

Abstract
Bladder cancer is the 10th most common cancer worldwide. For muscle‐invasive bladder cancer (MIBC), treatment includes radical cystectomy, radiotherapy, and chemotherapy; however, the outcome is generally poor. For non–muscle‐invasive bladder cancer (NMIBC), tumor recurrence is common. There is an urgent need for more effective and less harmful therapeutic approaches. Here, bladder cancer cell metabolic reprogramming to rely on aerobic glycolysis (the Warburg effect) and expression of associated molecular therapeutic targets by bladder cancer cells of different stages and grades, and in freshly resected clinical tissue, is investigated. Importantly, analyses indicate that the Warburg effect is a feature of both NMIBCs and MIBCs. In two in vitro inducible epithelial‐mesenchymal transition (EMT) bladder cancer models, EMT stimulation correlated with increased lactate production, the end product of aerobic glycolysis. Protein levels of lactate dehydrogenase A (LDH‐A), which promotes pyruvate enzymatic reduction to lactate, were higher in most bladder cancer cell lines (compared with LDH‐B, which catalyzes the reverse reaction), but the levels did not closely correlate with aerobic glycolysis rates. Although LDH‐A is expressed in normal urothelial cells, LDH‐A knockdown by RNAi selectively induced urothelial cancer cell apoptotic death, whereas normal cells were unaffected—identifying LDH‐A as a cancer‐selective therapeutic target for bladder cancers. LDH‐A and other potential therapeutic targets (MCT4 and GLUT1) were expressed in patient clinical specimens; however, positive staining varied in different areas of sections and with distance from a blood vessel. This intratumoral heterogeneity has important therapeutic implications and indicates the possibility of tumor cell metabolic coupling.
Keywords: epithelial‐mesenchymal transition, intratumoral heterogeneity, lactate dehydrogenase A, non–muscle‐invasive and muscle‐invasive bladder cancers, Warburg effect
This work identifies aerobic glycolysis (the Warburg effect) as a general feature of both non–muscle‐invasive and muscle‐invasive bladder cancers. We further show that the glycolytic enzyme lactate dehydrogenase A is a cancer‐selective therapeutic target for bladder cancers, but that there is intratumoral heterogeneity in expression of metabolic targets. This raises the possibility of intratumoral metabolic coupling that could be therapeutically targeted.

Abbreviations
- EMT
epithelial mesenchymal transition
- LDH‐A
lactate dehydrogenase A
- MIBC
muscle‐invasive bladder cancer
- NHUC
normal human urothelial cells
- NMIBC
non–muscle‐invasive bladder cancer
- OXPHOS
mitochondrial oxidative phosphorylation
- PDH
pyruvate dehydrogenase
- PDK
pyruvate dehydrogenase kinase
- TME
tumor microenvironment
1. INTRODUCTION
In 2018, there were an estimated 200 000 deaths worldwide from bladder cancer.1 A total of 25% of bladder cancer patients present with muscle‐invasive disease (MIBC), for which treatment includes radical cystectomy, radiotherapy, and/or platinum‐based chemotherapy.2 For patients with metastatic disease, median survival is less than 1 year. For the 75% of patients diagnosed with non–muscle‐invasive bladder cancer (NMIBC), tumor recurrence is common, and regular disease monitoring by cystoscopy is required because high‐grade tumors have a high risk of progression to MIBC.3 As well as impacting on patient quality of life, this has a significant cost burden for health providers. Until the recent FDA approval of the Fibroblast Growth Factor Receptor (FGFR) inhibitor erdafitinib and the immune checkpoint inhibitors pembrolizumab and atezolizumab, treatments for bladder cancer had not changed significantly for several decades.4 While these therapies are showing very promising effects in some patients, there is little or no improved response in others,4 and there remains a significant need for novel and more effective treatments for both MIBCs and NMIBCs.5
Alterations in the metabolism of cancer cells compared with normal, healthy cells are an emerging hallmark of most, if not all, cancers.6, 7 Following on from the discovery of oncogenic driver mutations in isocitrate dehydrogenase (IDH) in acute myeloid leukemia (AML) and low‐grade gliomas, two selective mutant IDH inhibitors ivosidenib and enasidenib have now been FDA‐approved for treatment of IDH‐mutant AML.8 There is considerable interest in the opportunities that cancer‐associated metabolic reprogramming may provide for targeted therapies that have fewer side effects than traditional cytotoxic chemotherapy.9, 10, 11, 12 Apart from targeting IDH‐mutant cancers, a number of other promising metabolic targets have been identified by in vitro and in vivo preclinical studies in different tumor types and genetic backgrounds.10, 11, 12, 13
Among different types of metabolic reprogramming that cancer cells may undergo, many cancer cells heavily depend upon glycolysis even in the presence of oxygen both for the generation of ATP and for macromolecule precursors that are required to support cancer cell growth and division.7, 14 This phenomenon of aerobic glycolysis, otherwise known as the Warburg effect, is directly linked to specific oncogene activation and tumor suppressor loss.15, 16, 17 While metabolic reprogramming is oncogene driven, it is generally poorly understood how different combinations of genetic lesions integrate to confer a particular metabolic preference or dependency, and the direct oncogenic mutation of metabolic enzymes as seen with IDH is rare.14 There is also growing evidence of the importance of nongenetic, cell‐extrinsic factors that also impact upon the metabolic phenotype of cancer cells.14, 18
While some cancer cell metabolic alterations are neutral for tumor growth, others are tumor enabling.14 The Warburg effect has been shown to be important for both tumor progression and maintenance and to contribute to cell transformation.19, 20, 21 Several enzymes directly involved in the Warburg effect, or its promotion, are essential for the survival of certain cancer cells, but importantly they appear to be dispensable in noncancer cells indicating a likely therapeutic window.19, 20, 21, 22, 23, 24
The purpose of this study was to investigate whether bladder cancers show the Warburg effect with the longer‐term goal that, if so, this may enable the development of new targeted therapies against bladder cancers. Given the heavier mutational signature of MIBCs,25 which includes genes associated with the Warburg effect, it was hypothesized that this would be a prominent feature of MIBCs but perhaps not of NMIBCs. The expression levels of key enzymes and transporters involved in the Warburg effect were also evaluated as potential therapeutic targets for further investigation.
