1) also developed adaptive tolerance by PR‐IgM. While mice immunized with 100:1 ratio were protected against diabetes already at the first booster immunization by d21, mice immunized with 0:1 ratio showed diabetes signs until d32 (Fig 4G). When these mice were subjected to a second booster immunization at d42, they showed weaker diabetes signs and reduced duration as compared to the first booster immunization (Fig 5F, compare Fig 4G). In agreement with the role of PR‐IgM in preventing diabetes, we observed elevated amounts of anti‐insulin IgM and no detectable anti‐insulin IgG at d49, one week after booster immunization (Fig 5F). Moreover, the increased levels of PR‐IgM also correlated with a decrease in serum pancreatic lipase levels (Appendix Fig S11).
These data show that PR‐IgM protects from destructive autoimmune responses and that the presence of monovalent autoantigen accelerates the development of PR‐IgM as compared with only complex autoantigen.
Higher antigen affinity for memory anti‐insulin‐IgM
Our data suggest that an increased amount of insulin‐reactive PR‐IgM as compared to insulin‐reactive IgG is key for protection from diabetes after booster immunization with InsA. To determine the autoreactive IgG to IgM ratio required for protection, we plotted the ratios of insulin‐reactive IgG to IgM obtained from ELISA measurements on a two‐dimensional graph against blood glucose levels or urine glucose levels. According to this analysis, the ratio of insulin‐reactive IgG to IgM must be below 0.1 (γ/µ < 0.1) to avoid the development of diabetes symptoms (Fig 6A).
Figure 6. High‐affinity memory IgM counteracts IgG‐mediated self‐destructive autoimmune responses in vivo .
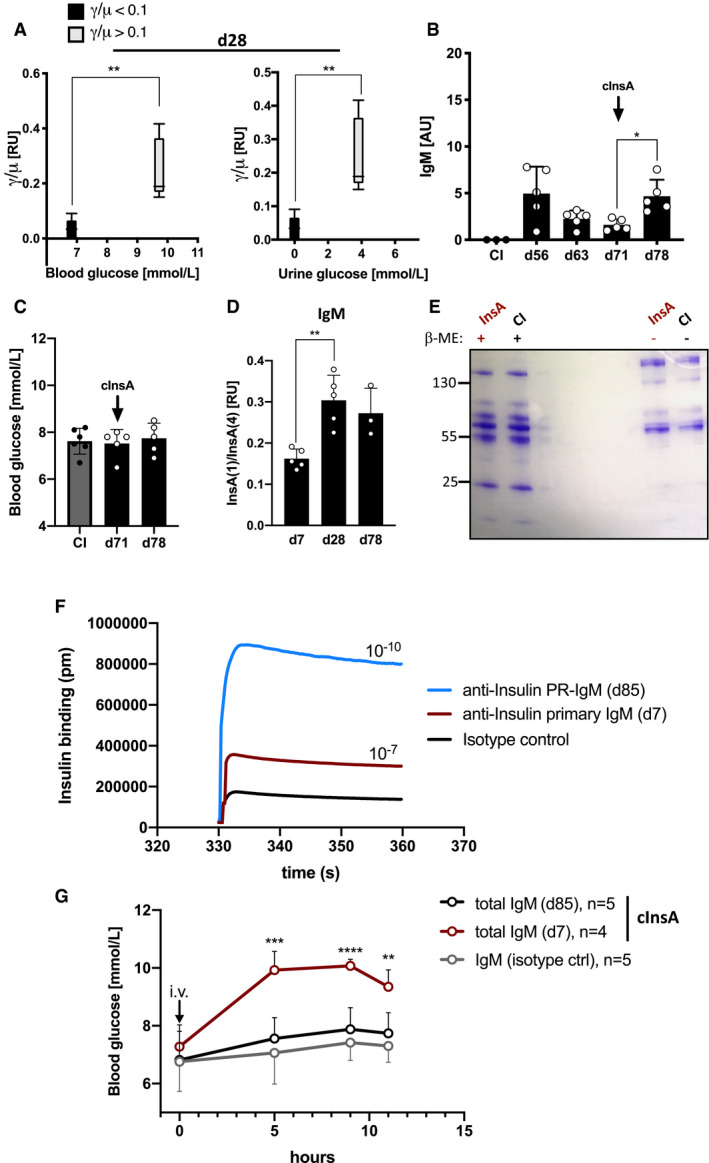
- Ratios of IgG to IgM derived from ELISA values plotted on a two‐dimensional graph against blood glucose levels (left panel) and urine glucose levels (right panel) for n = 5/group. Central bands: median, boxes: interquartile range, whiskers: max./min., statistical significance was calculated by using Mann–Whitney U‐test, **P < 0.01.
- Serum anti‐insulin‐IgM titers of mice immunized with InsA peptide with a γ/µ ratio < 0.1 (black, n = 5) and CI (gray, n = 3) mice measured by ELISA at indicated days (coating: insulin). Dots represent individual mice. Mean ± SD, statistical significance was calculated by using repeated measure ANOVA test, *P < 0.05.
- Blood glucose levels of InsA‐peptide‐immunized mice (γ/µ < 0.1, black, n = 5) and CI (gray, n = 6) mice were assessed at indicated days post‐immunization with a commercial blood glucose monitor device. Dots represent individual mice. Mean ± SD, statistical significance was calculated by using Kruskal–Wallis test, all comparisons were not significant.
- Insulin‐specific IgM affinity maturation of InsA‐peptide‐immunized mice at indicated days was measured by ELISA. Ratios of IgM binding to InsA(1) and InsA(4) (referring to molar antigen density) were calculated and plotted as relative units (n = 5 for d7, d28; n = 3 for d78). Dots represent individual mice. Mean ± SD, statistical significance was calculated by using repeated measure ANOVA test, **P < 0.01. Serum dilutions: 1:25.
- Coomassie‐stained SDS–PAGE showing purified serum IgM of complex InsA‐peptide (cInsA)‐immunized and CI mice under reducing (+β‐ME), and non‐reducing conditions. Representative of two independent experiments.
- Anti‐insulin affinity of PR‐IgM (blue), primary IgM (red), and isotype control (black) measured by bio‐layer interferometry. IgM binding to insulin was acquired in pm and used to calculate the dissociation constant shown in the graph (Kd = 1/Ka). Graph is showing antigen‐antibody association phase. Data are representative for three independent experiments.
- Blood glucose levels of intravenously injected mice with purified IgM from control immunization (CI, n = 5), complex InsA peptide (cInsA) immunization day 7 (total IgM d7, n = 4) and day 85 (total IgM d85, n = 5). Mean ± SD, statistical significance was calculated by using repeated measure ANOVA test showing comparison of red to black line, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Source data are available online for this figure.