2. MATERIALS AND METHODS
2.1. Cell lines and EMT induction
Twenty‐three human urothelial carcinoma cell lines were used, derived from bladder cancers of different pathological stages and grades, as previously described.26, 27, 28, 29 Cell lines and research identifiers (RRID) are detailed in Table S1 with all experiments performed using mycoplasma‐free cells. Known mutational status of the bladder cancer cell lines is shown in Table S2. Cell lines were authenticated by STR profiling28 and within 3 years of the presented cell line experiments. Cells were maintained at low passage in antibiotic‐free growth media, with all experiments conducted within 10 passages of authentication. Normal human urothelial cells (NHUCs) were derived from urothelium stripped from human ureters obtained at nephrectomy and were maintained in basal keratinocyte growth medium 2 (C20211, Promocell) containing growth factor supplements, 90 µM CaCl2, and 30 ng/ml cholera toxin (Sigma‐Aldrich).30 Telomerase‐immortalized (TERT) NHUCs (RRID CVCL_JX41) were also utilized.27 For EMT induction in FGFR1‐transduced 94‐10 urothelial carcinoma cells and in endogenously FGFR1‐expressing J82 cells, cells were treated with FGF2 (10 ng/mL) or heparin (10 µg/mL) and FGF2 (10 ng/mL) for 72 hours.31 Further details of treatment of transduced and transfected cell lines can be found in Supplementary Methods.
2.2. Metabolic analyses
Rates of cellular glucose consumption and lactate production were determined by quantification of changes in glucose and lactate levels in the culture media of cells over time using commercially available assays (Biovision Inc; #K607 [lactate]; #K606 [glucose]) as previously described.22 These were determined for cell lines in log phase growth at ~70% confluence with levels of lactate released into the media or glucose consumed assayed over a 6‐8‐hour period. Cell counts were performed using a Z2 Coulter analyzer (Beckman Coulter Ltd) at the assay endpoint. Rates of lactate released or glucose consumed were normalized to cell number and were determined from a minimum of three independent experiments. Cellular oxygen consumption was determined using a Clark‐type oxygen electrode (Oxytherm, Hansatech Instruments Ltd). Oxygen consumption rates were measured over a 20‐minute linear period and were normalized to cell number as described.22 To determine the contribution of mitochondrial oxidative phosphorylation (OXPHOS) to cellular oxygen consumption rates, cells were treated with 1 µmol/L of the mitochondrial complex I inhibitor rotenone to inhibit oxidative phosphorylation. The oxidative metabolic reserve of cell lines was determined using an XFp metabolic analyzer (Agilent) and calculation of the maximal increase in oxygen consumption rate above basal rates following exposure to a range of concentrations of uncoupling agent FCCP (0.125‐2 µmol/L; Cell Mito Stress Assay [Agilent]).32
2.3. Real‐time quantitative reverse transcription PCR (qRT‐PCR)
Total RNA was isolated from cells using an RNeasy mini kit (Qiagen) and quantitated by UV spectroscopy as described.33 For real‐time qRT‐PCR, 50 ng of total RNA were used per reaction with Quantitect SYBRGreen RT‐PCR kit (Qiagen). LDH‐A– and LDH‐B–specific primers and cycling conditions were as described.22
2.4. Gene expression array profiling
Whole‐genome expression profiling was performed using GeneChip Human Genome U133 Plus 2.0 Arrays (Affymetrix) of total RNA from the panel of bladder cancer cell lines (Hurst et al, manuscript in preparation). mRNA levels for pyruvate dehydrogenase kinases (PDKs) 1‐4 are presented in Figure S4. Results are shown for two different mRNA probe sets for each PDK. Affymetrix probe set identifiers are as follows: PDK1 probe 1: 206686_at, probe 2: 226452_at; PDK2 probe 1: 202590_s_at, probe 2: 213724_s_at; PDK3 probe 1: 206348_s_at, probe 2: 221957_at; PDK4 probe 1: 205960_at, probe 2: 225207_at.
2.5. Immunoblotting
Cell lysates were prepared as described34 and protein concentrations determined by the BCA assay. Equivalent protein amounts were resolved by SDS‐PAGE and electroblotted onto nitrocellulose. Primary antibodies were: anti‐LDH (Epitomics; antibody detects both LDH‐A (lower band) and LDH‐B (upper band) and has been verified for its specificity by RNAi),22 anti‐phosphorylated Y10 LDH‐A,35 anti‐aurora A and anti‐E‐cadherin (Cell Signaling Technology), anti‐PDH‐E1a (Abcam), anti‐phosphorylated S293 PDH‐E1a (Novus Biologics), anti‐phosphoERK, anti‐ERK and anti‐FGFR3 clone B9 (Santa Cruz Biotechnology), and anti‐actin (Chemicon International). Quantification of protein bands was done by densitometry analysis using Image J software (https://imagej.nih.gov) with levels normalized relative to actin as a loading control.
2.6. siRNA transfection, LDH inhibition, and apoptotic quantification
NHUC, 97‐7, RT112, UMUC3, and J82 cells were transiently transfected with siRNAs 24 hours post seeding by formulating HPLC‐purified synthetic siRNAs into liposomes using oligofectamine (Invitrogen) as previously described.22, 33, 36 LDH‐A, LDH‐B, and SIRT1 siRNA sequences have been previously validated as described.22 Representative phase contrast cell images were taken 72 hours post transfection, and protein lysates were prepared to confirm target protein knockdown. For quantification of apoptotic cell death, harvested cells were stained with annexin V and propidium iodide for 15 minutes at 37°C and analyzed by image cytometry as previously described.32 Other cell lines were treated with the LDH inhibitor NHI‐222 (15 μmol/L, Sigma) for 48 hours, and effects on cell number were compared with vehicle control–treated cells.