Next, we addressed the question whether PR‐IgM responses resemble memory responses by providing long‐term tolerance. To this end, we monitored the decline of anti‐insulin IgM levels over time followed by anti‐insulin recall responses (Fig 6B). We found that anti‐insulin IgM persists for weeks and that booster immunization with cInsA at day 71 induces only IgM, but neither IgG nor signs of dysregulated glucose metabolism in blood or urine (Fig 6B and C, and Appendix Fig S10B and C). IgM memory responses are associated with increase of antibody‐affinity toward antigen which can be monitored by determining the antibody binding capacity to low‐valence antigen as compared to high‐valence counterpart (Shimizu et al, 2004). Therefore, we performed ELISA experiments using monovalent InsA (1) or polyvalent InsA (4) as antigen for coating of ELISA plates. Increased binding to monovalent InsA indicates increased affinity of the insulin‐specific antibodies. Interestingly, IgM generated after booster InsA immunization shows higher anti‐insulin affinity compared to the primary IgM collected on day 7 (Fig 6D). To further examine the role of autoreactive memory PR‐IgM generated during immune responses, we purified total IgM from control immunization or InsA‐immunized mice (Fig 6E). To confirm that PR‐IgM (d85) has an increased affinity to insulin, we performed interferometric assays for affinity characterization (Kumaraswamy & Tobias, 2015). We found that purified PR‐IgM shows a highly increased binding affinity to insulin compared to the primary IgM or isotype control (Fig 6F).
When administered intravenously into naïve wild‐type mice, insulin‐reactive memory PR‐IgM from InsA‐boosted mice, collected at d85, showed no effect on blood glucose levels and behaved similar to control IgM suggesting that PR‐IgM binds insulin without inducing insulin depletion (Fig 6G). However, primary insulin‐reactive IgM, collected at d7, led to increased blood glucose levels by 5 h after intravenous injection (Fig 6G).
Together, autoreactive memory PR‐IgM does not induce dysregulation in blood glucose levels despite its increased anti‐insulin affinity.
Protective memory anti‐insulin‐IgM is monospecific
The results presented above point toward an unexpected fundamental difference between autoreactive primary IgM and PR‐IgM. In fact, primary anti‐insulin‐IgM induced diabetes symptoms although produced at much lower quantity as compared to memory PR‐IgM which possesses a higher insulin affinity but did not induce pathology. To directly test the protective function of autoreactive memory PR‐IgM against destructive autoimmunity, mice were immunized with cInsA alone or cInsA together with intravenous injections of 50 µg total IgM containing 5 µg of anti‐insulin memory PR‐IgM every 48 h starting from d0 (Fig 7A and B). Interestingly, the presence of insulin‐specific PR‐IgM mitigated autoimmune dysglycemia and completely prevented glycosuria on day 7 as compared to mice immunized with cInsA alone (Fig 7B). To exclude that PR‐IgM injections neutralized injected cInsA, we performed anti‐carrier (KLH) ELISA and found no difference in anti‐KLH‐IgM levels between the two groups at day 7 (Appendix Fig S12). Similarly, intravenous injections of anti‐insulin PR‐IgM together with total IgG pulldown from cInsA immunized mice prevented hyperglycemia induced by anti‐insulin IgG in the pulldown (Fig 7C).
Figure 7. Differences in the affinity and specificity of primary versus memory IgM control autoimmune responses.
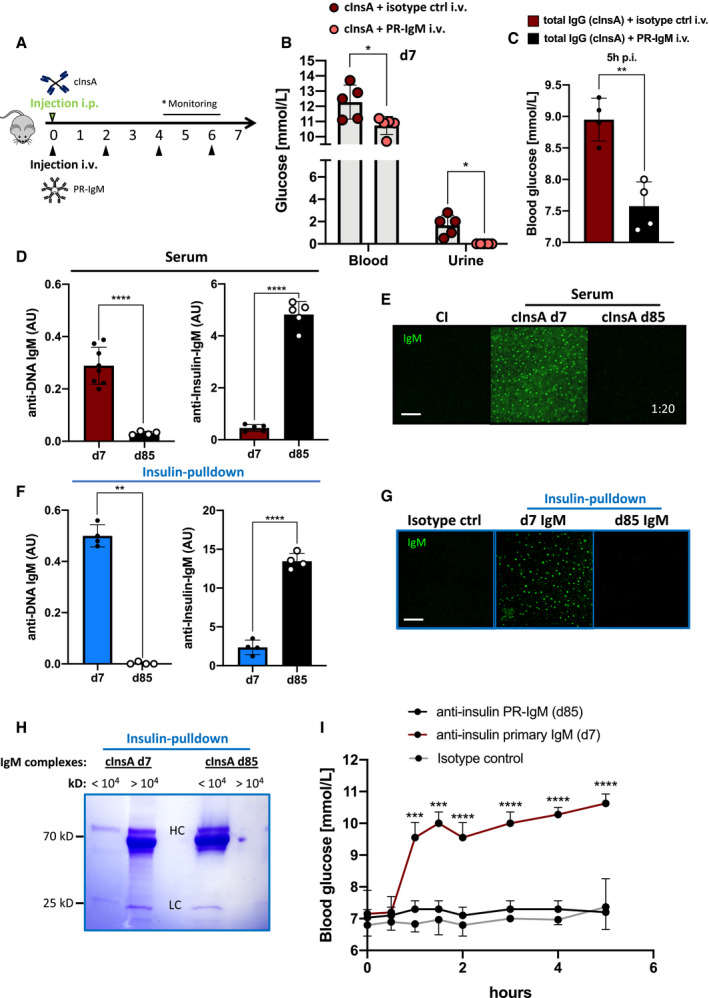
-
ASchematic illustration of immunization schedule with complex InsA peptides (cInsA) intraperitoneally and insulin‐specific protective IgM (PR‐IgM) in 48 h cycles intravenously (i.v.). *monitoring: diabetes symptoms were only observed within cInsA only group.
-
BBlood and urine glucose levels of mice immunized with complex InsA peptides (cInsA) (red, n = 5) and cInsA plus intravenously injected (i.v.) pIgM (salmon, n = 5) at day 7. Dots represent individual mice. Mean ± SD, statistical significance was calculated by using Mann–Whitney U‐test, *P < 0.05.
-
CBlood glucose levels of mice intravenously injected with total IgG pulldown of cInsA immunized mice (red, n = 4), or total IgG (cInsA) together with anti‐insulin PR‐IgM (black, n = 4). Measurements were done 5 h post‐intravenous injection. Data were collected from two independent experiments. Mean ± SD, statistical significance was calculated by using Mann–Whitney U‐test, **P < 0.01.
-
D, F(D) Serum (dilution: 1:50) and (F) insulin‐specific IgM (concentration: 500 ng/ml) tested for dsDNA (left panel, coating: calf thymus dsDNA) and insulin (right panel, coating: insulin) reactivity measured by ELISA. Mice were immunized with cInsA and serum was collected on day 7 (D: n = 8, left; n = 5, right; F: n = 4) and day 85 (D: n = 4, left; n = 5, right, F: n = 4) post‐immunization. Dots represent individual mice. Mean ± SD, statistical significance was calculated by using Mann–Whitney U‐test, **P < 0.01, ****P < 0.0001.
-
E, GAnti‐nuclear‐IgM (ANA) of control‐immunized (CI, n = 3) and complex InsA‐peptide(cInsA)‐immunized mice on day 7 (n = 3) and day 85 (n = 3) analyzed by indirect immunofluorescence. We used (E) total serum (dilutions: 1:20) or (G) insulin‐specific IgM (concentration: 500 ng/ml) purified from serum at indicated days on commercial HEp‐2 slides. Scale bar: 10 µm. Green fluorescence indicates IgM bound to nuclear structures. Data are representative for three independent experiments with isotype control: n = 3, day 7: n = 3, day 85: n = 3.
-
HCoomassie‐stained SDS–PAGE showing primary (cInsA d7) and memory (cInsA d85) insulin‐specific purified IgM pre‐incubated with insulin and calf thymus dsDNA. Samples were loaded onto the gel after size exclusion with a cut‐off at 10,000 kD (fractions referring to >/< 104 kD). IgM heavy chain (HC): 69 kD, IgM light chain (LC): 25 kD, J‐chain: 15 kD. Data shown are representative of three independent experiments.
-
IBlood glucose levels of mice intravenously injected with either IgM isotype ctrl (n = 6), anti‐insulin PR‐IgM (d85, n = 5), anti‐insulin primary IgM (d7, n = 4) after insulin‐specific pulldown. Mean ± SD, statistical significance was calculated by using repeated measure ANOVA test showing comparison of red and black line, ***P < 0.001, ****P < 0.0001.
These data suggest that memory anti‐insulin PR‐IgM directly prevents the depletion of insulin by primary anti‐insulin IgM as well as anti‐insulin IgG thereby blocking diabetes development.
One explanation for the differences between the autoreactive primary and memory PR‐IgM might be that primary IgM is polyreactive and might be produced by B1 B cells as a first line of immune protection (Benedict & Kearney, 1999; McHeyzer‐Williams, 2003; Lobo, 2016; Kreslavsky et al, 2018). Presumably, this polyreactivity results in joint immune complexes with a high molecular weight containing multiple autoantigens allowing elimination by phagocytes thereby depleting the bound insulin. In contrast, autoreactive memory PR‐IgM might be monospecific for autoantigen and may therefore release the autoantigen after binding without formation of immune complexes. To test this, we analyzed the polyreactive potential of primary IgM as compared to memory PR‐IgM. Anti‐DNA ELISA (Fig 7D) and indirect immune fluorescence using HEp‐2 slides (Fig 7E) showed that in contrast to primary IgM, memory PR‐IgM is not polyreactive but specifically binds to insulin (Fig 7D and E).
To show that anti‐insulin IgM is specifically responsible for the observed effects, we performed insulin‐specific pulldown assays using sera from InsA‐immunized mice. The pulldown resulted in pure insulin‐specific IgM as revealed by Western blot analysis. We blotted insulin‐specific purified IgM onto membranes and examined the presence of IgM and IgG heavy chains. As expected, we could only detect IgM heavy chains indicating an IgG‐free pulldown (Appendix Fig S13A). We also performed anti‐DNA ELISA (Fig 7F, Appendix Fig S13B) and indirect immune fluorescence on HEp‐2 slides (Fig 7G) using purified primary anti‐insulin IgM or memory anti‐insulin PR‐IgM. The results confirm the finding that in contrast to primary IgM, purified anti‐insulin PR‐IgM is not polyreactive and specifically binds to insulin (Fig 7F and G). In addition, we performed ELISA‐based competition assays by using soluble dsDNA as competitor. The results show that dsDNA is able to heavily interfere with primary IgM binding to insulin, but has only a slight interfering effect on PR‐IgM (Appendix Fig S14). These results support the notion that PR‐IgM is monospecific to insulin, whereas primary IgM remains polyreactive.
To directly test the hypothesis that primary anti‐insulin IgM forms large immune complexes whereas PR‐IgM does not, we incubated anti‐insulin primary IgM or PR‐IgM with insulin and DNA and determined the formation of immune complexes using size exclusion spin columns. In contrast to PR‐IgM, we found that primary anti‐insulin IgM forms mainly large complexes of > 104 kD (Fig 7H). To show that the purified primary anti‐insulin IgM is responsible for the dysregulation of glucose metabolism, we injected 5 µg of purified anti‐insulin primary IgM or PR‐IgM intravenously and monitored blood glucose. In contrast to PR‐IgM, we observed a vigorous increase in blood glucose after injection of purified primary anti‐insulin IgM (Fig 7I). Interestingly, the increase in blood glucose emerged faster after injection of purified anti‐insulin primary IgM as compared to total primary IgM (compare Fig 6G with Fig 7I).
In summary, these data suggest that increased specificity to autoantigen is important for autoreactive memory PR‐IgM to be protective during immune responses (graphical abstract). Moreover, the induced generation of autoreactive PR‐IgM is most likely a critical step in B‐cell tolerance.
Discussion
A critical aspect for understanding autoreactive immune responses is the design of experimental systems that resemble physiological situation. A key characteristic of the present study is the use of wild‐type animals expressing a normal B‐cell repertoire combined with insulin as a widely studied autoantigen which regulates glucose metabolism (Tokarz et al, 2018). Moreover, anti‐insulin antibodies represent an essential part of the diabetes mellitus metabolic disorder characterized by high blood glucose and associated complications (Uchigata et al, 1994, 2010; Pavithran et al, 2016; Hu & Chen, 2018). Thus, in contrast to transgenic mouse models expressing monospecific transgenic BCRs and cognate antigen, our experimental system uses a physiological B‐cell repertoire in wild‐type mice and a disease‐related autoantigen.
Notably, the diabetes symptoms used as readout for the generation of anti‐insulin antibodies are similar to exogenous insulin antibody syndrome (EIAS), which is a clinical syndrome associated with insulin antibodies induced by exogenous insulin in diabetic patients (Hu & Chen, 2018). Indeed, about half of the patients receiving insulin during diabetes therapy acquire anti‐insulin antibodies that are associated with unpredictable phases of glucose metabolism dysregulation referred to as dysglycemia (Berson et al, 1956; Uchigata et al, 1994; Lahtela et al, 1997; Hu & Chen, 2018). In agreement with our data, the most abundant class of anti‐insulin antibodies in EIAS patients is IgG (Fineberg et al, 2005) and these autoantibodies are sufficient for induction of insulin resistance (Ganz et al, 1990; Asai et al, 2003; Koyama et al, 2005). On the other hand, it has also been observed that some anti‐insulin antibodies seem to act as carrier that bind to insulin and prolong its action in a kind of “reservoir‐like” effect (Hu & Chen, 2018). It is tempting to speculate that the protective anti‐insulin IgM discovered in our experiments serves as carrier that releases endogenous insulin in controlled manner and at the same time prevents IgG binding and subsequent destruction. Thus, understanding the function of protective anti‐insulin IgM may not only help to protect endogenous insulin but might also lead to optimized release of the substituted insulin via the reservoir function.