2.7. Patient tissue samples and immunohistochemistry
Freshly resected patient tissue samples were obtained from transurethral resection samples with the approval of the Leeds East Research Ethics Committee and informed patient consent. Specimens were weighed on parafilm, washed in fresh DMEM to remove residual lactate, and then cultured overnight at 37°C in 5% CO2 in a small volume of fresh DMEM containing 10% FBS. A small volume of culture medium was removed after 4 hours and after overnight culture for determination of lactate levels as described.22 The specimens were histologically graded and staged according to the 1973 World Health Organization recommendations and TNM classification by a trained pathologist. For immunohistochemistry, serial sections of fixed tissue specimens were blocked following antigen retrieval and then stained for target antigens using standard IHC methodology as previously described.37 Antibodies used were: GLUT1 (ab15309, Abcam), LDH‐A (19987‐1‐AP, Proteintech), MCT4 (H90, Santa Cruz Biotechnology), and MCT1 (AB3538P, Merck Millipore). Total RNA from a panel of 263 urothelial tumors was analyzed for mRNA expression levels of LDH‐A, LDH‐B, GLUT1, MCT4, and MCT1 (Supplementary Methods) (Hurst et al, manuscript in revision).
3. RESULTS
3.1. Warburg effect occurrence in bladder cancer cell lines derived from stage Ta‐T4 tumors
The Warburg effect is defined as the cellular metabolism of glucose to lactate in the presence of oxygen.10, 38 Analysis of extracellular lactate revealed that all 23 human bladder cancer cell lines tested performed aerobic glycolysis with rates of cellular lactate release differing up to ~8‐fold across the cell line panel (Figure 1A). Proliferating NHUC in culture also released lactate (Figure 1A, green dotted line) consistent with other studies showing that proliferating normal cells use aerobic glycolysis to support their growth.39 However, in confluent NHUC, which is more representative of nonproliferative, differentiated tissue, rates of lactate release were halved (Figure 1A, red dotted line).
FIGURE 1.
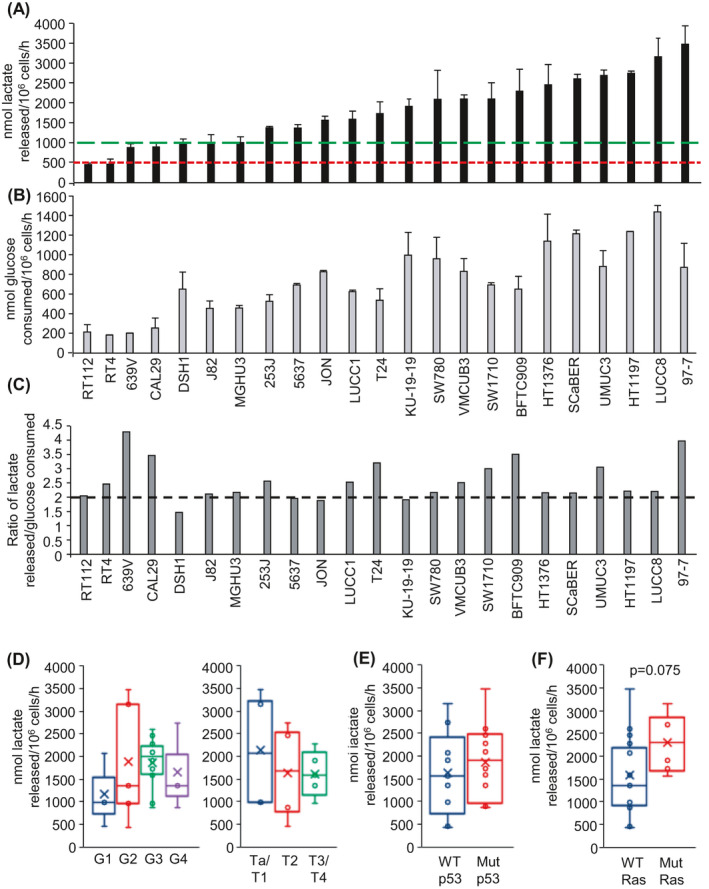
Aerobic glycolysis of bladder cancer cell lines. A, Rate of cellular lactate release into the culture media of the indicated bladder cancer cell lines (nmoles of lactate released into the cell culture media per 106 cells per hour). Values presented represent the mean ± SD from a minimum of three independent experiments. Green dotted line represents rate of lactate release by proliferating normal human urothelial cells (NHUC), and the red dotted line represents rate of lactate release by confluent NHUC. B, Rate of cellular glucose consumption from the culture media of the indicated bladder cancer cell lines (nmoles of glucose consumed per 106 cells per hour). Values presented represent the mean ± SD from a minimum of three independent experiments. C, Ratio of the rate of lactate release to the rate of glucose consumption. The ratios are calculated from the mean rates shown in (A) and (B), respectively. Dotted line indicates two molecules of lactate released per glucose consumed. D, Box and whisker plot showing the median, mean (x), and minimum and maximum rates of cellular lactate release by cell lines clustered by grade (left) or stage (right). E, Box and whisker plot showing the median, mean (x), and minimum and maximum rates of cellular lactate release by cell lines clustered by p53 status. F, Box and whisker plot showing the median, mean (x), and minimum and maximum rates of cellular lactate release by cell lines clustered by Ras mutational status
Rates of cellular glucose consumption were also determined (Figure 1B) because lactate can also be generated by glutaminolysis.40 The ratio of cellular rate of lactate release to that of glucose consumption was close to 2:1 for the majority of the cell lines (Figure 1C), consistent with lactate generation by aerobic glycolysis (this generates two molecules of lactate per molecule of glucose consumed). Several of the cell lines (eg, 639V, CAL29, BTC909, 97‐7) showed a lactate to glucose ratio greater than 2:1 suggesting some lactate generation by glutaminolysis.