The current results reveal an unexpected regulatory role of soluble monovalent antigen. Although by itself unable to mount considerable antibody responses, monovalent antigen modulates immune responses induced by multivalent antigen toward the generation of IgM. This occurs at the expense of IgG, and since autoreactive IgG is a hallmark of autoimmune diseases, monovalent antigen might prevent the outbreak of autoimmune diseases by shifting the immune responses toward protective IgM. Together with previous findings suggesting that IgD‐BCR controls the responsiveness of mature B cells by sensing the antigen valency, it is conceivable that monovalent antigen modulates B‐cell responsiveness and IgG antibody generation via the IgD‐class BCR (Übelhart et al, 2015). As a consequence, autoimmune diseases might develop when the ratio of autoantigen complexes exceeds a distinct threshold which then leads to increased autoreactive IgG as compared to IgM. In fact, our data show that an increased ratio of multivalent to monovalent antigen is associated with increased ratio of IgG to IgM in the immune responses. This relationship is independent of whether the antigen is self‐derived or foreign. Therefore, anti‐insulin autoantibodies of IgM and IgG type were easily generated in wild‐type mice after injection of insulin complexes suggesting that autoreactive B cells recognizing insulin are present in the periphery. Similarly, B cells recognizing the C‐peptide, as an example for low‐abundance autoantigen, are also present and induce specific autoantibodies upon encountering multivalent C‐peptide. This suggests that the peripheral repertoire of B cells of wild‐type animals contains autoreactive specificities for most likely every autoantigen. These autoreactive B cells are regulated by the relative amounts of antigen forms as shown for insulin in this study. The resulting model has fundamental consequences for B‐cell selection, antibody responses after vaccination, B‐cell tolerance, and control of harmful autoimmune responses. By proposing a dynamic regulation of B‐cell responses including the inducible production of autoantigen‐specific protective IgM, this model questions the central B‐cell tolerance concept, which suggests that the elimination of autoreactive B‐cell specificities in early stages of development is crucial for avoiding harmful autoreactive responses (Goodnow et al, 1988, 1989; Nemazee & Buerki, 1989; Nemazee & Bürki, 1989; Hartley et al, 1991; Brink et al, 1992). Given the ample generation of autoreactive antibodies and the protective role of autoreactive IgM shown here, it is likely that a highly diverse autoreactive primary B‐cell repertoire is essential for the unrestricted generation of protective IgM to control harmful autoimmune responses. This might explain why immune deficiencies are often associated with autoimmune diseases as deficiencies might interfere with the generation of a diverse B‐cell repertoire (Gupta & Gupta, 2017). According to this scenario, the secondary rearrangements of the immunoglobulin genes including receptor editing represent mechanisms that expand the B‐cell repertoire and thus the ability to manage the potential changes in foreign or self‐antigen. The important role of autoreactive B‐cell specificities for B‐cell development is exemplified by the autoreactive behavior of the precursor‐BCR, which is expressed on developing early B cells (Köhler et al, 2008; Herzog & Jumaa, 2012).
A diverse B‐cell repertoire ensures the generation of protective IgM to the same degree of diversity as that of generating pathogen‐specific antibodies. The main difference is that IgG isotypes are characteristic for effective, long‐term, pathogen‐specific antibody responses. Interestingly, our data demonstrate that protective IgM is induced by recall immunizations and shows affinity maturation typical for memory responses. Therefore, it is likely that the control of destructive autoimmune reactions involves the inducible generation of autoreactive IgM as part of an adaptive tolerance memory that prevents self‐destructive responses. Consequently, it is reasonable to consider the generation of memory IgM as important sign of autoreactive processes as compared with memory IgG as a standard indication for efficient pathogen‐specific immunizations. These characteristics might be of great importance for vaccination strategies as increased ratio of memory IgM may be indicative of autoimmune processes. Given the autoreactive potential of B cells shown here and the possibility that certain individuals under yet unknown conditions cannot produce protective IgM and can therefore not control the autoimmune processes, determining the ratio of memory IgM and IgG might be a crucial step for vaccine design and assessment of vaccination results.
Thus, our study has important consequences for vaccine design and application as the development of vaccines relies on the current model of B selection proposing that central tolerance mechanisms remove most autoreactive B‐cell specificities resulting in a peripheral B‐cell repertoire mostly devoid of autoreactive potential. Accordingly, the proposed absence of autoreactive specificities would prevent vaccines from inducing autoreactive responses. However, our finding that the normal peripheral B‐cell repertoire is capable of inducing broad autoreactive responses suggests that caution is required when designing and applying new vaccines.
Similarly, the use of sulfhydryl group‐containing drugs was reported to be associated with dysglycemia and the development of anti‐insulin autoantibodies (Wintersteiner, 1933; Pavithran et al, 2016). According to our data, it is conceivable that sulfhydryl‐groups cause complex formation, possibly by mediating disulfide bonds, of autoantigens including insulin thereby triggering autoreactive B cells to produce insulin‐specific antibodies. Given the autoreactive potential of normal B cells, medications, nutrients, protein degradation or cell death might contribute to the formation of autoantigen complexes and thus induce autoreactive responses.
Notably, natural IgM produced by B1 cells has been proposed to play a protective role in other human diseases associated with immune cell activation (Shapiro‐Shelef & Calame, 2005; Cantrell, 2015) by altered self‐structures. In arthrosclerosis for instance, natural IgM is proposed to bind to oxidized self‐structures thereby preventing the activation of innate immune cells and the subsequent inflammation (Benedict & Kearney, 1999; Amir et al, 2012; Tsiantoulas et al, 2012). However, in contrast to the protective memory IgM described here, natural antibodies are produced in the absence of previous exposure to antigen and do not undergo affinity maturation as they usually lack mutations in their Ig genes (Nakamura et al, 1988).
The anti‐insulin protective IgM (PR‐IgM), which is produced during secondary responses, is likely to be memory IgM for several reasons. First, serum titers of anti‐insulin IgM were greatly increased after boost immunization, which is typical of a memory response (Weill & Reynaud, 2020). Second, antigen affinity of IgM was increased from primary (d7) to secondary (d28) immune responses after the first boost at d21 and was maintained for long time after immunization. Third, the number of IgM+ insulin‐reactive B cells was considerably increased compared at d85 as compared with d7. Fourth, IgM on d85 is not polyreactive, and therefore, it is unlikely that PR‐IgM is produced by recall primary responses which are clearly polyreactive according to our data.
The primary IgM, which induces diabetes symptoms in our study, most likely belongs to natural antibodies suggesting that polyreactive IgM may also induce harmful autoimmune responses. Currently, it is unclear how the high‐affinity anti‐insulin memory IgM protects insulin from removal by IgG or primary low‐affinity IgM. It is possible that low‐affinity anti‐insulin IgM, due to reduced specificity for insulin, binds other autoantigens leading to higher molecular weight immune complexes that are bound and digested by phagocytes. Anti‐insulin IgM possessing high specificity might prevent this scenario. Thus, an important consequence of the present study is the opportunity of inducing or restoring immune tolerance by characterization of the involved autoantigen and the affinity of antibody binding. Using insulin as an example for a common autoantigen shows that the ratio between monovalent antigen and complex multivalent antigen is crucial for regulating immune responses. Shifting the balance of autoantigen toward multivalent forms leads to autoreactive IgG antibodies associated with autoimmune diseases (Kaneko et al, 2006).
The induction of protective autoreactive memory IgM (PR‐IgM) by ratios of soluble and complex antigen prevents the development of autoreactive IgG causing harmful autoreactive responses. It is conceivable that this process protects soluble as well as membrane‐bound autoantigens, thereby preventing autoimmune targeting and destruction of respective cells/autoantigens. Based on molecular characterization of the self‐structures involved in autoimmune diseases, the regulated induction of protective IgM might provide a new concept for the treatment of already manifest autoimmune diseases or for preventing their development.
Taken together, this study is a paradigm shift from a static model of B‐cell development, in which absolute selection mechanisms are proposed to prevent initial generation of autoreactive specificities, toward a dynamic model of development, in which immune responses are regulated by relative amounts of antigen forms and antibody isotypes allowing an unrestricted potential of adaptive immune responses that are regulated by adaptive tolerance memory.
Materials and Methods
Mice
Eight‐ to 12‐week‐old female C57BL/6 mice and female B‐cell‐deficient mice (Hobeika et al, 2006) were immunized intraperitoneally (i.p.) with a mixture of 13–50 µg antigen with 50 µg CpG‐ODN1826 (Biomers) in 1× PBS for the first injection. Following injections (boosts) were done without CpG‐ODN1826. Control immunization (CI) mice received PBS and CpG‐ODN1826 (50 µg/mouse). Long‐term immune responses of immunized mice were monitored up to an age of over 30 weeks. Native biotinylated murine insulin was purchased from BioEagle. Animal experiments were performed in compliance with license 1,484 for animal testing at the responsible regional board Tübingen, Germany. All mice used in this study were bred and housed within the animal facility of Ulm University under specific‐pathogen‐free conditions or obtained from Jackson at the age of 6 weeks.
Peptides
C‐Peptide peptides (RoyoBiotech, Shanghai) and insulin peptides (Peptides & Elephants, Berlin) were dissolved according to their water solubility in pure water or 1% Dimethylformamide (DMF). For covalent coupling of peptides to key hole limpet hemocyanin (KLH), a N‐terminal cysteine was added. Coupling of peptides to streptavidin (SAV, Thermo Scientific) was done by addition of biotin to the N‐terminus. The C‐terminus was left unmodified with an OH‐group for better handling.
Flow cytometry
Cell suspension was Fc‐receptor blocked with polyclonal rat IgG‐UNLB (2,4G2; BD) and stained according to standard protocols. Biotin‐conjugated peptides/antibodies were detected using streptavidin Qdot605 (Molecular Probes; Invitrogen). Viable cells were distinguished from dead cells by usage of Fixable Viability Dye eFluor780 (eBioscience). Cells were acquired at a Canto II Flow Cytometer (BD). If not stated otherwise numbers in the dot plots indicate percentages in the respective gates whereas numbers in histograms state the mean fluorescence intensity (MFI).
Antibody specificity, conjugate, clone, supplier catalog number, dilution: anti‐CD45R/B220 (PE‐Cy7 RA3‐6B2 Invitrogen, 25045282, 1:200); Anti‐CD19, (PerCP‐Cy5.5, 1D3, BD Biosciences, 551001, 1:200); Anti‐CD11b (FITC, M1/70, BD Biosciences, 557396, 1:200); Anti‐IgD (Biotin, southern Biotech, 1120‐08, 1:300); Anti‐IgG (APC, Poly4053, BioLegend, 405308, 1:400); Anti‐IgM (eFluor 450, eB121‐15F9, Invitrogen, 48‐5890‐82, 1:300); streptavidin (Qdot 605, Invitrogen, Q10001MP, 1:400); Anti‐CD138 (PE, 281‐2, BD Biosciences, 553714, 1:300); Anti‐CD23 (PE, B384, BD Biosciences, 553139, 1:300); Anti‐Ly6G (PE‐Cy7, 1A8, BD Biosciences, 560601, 1:200); Anti‐CD16/CD32 (Unlabeled, 2.4G2, BD Pharmingen, 553142, 1:100); Anti‐GL‐7 FITC (09054D, BD Biosciences, 553666, 1:100).
Enzyme‐linked immunosorbent assay (ELISA)
A 96‐Well plates (Nunc, MaxiSorp) were coated either with, native insulin (Sigma‐Aldrich, Cat. 91077C), streptavidin (Thermo Scientific, Cat. 21125), or calf thymus DNA (Thermo Scientific, Cat.15633019), with 10 µg/ml, or anti‐IgM, anti‐IgG‐antibodies (SouthernBiotech). Loading with a biotinylated peptide (2.5 µg/ml) of SAV‐plates and blocking was done in 1% BSA blocking buffer (Thermo Fisher). Serial dilutions of 1:3 IgM or IgG antibodies (SouthernBiotech) were used as standard. The relative concentrations stated as arbitrary unit (AU), were determined via detection by Alkaline Phosphatase (AP)‐labeled anti‐IgM/anti‐IgG (SouthernBiotech), respectively. The p‐nitrophenylphosphate (pNPP; Genaxxon) in diethanolamine buffer was added and data were acquired at 405 nm using a Multiskan FC ELISA plate reader (Thermo Scientific). All samples were measured in duplicates.
For analysis of affinity maturation, results from plates coated with either peptide (1) or peptide (4) were calculated by dividing peptide (1) by peptide (4). Thus, results were stated as relative units [RU] within the figures.
For determination of serum levels of pancreatic lipase (PL), we used a mouse PL kit (MyBioSource, MBS733780) according to the manufacturer’s protocol.
Antibody specificity, host/isotype, conjugate clone, class, supplier catalog number, dilution: anti‐mouse IgM (Goat, IgG, Unlabeled, polyclonal, SouthernBiotech, 1020‐01); anti‐mouse IgG (Goat, IgG, Unlabeled, polyclonal, SouthernBiotech, 1030‐01); Mouse IgM (Unlabeled, polyclonal, SouthernBiotech, 0101‐01); Mouse IgG (Unlabeled, polyclonal, SouthernBiotech, 0107‐01); anti‐mouse IgM (Goat, AP, polyclonal, SouthernBiotech, 0120‐04); anti‐mouse IgG (Goat, AP, polyclonal, SouthernBiotech, 0130‐04).