Clustering of the cell lines based on the grade and stage of the tumors from which they were derived (Table S1; Ta and T1 are NMIBC; ≥T2 are MIBC) failed to indicate any statistically significant correlations with rates of cellular lactate release (Figure 1D). Cell clustering based on TP53 mutational status was also performed, as loss of p53 tumor suppressor function is reported to promote the Warburg effect;16 however, no correlation was observed with cellular rates of lactate release (Figure 1E). Another common genetic lesion in bladder cancers associated with the Warburg effect is oncogenic mutation of RAS genes.15, 17 Interestingly, median and mean rates of cellular lactate release were higher in the mutant Ras cell cluster (P = .075, Figure 1F).
In addition to mutation of TP53 and RAS genes, a high proportion of urothelial carcinomas show upregulation of FGFR1 signalling,41 and activating mutations in FGFR3 are frequently found in Ta bladder cancers.41, 42 Interestingly, FGFR1 has been reported to phosphorylate the glycolytic enzyme LDH‐A at Y10 and enhance its activity in a number of different cancer types.35 It was therefore assessed whether increased FGFR1 or FGFR3 signaling in bladder cancers has any effect on the phosphorylation of LDH‐A or rates of cellular lactate production. Using complementary knockdown and overexpression approaches, no effects were observed either on LDH‐A phosphorylation or lactate release (Figure S1). Cell line clustering based on FGFR3 mutational status showed that mean and median rates of lactate release were both lower in the mutated FGFR3 cell cluster, but no statistically significant correlation was observed (Figure S1G).
3.2. Cellular oxygen consumption rates are independent of rates of lactate release
Cellular oxidative metabolism was assessed through quantification of oxygen consumption rates for fourteen of the bladder cancer cell lines (Figure 2A). It was hypothesized that cell lines with the highest rates of lactate release may show reduced levels of oxidative metabolism and vice versa, consistent with the notion of a metabolic switch away from glucose oxidation and OXPHOS toward glycolysis.43 For RT112 and RT4 cells, which show the lowest rates of lactate release (Figure 1A), this notion loosely applies with these two cell lines having a much higher ratio of oxygen consumption to lactate release (Figure 2B) or glucose consumption (Figure S2). However, considering all the cell lines analyzed, there was no inverse correlation of oxygen consumption rates either with lactate release (Figure 2C) or glucose consumption (Figure 2D). Wild‐type p53, via SCO2, promotes OXPHOS;44 however, there was no correlation with cellular p53 or Ras status or with cell line tumor grade (Figure 2E‐G). Treatment with mitochondrial complex I inhibitor rotenone45 inhibited oxygen consumption by ~75%‐96% in the four cell lines tested (Figure S2B) indicating that the majority of oxygen consumption in these cells is due to OXPHOS rather than fatty acid oxidation.
FIGURE 2.
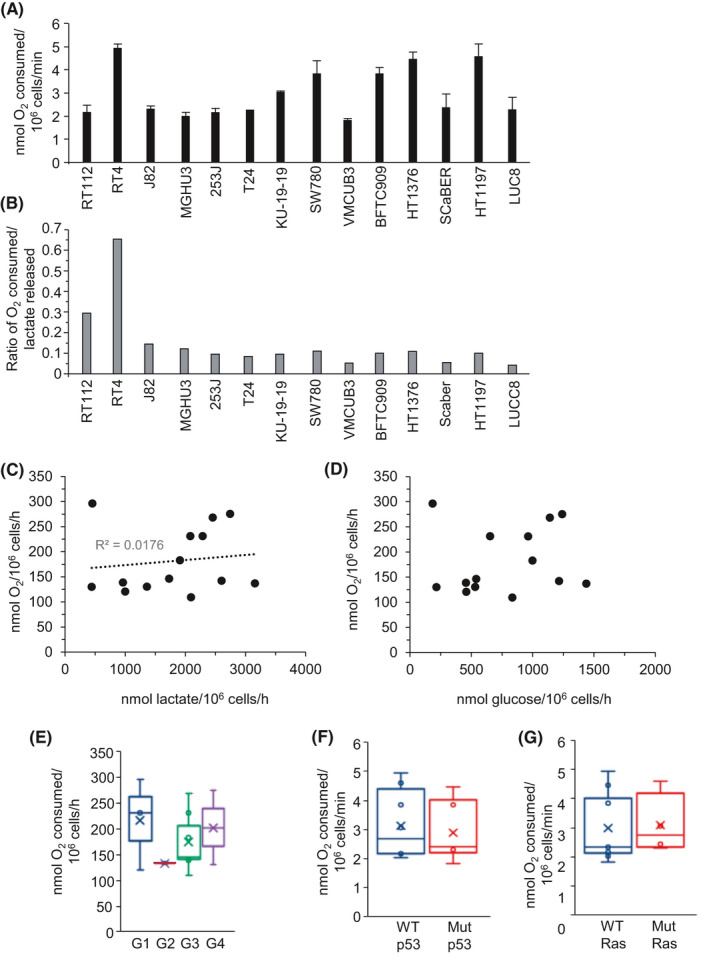
Oxygen consumption rates of bladder cancer cell lines are independent of aerobic glycolysis and cellular rates of lactate production. A, Rate of cellular oxygen consumption of the indicated human bladder cancer cell lines (nmoles of oxygen consumed per 106 cells per minute). Values presented represent the mean ± SD from three independent experiments. Cell lines are ordered from left to right on the basis of cellular lactate release (low to high rate of lactate release). B, Ratio of the rate of cellular oxygen consumption to the rate of cellular lactate release for the indicated human bladder cancer cell lines. The ratios are calculated from the mean rates shown in Figure 2A (O2 consumed) and Figure 1A (lactate released). Scatter plots of cellular oxygen consumption rate against the rate of cellular lactate release (C), or glucose consumption rate (D) with the mean rates for each individual cell line indicated by a black circle. Box and whisker plots showing the median, mean (x), and minimum and maximum rates of cell line oxygen consumption following cell line clustering by grade (E), p53 status (F), or Ras status (G)
3.3. mRNA and protein expression of key enzymes involved in the Warburg effect
The enzyme lactate dehydrogenase which is composed of LDH‐A and/or LDH‐B subunits, catalyzes the reduction of pyruvate to lactate, the last step of aerobic glycolysis. While predominance of LDH‐A subunits over LDH‐B in the LDH tetrameric complex promotes pyruvate reduction to lactate, LDH‐B typically favors the reverse reaction.46, 47, 48 Expression of LDH‐A and LDH‐B mRNA differed across the 23 bladder cancer cell lines ~17‐fold and ~64‐fold, respectively, indicating considerable heterogeneity in their expression (Figure S3A,B). The relative expression of LDH‐A compared with LDH‐B, which is predicted to influence the directionality of pyruvate/lactate interconversion, varied as much as 52‐fold across the cell line panel (Figure S3C). Relative to RT112 cells, which showed the lowest rate of lactate release of the bladder cancer cell lines, most of the cell lines expressed increased LDH‐A mRNA relative to LDH‐B, consistent with increasing rates of cellular lactate release (Figure S3D).