Enzyme‐linked immuno‐spot assay (ELISpot)
Total splenocytes were measured in triplicates with 300,000 cells/well. ELISpot plates were pre‐coated with either native insulin (Sigma‐Aldrich, Cat. 91077C), or C‐peptide (RoyoBiotech). After 12–24 h incubation of the cells at 37°C, antigen‐specific IgM or IgG was detected via anti‐IgM‐bio:SAV‐AP or anti‐IgG‐bio:SAV‐AP (Mabtech). Handling of the plates and antibody concentrations was done according to the manufacturer’s recommendations.
HEp‐2 slides and fluorescence microscopy
HEp‐2 slides (EUROIMMUN, F191108VA) were used to asses reactivity of serum IgM or insulin‐specific IgM to nuclear antigens (ANA). Sera of insulin‐A‐peptide‐immunized mice on days 7 and 85 post‐immunization were diluted to an equal concentration of IgM (approx. 500 ng/ml anti‐Insulin‐IgM in each sample) and applied onto the HEp‐2 slides. Anti‐IgM‐FITC (eBioscience, Cat. 11‐5790‐81) was used for detection of ANA‐IgM. Stained HEp‐2 slides were analyzed using fluorescence microscope Axioskop 2 (Zeiss) and DMi8 software (Leica).
Monitoring of blood and urine glucose levels
Assessment of urine glucose levels was done using Combur 10 M Test stripes (Roche Diagnostics, Mannheim). Sterile stripes were used during daily mouse handling and the displayed color change after testing was compared to the manufacturer’s standard of glucose levels in mmol/l. AccuCheck (Roche Diagnostics, Mannheim) blood glucose monitor was used to measure blood glucose levels of mice. Blood was taken from the tail vein from ad libitum fed mice and transferred onto sterile test stripes. Glucose levels were measured in mmol/l at days stated in the figures for each mouse per group. Control mice were tested with immunized littermates and measured at similar times of the day.
SDS–PAGE, Coomassie, and Western blot
Organs were taken immediately after sacrifice and lysed in RIPA buffer containing protease and phosphatase inhibitors (50 mM Tris–HCl, pH 7.4, 1% NP‐40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA (pH 8)), 1 mM sodium orthovanadate, 1 mM NaF, protease inhibitor cocktail (Sigma‐Aldrich). Samples were separated on 10–20% SDS–polyacrylamide gels and either blotted onto PVDF membranes (Millipore) or incubated with Coomassie (Coomassie brilliant blue R‐250, Thermo Fisher) for 45 min and subsequently de‐stained. Membranes were blocked for 1 h at room temperature in 5% BSA PBS with constant agitation. Primary antibodies were diluted in 5% BSA PBS (BIOMOL Research Laboratories). Secondary antibodies were diluted in 5% BSA PBS. Development of the membrane and recording of the data were done with an optical system Fusion SL (Vilber).
Antibody specificity, host/isotype, conjugate, clone, class, supplier catalog number, dilution: anti‐mouse‐IgG (IgG1, HRP, Human, cell signaling, 7076 1: 10,000); anti‐beta‐actin (IgG, Rabbit, D6A8, cell signaling, 8457, 1:500); anti‐rabbit‐IgG (IgG, Goat, cell signaling, 7074, 1:10,000).
Pulldown of total serum immunoglobulins
Sera of immunized mice were taken immediately after euthanasia and either IgM or IgG were purified. Removal of antigen bound to antibodies was achieved by repeated freeze‐thaw cycles of the serum and pH‐shift during elution (Reverberi & Reverberi, 2007). For IgG protein G sepharose beads (Thermo Fisher) were used according to the manufacturer’s protocol and dialyzed overnight in 10 times sample volume in 1× PBS. For IgM, HiTrap IgM columns (GE Healthcare, Sigma‐Aldrich) were used according to the manufacturer’s protocol and dialyzed overnight in 10 times sample volume 1× PBS. Quality control of the isolated immunoglobulins was addressed via SDS–PAGE and Coomassie and the amount of insulin‐specific immunoglobulins determined via ELISA. Finally, 20–50 µg (1–10 µg insulin‐specific‐Ig) were injected intravenously.
Isolation of insulin‐specific serum immunoglobulins
Sera of cInsA (complex insulin‐A peptide) and control‐immunized mice were taken immediately after euthanasia and prepared for insulin‐specific immunoglobulin isolation. Streptavidin bead columns (Thermo Scientific, Cat. 21115) were loaded with 10 µg bio‐insulin (BioEagle). The sera were incubated for 90 min at room temperature to ensure binding of insulin‐specific antibodies to the beads. Isolation of the insulin antibodies was done by pH‐shift using the manufacturer’s elution and neutralization solutions. Quality of the isolated immunoglobulins was examined via Coomassie and Western blot analysis using anti‐IgM heavy chain (Thermo Scientific, Cat. 62‐6820) and anti‐IgG heavy chain (Cell Signaling Technologies, Cat. 7076) antibodies. For further in vivo experiments, the isolated antibodies were dialyzed overnight (see above).
Bio‐layer interferometry (BLI)
Bio‐Layer Interferometric assays (BLItz device, ForteBio) were used to determine the affinity of antigen‐antibody interactions (Kumaraswamy & Tobias, 2015). Here, we used insulin‐specific IgM (see isolation of insulin‐specific immunoglobulins) and insulin‐bio (Thermo Fisher) as target. Targets were loaded onto streptavidin biosensors (ForteBio). Binding affinities of IgM to insulin were acquired in nm. Subsequently, the calculated affinity value (Ka) was used to determine the dissociation constant (Kd): Kd = 1/Ka. Following protocol was used: 30‐s baseline, 30‐s loading, 30‐s baseline, 240‐s association, 120‐s dissociation. For buffering of samples, targets, and probes, the manufacturer’s sample buffer (ForteBio) was used.
Flow Cytometric Bead Array for mouse inflammatory cytokines
To determine pancreas supernatant inflammatory cytokine levels of mice immunized with cInsulin or control immunization, we performed a BD Cytometric Bead Array (Mouse Inflammation, BD Biosciences, Cat.: 552364, Lot.: 005197). Samples were diluted according to the manufacturer’s protocol. IL‐12p70, TNF‐α, IFN‐γ, MCP‐1, IL‐10, and IL‐6 APC‐labeled beads were used together with PE‐labeled detector reagent. The assay was measured at a FACS Canto II and analyzed via FlowJo10 software. Relative cytokine levels correlate to the mean fluorescence intensity of each cytokine bead within the PE channel.
Statistical analysis
Graphs were created and statistically analyzed by using GraphPad Prism (version 6.0h) software. The numbers of individual replicates or mice (n) are stated within the figure or figure legends. Data sets were analyzed by D'Agostino & Pearson omnibus normality test and/or Shapiro–Wilk normality test in GraphPad Prism software to determine whether they are normally distributed. If one of the data sets was not normally distributed or the sample number n was too small to perform the normality tests, non‐parametric tests were used to calculate P‐values. Statistical tests used to calculate P‐values are stated within the figure legends. P‐values > 0.05 were considered to be statistically significant (*P < 0.05; **P < 0.01; ***P < 0.001, ****P < 0.0001).