Similar to mRNA levels, protein expression of LDH‐A and LDH‐B varied both in absolute expression levels and in the relative levels of LDH‐A and LDH‐B across the cell line panel (Figure 3A). mRNA and protein levels of LDH‐A and LDH‐B within individual cell lines were only partially correlated, consistent with post‐transcriptional regulation of gene expression (Figure 3B,C). LDH‐A/LDH‐B mRNA and protein ratios were positively correlated (R 2 = .7809) (Figure 3D). Analysis of LDH‐A/LDH‐B protein ratio as a function of cellular lactate release rate showed that only seven of the 23 cell lines express more LDH‐B protein than LDH‐A, and that these were among the slowest lactate producers (Figure 3E). However, there was no positive linear correlation between LDH‐A/LDH‐B protein ratio of cell lines and their cellular lactate release rates (Figure 3E).
FIGURE 3.
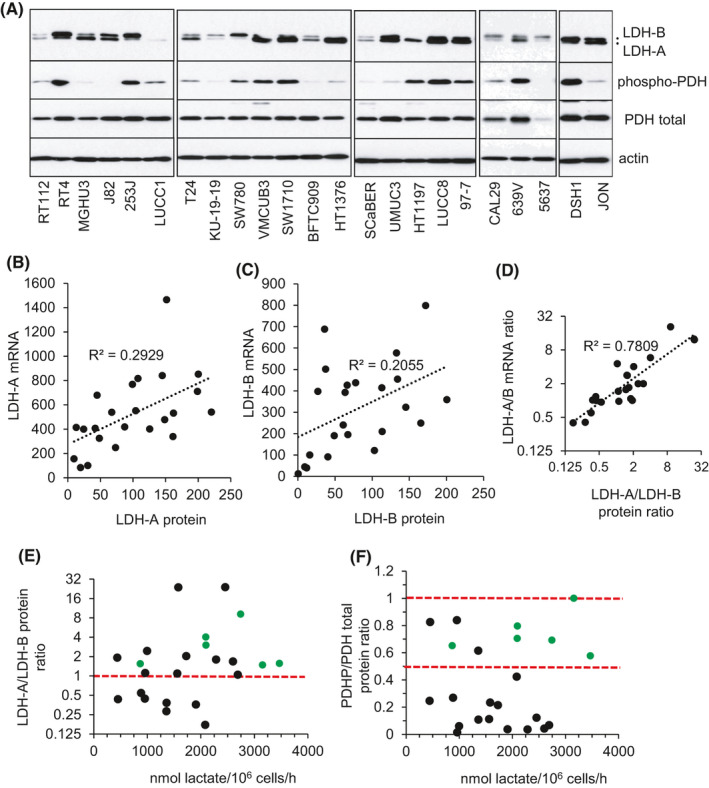
Protein expression of LDH‐A, LDH‐B, and phosphorylated pyruvate dehydrogenase (PDH) and correlation analysis with cell line rates of extracellular lactate release. A, Immunoblots showing relative protein expression levels of LDH‐A, LDH‐B, phosphorylated PDH (S293 PDH‐E1a), and total PDH in the indicated bladder cell lines. Actin was used as a loading control with equivalent exposures (normalized to RT112) for all cell line panels. Correlation analysis of cell line mRNA and protein levels for LDH‐A (B) and LDH‐B (C). Mean value for each cell line is represented by a black circle. D, Scatter plot of cell line LDH‐A/LDH‐B mRNA ratio relative to LDH‐A/LDH‐B protein ratio for each cell line. Mean value for each cell line is represented by a black circle; R 2 as indicated. E, F, Scatter plots of cell line LDH‐A/LDH‐B protein ratios and PDHP/PDH protein ratios relative to the mean rate of cellular lactate release (Figure 1A), with each individual cell line represented by a black or green circle. For the LDH‐A/LDH‐B protein ratio scatter plot, cell lines above the horizontal red dotted line express more LDH‐A protein relative to LDH‐B. PDHP/PDH total protein ratio is expressed relative to the LUCC8 cell line which showed the highest proportion of phosphorylated PDH relative to total PDH levels (LUCC8 PDHP/PDH ratio arbitrarily set at 1). Cell lines within the two horizontal red dotted lines in (F) represent cell lines with ≥50% of total PDH being phosphorylated at S293 assuming 100% phosphorylation of LUCC8 total PDH. Green circles in both scatter plots represent cell lines which have an LDH‐A/LDH‐B ratio ≥1 and a PDHP/PDH ratio ≥0.5
The Warburg effect is also promoted by inhibition of pyruvate entry into the TCA cycle through the phosphorylation and inactivation of pyruvate dehydrogenase (PDH) by PDKs 1‐4.38, 49 Like LDH‐A and LDH‐B, mRNA expression of PDKs 1‐4 differed considerably between cell lines (Figure S4), and phosphorylated PDH levels (PDHP) similarly varied (Figure 3A). Analysis of the ratio of PDHP to total PDH failed to show a clear correlation with cellular rates of lactate release (Figure 3F). These expression results (LDH, PDHP) highlight the complexity of regulation of the Warburg effect and the limitations of using a surrogate expression marker and indicate the need to measure metabolic flux directly for an accurate readout of the magnitude of any Warburg effect.
3.4. LDH‐A silencing impairs growth and survival of bladder cancer cell lines
As LDH‐A was detectable in all of the bladder cancer cell lines, albeit at different expression levels (Figure 3A), the effects of its depletion by RNAi were analyzed. Studies in other cancer types have shown LDH‐A dependency.19, 20, 22 Using a previously validated LDH‐A siRNA,22 immunoblots indicated selective depletion of LDH‐A protein (Figure 4A; NHUC and RT112 LDH blots are overexposed relative to the 97‐7 and UMUC3 blots for ease of comparison of knockdown levels). LDH‐A silencing had no discernible effect on NHUCs (Figure 4B). In bladder cancer cell lines (97‐7: highest rate of lactate release; RT112: lowest rate of lactate release [Figure 1A]; UMUC3; and J82 cells), cell numbers were significantly reduced by LDH‐A depletion which correlated with increased apoptotic cell death (Figure 4B,C; Figure S5). Unexpectedly, in J82 cells LDH‐B silencing induced higher levels of apoptosis than LDH‐A silencing (Figure S5). Interestingly, further preliminary investigation showed that J82 cells express Aurora A kinase (Figure S5), which has recently been shown to phosphorylate LDH‐B resulting in LDH‐B promoting aerobic glycolysis.48 Against HT1197 and HT1376 bladder cancer cells, LDH inhibitor NHI‐222 adversely affected cell growth compared with normal urothelial cells and resulted in a ~2.5‐fold increase in the proportion of late‐stage apoptotic cells in the HT1376 cells (Figure S5).
FIGURE 4.
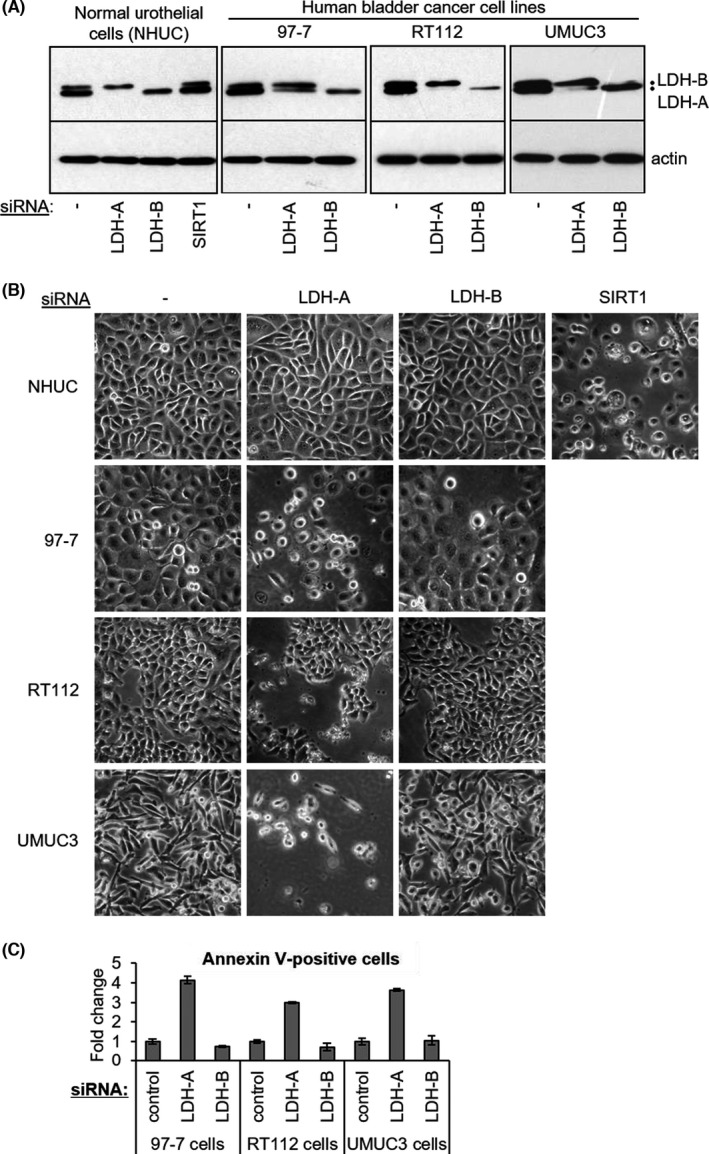
LDH‐A knockdown induces apoptotic cell death of bladder cancer cell lines but not of normal human urothelial cells. A, Immunoblots showing selective depletion of LDH‐A protein by LDH‐A siRNA in the indicated cell lines and effects of LDH‐B siRNA at 72 h following siRNA transfection. LDH‐A/LDH‐B blots for normal human urothelial cells (NHUC) and RT112 bladder cancer cells are overexposed relative to the 97‐7 and UMUC3 blots in order to obtain similar LDH‐A/B protein levels in control‐transfected cells (no siRNA) for all three lines to facilitate comparison of extent of LDH‐A knockdown between the cell lines. Actin was used as a loading control. As a positive control for cell death induction in NHUC, these cells were additionally treated with SIRT1 siRNA. B, Representative phase contrast images of 97‐7, RT112, and UMUC3 human bladder cancer cells and of normal human urothelial cells (NHUCs) 72 h post transfection with the indicated siRNAs. C, Quantification of apoptotic cell death induced by LDH‐A and LDH‐B siRNAs 72 h post transfection as determined by the proportion of annexin V–positive cells; values are expressed as a fold change in the proportion of annexin V–positive cells relative to background levels in control cells
Further metabolic analyses in several of the bladder cancer cell lines show a lack of any, or little, oxidative metabolic reserve compared with normal urothelial cells (Figure S6). This suggests that while normal urothelial cells may be able to metabolically compensate to targeting of aerobic glycolysis (eg, via LDH‐A), the bladder cancer cell lines have little oxidative metabolic reserve to be able to energetically compensate to sustain their growth and survival.