Author contributions
TA performed all experiments, analyzed and interpreted data, prepared the figures together with HJ and contributed to the writing of the manuscript. HJ designed the study, proposed experiments, supervised the work, and wrote the manuscript. Both authors read and discussed the manuscript.
Conflict of interest
HJ has applied for a patent for the use of PR‐IgM to protect autoantigens.
Supporting information
Review Process File
Appendix
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 6
Acknowledgements
We thank C. Setz and M. Young for assistance in performing experiments, O. El Ayoubi for serum immunoglobulin pulldown, G. Allies for injections of mice and SDS–PAGE We thank M. Reth for critical reading of the manuscript. This work was supported by the DFG through TRR130 (B cells and beyond) project 01, SFB1074 (Experimental Models and Clinical Translation in Leukemia), SFB 1279 (Exploration of the Human Peptidome). Open Access funding enabled and organized by Projekt DEAL.
The EMBO Journal (2021) 40: e107621.
Data availability
This study includes no data deposited in external repositories.
References
- Amir S, Hartvigsen K, Gonen A, Leibundgut G, Que X, Jensen‐Jarolim E, Wagner O, Tsimikas S, Witztum JL, Binder CJ (2012) Peptide mimotopes of malondialdehyde epitopes for clinical applications in cardiovascular disease. J Lipid Res 53: 1316–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai M, Kodera T, Ishizeki K, Uebori S, Kashiwaya T, Itoh H, Makino I (2003) Insulin lispro reduces insulin antibodies in a patient with type 2 diabetes with immunological insulin resistance. Diabetes Res Clin Pract 61: 89–92 [DOI] [PubMed] [Google Scholar]
- Benedict CL, Kearney JF (1999) Increased junctional diversity in fetal B cells results in a loss of protective anti‐phosphorylcholine antibodies in adult mice. Immunity 10: 607–617 [DOI] [PubMed] [Google Scholar]
- Benschop RJ, Brandl E, Chan AC, Cambier JC (2001) Unique signaling properties of B Cell antigen receptor in mature and immature B Cells: implications for tolerance and activation. J Immunol 167: 4172–4179 [DOI] [PubMed] [Google Scholar]
- Berson SA, Yalow RS, Bauman A, Rothschild MA, Newerly K (1956) Insulin‐I131 metabolism in human subjects: demonstration of insulin binding globulin in the circulation of insulin treated subjects. J Clin Invest 35: 170–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brink R, Goodnow CC, Crosbie J, Adams E, Eris J, Mason DY, Hartley SB, Basten A (1992) Immunoglobulin M and D antigen receptors are both capable of mediating B lymphocyte activation, deletion, or anergy after interaction with specific antigen. J Exp Med 176: 991–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantrell D (2015) Signaling in lymphocyte activation. Cold Spring Harb Perspect Biol 7: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GS, Stroehla BC (2003) The epidemiology of autoimmune diseases. Autoimmun Rev 2: 119–125 [DOI] [PubMed] [Google Scholar]
- Cyster JG, Hartley SB, Goodnow CC (1994) Competition for follicular niches excludes self‐reactive cells from the recirculating B‐cell repertoire. Nature 371: 389–395 [DOI] [PubMed] [Google Scholar]
- Fineberg SE, Kawabata T, Finco‐Kent D, Liu C, Krasner A (2005) Antibody response to inhaled insulin in patients with type 1 or type 2 diabetes. An analysis of initial phase II and III inhaled insulin (Exubera) trials and a two‐year extension trial. J Clin Endocrinol Metab 90: 3287–3294 [DOI] [PubMed] [Google Scholar]
- Ganz MA, Unterman T, Roberts M, Uy R, Sahgal S, Samter M, Grammer LC (1990) Resistance and allergy to recombinant human insulin. J Allergy Clin Immunol 86: 45–51 [DOI] [PubMed] [Google Scholar]
- Gay D, Saunders T, Camper S, Weigert M (1993) Receptor editing: An approach by autoreactive B cells to escape tolerance. J Exp Med 177: 999–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez D, Addison A, De Rosa A, Brooks A, Cameron IC (2012) Retrospective study of patients with acute pancreatitis: Is serum amylase still required? BMJ Open 2: e001471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith‐Gill SJ, Brink RA, Pritchard‐Briscoe H, Wotherspoon JS, Loblay RH, Raphael Ket al (1988) Altered immunoglobulin expression and functional silencing of self‐reactive B lymphocytes in transgenic mice. Nature 334: 676–682 [DOI] [PubMed] [Google Scholar]
- Goodnow CC, Crosbie J, Jorgensen H, Brink RA, Basten A (1989) Induction of self‐tolerance in mature peripheral B lymphocytes. Nature 342: 385–391 [DOI] [PubMed] [Google Scholar]
- Grönwall C, Vas J, Silverman GJ (2012) Protective roles of natural IgM antibodies. Front Immunol 3, 66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Gupta A (2017) Selective IgM deficiency‐An underestimated primary immunodeficiency. Front Immunol 8: 1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley SB, Crosbie J, Brink R, Kantor AB, Basten A, Goodnow CC (1991) Elimination from peripheral lymphoid tissues of self‐reactive B lymphocytes recognizing membrane‐bound antigens. Nature 353: 765–769 [DOI] [PubMed] [Google Scholar]
- Herzog S, Jumaa H (2012) Self‐recognition and clonal selection: Autoreactivity drives the generation of B cells. Curr Opin Immunol 24: 166–172 [DOI] [PubMed] [Google Scholar]
- Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, Reth M (2006) Testing gene function early in the B cell lineage in mb1‐cre mice. Proc Natl Acad Sci U S A 103: 13789–13794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Chen F (2018) Exogenous insulin antibody syndrome (EIAS): A clinical syndrome associated with insulin antibodies induced by exogenous insulin in diabetic patients. Endocr Connect 7: R47–R55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko Y, Nimmerjahn F, Ravetch JV (2006) Anti‐inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 313: 670–673 [DOI] [PubMed] [Google Scholar]
- Köhler F, Hug E, Eschbach C, Meixlsperger S, Hobeika E, Kofer J, Wardemann H, Jumaa H (2008) Autoreactive B Cell receptors mimic autonomous pre‐B cell receptor signaling and induce proliferation of early B cells. Immunity 29: 912–921 [DOI] [PubMed] [Google Scholar]
- Koyama R, Nakanishi K, Kato M, Yamashita S, Kuwahara H, Katori H (2005) Hypoglycemia and hyperglycemia due to insulin antibodies against therapeutic human insulin: treatment with double filtration plasmapheresis and prednisolone. Am J Med Sci 329: 259–264 [DOI] [PubMed] [Google Scholar]
- Kreslavsky T, Wong JB, Fischer M, Skok JA, Busslinger M (2018) Control of B‐1a cell development by instructive BCR signaling. Curr Opin Immunol 51: 24–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaraswamy S, Tobias R (2015) Label‐free kinetic analysis of an antibody‐antigen interaction using biolayer interferometry. In Protein‐protein interactions: methods and applications, Meyerkord CL, Fu H (eds), 2 nd edn, pp 165–182. New York: Springer; [DOI] [PubMed] [Google Scholar]
- Lahtela JT, Knip M, Paul R, Antonen J, Salmi J (1997) Severe antibody‐mediated human insulin resistance: Successful treatment with the insulin analog lispro: a case report. Diabetes Care 20: 71–73 [DOI] [PubMed] [Google Scholar]
- Lobo PI (2016) Role of natural autoantibodies and natural IgM anti‐leucocyte autoantibodies in health and disease. Front Immunol 7, 198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHeyzer‐Williams MG (2003) B cells as effectors. Curr Opin Immunol 15: 354–361 [DOI] [PubMed] [Google Scholar]
- Nakamura M, Burastero SE, Ueki Y, Larrick JW, Notkins ALCP (1988) Probing the normal and autoimmune B cell repertoire with Epstein‐Barr virus. Frequency of B cells producing monoreactive high affinity autoantibodies in patients with Hashimoto’s disease and systemic lupus erythematosus. J Immunol 141, 4165–4172 [PubMed] [Google Scholar]
- Nemazee D, Buerki K (1989) Clonal deletion of autoreactive B lymphocytes in bone marrow chimeras. Proc Natl Acad Sci U S A 86: 8039–8043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemazee DA, Bürki K (1989) Clonal deletion of B lymphocytes in a transgenic mouse bearing anti‐MHC class I antibody genes. Nature 337: 562–566 [DOI] [PubMed] [Google Scholar]
- Nossal GJV, Pike BL (1978) Mechanisms of clonal abortion tolerogenesis: I. Response of immature hapten‐specific B lymphocytes. J Exp Med 148: 1161–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nossal GJV, Pike BL (1980) Clonal anergy: Persistence in tolerant mice of antigen‐binding B lymphocytes incapable of responding to antigen or mitogen. Proc Natl Acad Sci U S A 77: 1602–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill SK, Getahun A, Gauld SB, Merrell KT, Tamir I, Smith MJ, Dal Porto JM, Li QZ, Cambier JC (2011) Monophosphorylation of CD79a and CD79b ITAM motifs initiates a SHIP‐1 phosphatase‐mediated inhibitory signaling cascade required for B cell anergy. Immunity 35: 746–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padoa CJ, Crowther NJ, Thomas JW, Hall TR, Bekris LM, Torn C, Landin‐Olsson M, Ortqvist E, Palmer JP, Lernmark Aet al (2005) Epitope analysis of insulin autoantibodies using recombinant Fab. Clin Exp Immunol 140: 564–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavithran PV, Bhavani N, Jayakumar RV, Menon AS, Kumar H, Menon VU, Nair V, Abraham N (2016) Autoantibodies to insulin and dysglycemia in people with and without diabetes: an underdiagnosed association. Clin Diabetes 34: 164–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reverberi R, Reverberi L (2007) Factors affecting the antigen‐antibody reaction. Blood Transfus 5: 227–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setz CS, Khadour A, Renna V, Iype J, Gentner E, He X, Datta M, Young M, Nitschke L, Wienands Jet al (2019) Pten controls B‐cell responsiveness and germinal center reaction by regulating the expression of IgD BCR. EMBO J 38, e100249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro‐Shelef M, Calame KC (2005) Regulation of plasma‐cell development. Nat Rev Immunol 5: 230–242 [DOI] [PubMed] [Google Scholar]
- Shimizu T, Kozono Y, Kozono H, Oda M, Azuma T (2004) Affinity maturation of secreted IgM pentamers on B cells. Int Immunol 16: 675–684 [DOI] [PubMed] [Google Scholar]
- Smith RC, Southwell‐Keely J, Chesher D (2005) Should serum pancreatic lipase replace serum amylase as a biomarker of acute pancreatitis? ANZ J Surg 75: 399–404 [DOI] [PubMed] [Google Scholar]
- Suurmond J, Diamond B (2015) Autoantibodies in systemic autoimmune diseases: Specificity and pathogenicity. J Clin Invest 125: 2194–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiegs SL, Russell DM, Nemazee D (1993) Receptor editing in self‐reactive bone marrow B cells. J Exp Med 177: 1009–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokarz VL, MacDonald PE, Klip A (2018) The cell biology of systemic insulin function. J Cell Biol 217: 2273–2289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiantoulas D, Gruber S, Binder CJ (2012) B‐1 cell immunoglobulin directed against oxidation‐specific epitopes. Front Immunol 3: 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Übelhart R, Hug E, Bach MP, Wossning T, Dühren‐von Minden M, Horn AHC, Tsiantoulas D, Kometani K, Kurosaki T, Binder CJet al (2015) Responsiveness of B cells is regulated by the hinge region of IgD. Nat Immunol 16: 534–543 [DOI] [PubMed] [Google Scholar]
- Uchigata Y, Eguchi Y, Takayama‐Hasumi S, Omori Y (1994) Insulin autoimmune syndrome (Hirata Disease): clinical features and epidemiology in Japan. Diabetes Res Clin Pract 22: 89–94 [DOI] [PubMed] [Google Scholar]
- Uchigata Y, Hirata Y, Iwamoto Y (2010) Insulin autoimmune syndrome (Hirata disease): epidemiology in Asia, including Japan. Diabetol Int 1: 21–25 [Google Scholar]
- Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC (2003) Predominant autoantibody production by early human B cell precursors. Science 301: 1374–1377 [DOI] [PubMed] [Google Scholar]
- Weill JC, Reynaud CA (2020) IgM memory B cells: specific effectors of innate‐like and adaptive responses. Curr Opin Immunol 63: 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wintersteiner O, Abramson HA (1933) The isoelectric point of insulin. J Biol Chem 99: 741–753 [Google Scholar]
- Yang Z‐W, Meng X‐X, Xu P (2015) Central role of neutrophil in the pathogenesis of severe acute pancreatitis. J Cell Mol Med 19: 2513–2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zemlin M, Wang YH, Munfus D, Huye LE, Findley HW, Bridges SL, Roth DB, Burrows PD, Cooper MD (2003) Contribution of VH gene replacement to the primary B cell repertoire. Immunity 19: 21–31 [DOI] [PubMed] [Google Scholar]
- Zhu S, Larkin D, Lu S, Inouye C, Haataja L, Anjum A, Kennedy R, Castle D, Arvan P (2016) Monitoring C‐peptide storage and secretion in islet b‐cells in vitro and in vivo . Diabetes 65: 699–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Appendix
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 6
Data Availability Statement
This study includes no data deposited in external repositories.