3.5. Cellular lactate production is increased in response to FGFR1‐induced EMT
Ectopic expression and activation of FGFR1 in the urothelial carcinoma cell line 94‐10 induces epithelial‐mesenchymal transition (EMT).31 As increased extracellular lactate has been reported to promote loss of cell‐cell adhesion that is important for EMT and cancer cell migration through extracellular acidification,50, 51, 52 it was investigated what effect EMT induction via FGFR1 had on cellular lactate production. FGFR1 activation in transfected 94‐10 cells, by FGF2, or heparin and FGF2,31 induced ERK phosphorylation, which is downstream of FGFR1 signaling, loss of E‐cadherin (Figure 5A), and a change in cell morphology from epithelial to mesenchymal‐like (Figure 5B). Significantly, the cells induced to undergo EMT also displayed increased rates of lactate production (Figure 5C, P < .001). Similar results were also observed in J82 cells induced to undergo EMT (Figure S7). Future studies will investigate this further and the potential of targeting lactate release to suppress EMT.
FIGURE 5.
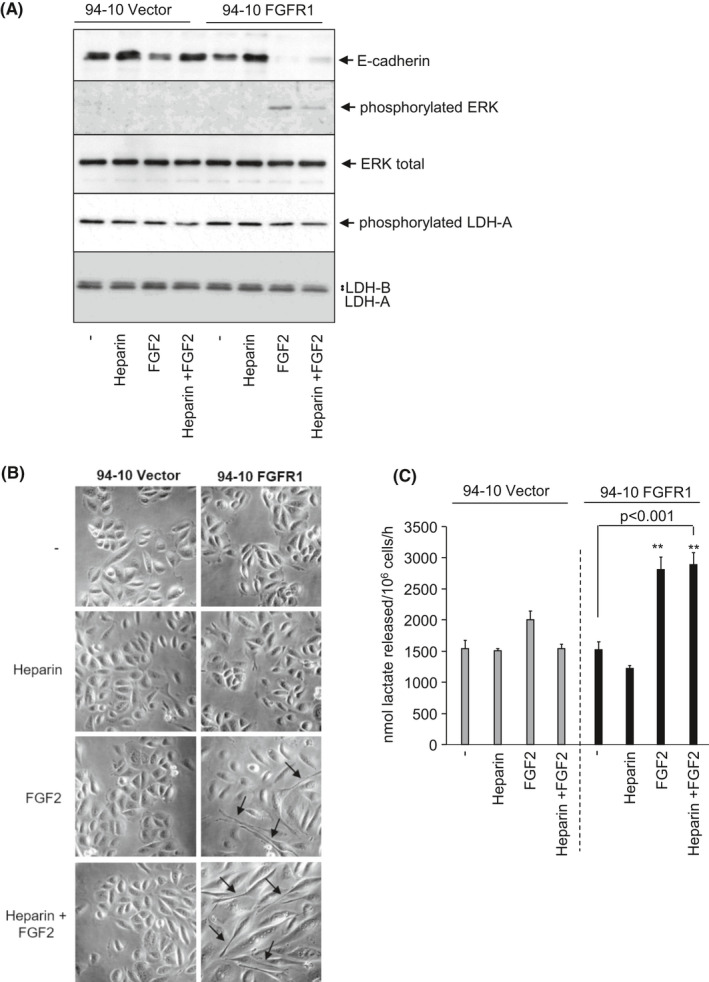
Enhanced rates of lactate release following epithelial mesenchymal transition (EMT) induction in an in vitro FGFR1‐inducible bladder cancer model of EMT. A, Immunoblots showing the induction of FGFR1 signaling in FGFR1‐expressing 94‐10 human bladder cancer cells by FGF2, or FGF2 plus heparin, as indicated by increased phosphorylated ERK and increased EMT as indicated by decreased E‐cadherin expression. Effect of FGFR1‐induced EMT on LDH‐A, LDH‐B, and LDH‐A phosphorylation levels are also shown. B, Phase contrast images showing the induction of a mesenchymal‐like morphology (indicated by black arrows) following treatment of FGFR1‐expressing 94‐10 human bladder cancer cells with FGF2 or FGF2 plus heparin, consistent with EMT. C, Effect of EMT induction on the rate of cellular lactate release (nmoles lactate released/106 cells/hour). **P < .001, Student's t‐test
3.6. Lactate release by freshly resected Ta, T1, and T2 bladder cancer tissue and heterogeneous intratumoral expression of key enzymes and transporters important for the Warburg effect
To assess whether in vitro cell line observations of the Warburg effect are relevant in a clinical setting, freshly resected bladder cancer tissue was analyzed for lactate release. All the resected bladder cancer tissues (Ta, T1, T2) released lactate into the cell culture media (Figure 6A). Ta clinical specimens displayed higher rates of lactate release than T1 or T2 specimens (P < .05 and P < .01) although the number of specimens analyzed of different stages is low, and this should therefore be regarded as preliminary data (Figure 6B). Serial sections of tissue specimens analyzed for lactate release were assessed for expression of potential therapeutic targets linked to the Warburg effect (Figure 6C). Positive cytoplasmic staining was observed for LDH‐A in Ta, T1, and T2 specimens. For glucose transporter GLUT1 and lactate export transporter MCT4, membranous and/or cytoplasmic staining was obtained. Expression of all three potential targets (LDH‐A, MCT4, GLUT1) was heterogeneous across sections. For example, in the T1 G3 “2165” specimen (Figure 6C), increased expression and colocalization of LDH‐A, GLUT1, and MCT4 are evident further away from a blood vessel, which is consistent with reduced oxygen supply to these cells and increased dependency on aerobic glycolysis. Similar heterogeneous patterns of expression were also observed in the other specimens with some areas of sections showing only weak positive staining for these targets (Figure 6C). Staining for the lactate import transporter MCT153 was mostly negative; however, a small number of cells within sections did positively stain for MCT1 (eg, T2 G3 “2143” specimen; Figure 6C). This raises the possibility of metabolic coupling between bladder cancer cells in the context of the pathophysiological tumor microenvironment (TME); for example, glycolytic cancer cells exporting lactate that is imported by MCT1‐expressing cancer cells for lactate‐fueled OXPHOS.
FIGURE 6.
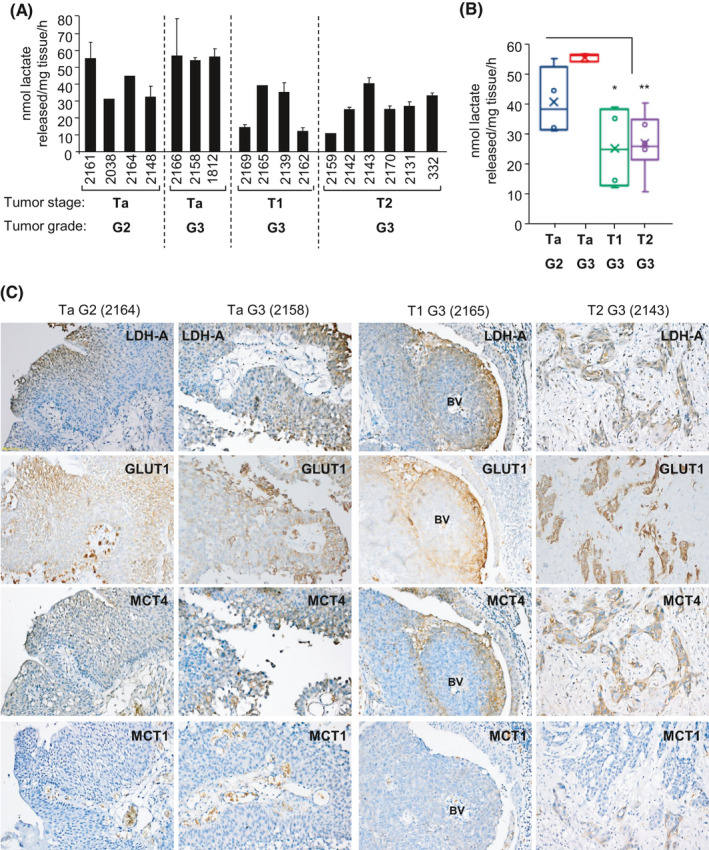
Lactate release by freshly resected bladder cancer tissue and intratumoral heterogeneous expression of key targets associated with the Warburg effect. A, Mean rate of lactate release into fresh growth medium per milligram of tumor tissue per hour. Stage and grading of tumor tissue as indicated. Freshly resected TURBTs were weighed prior to overnight (16 h) incubation in 500 µL of fresh growth media at 37°C in a 5% CO2 incubator and determination of levels of lactate released into the medium during the incubation period (see "Methods” section). Four‐ (or three‐) digit numbers indicate anonymized clinical specimen identifiers for different patient samples. Mean ± SD from three independent lactate determinations. B, Box plot of the mean rates of cellular lactate release by clinical specimens clustered by stage and grade, *P < .05, **P < .01, Student's t‐test. C, Tissue sections of the indicated clinical specimens (analyzed in [A] for lactate release) that have been immunostained for the indicated metabolic targets. Representative IHC images are shown; location of blood vessel (BV) indicated for 2165 specimens
A panel of 263 urothelial tumors was further analyzed according to stage for mRNA expression of Warburg effect–associated putative targets LDH‐A, GLUT1, and lactate export transporter MCT4 as well as for LDH‐B and lactate import transporter MCT1 (Figure 7). Mean and median LDH‐A expression was higher in MIBCs than Ta and T1 tumors, and MIBCs showed an increased LDH‐A/LDH‐B ratio. However, GLUT1 expression was higher in Ta tumors, while there was no significant difference in expression of lactate export transporter MCT4 across the tumor stages (Figure 7). Overall, the analyses do not clearly indicate predominance of the Warburg effect in any tumor stage but rather that all have upregulation, suggesting that this may be an early requirement during tumor development. Expression of lactate import transporter MCT1 was modestly increased in T1 and some MIBCs compared with Ta, which may account for decreased lactate release in some specimens analyzed compared with Ta (Figure 6A) and is further supportive of metabolic coupling in the TME. Figure S8 shows association of expression of the different putative targets with patient overall survival following analysis of the publicly available urothelial cancer TCGA dataset (n = 408; R2 Genomics Analysis Platform, http://r2.amc.nl) with higher expression generally associated with worse overall survival (except for LDH‐B).
FIGURE 7.
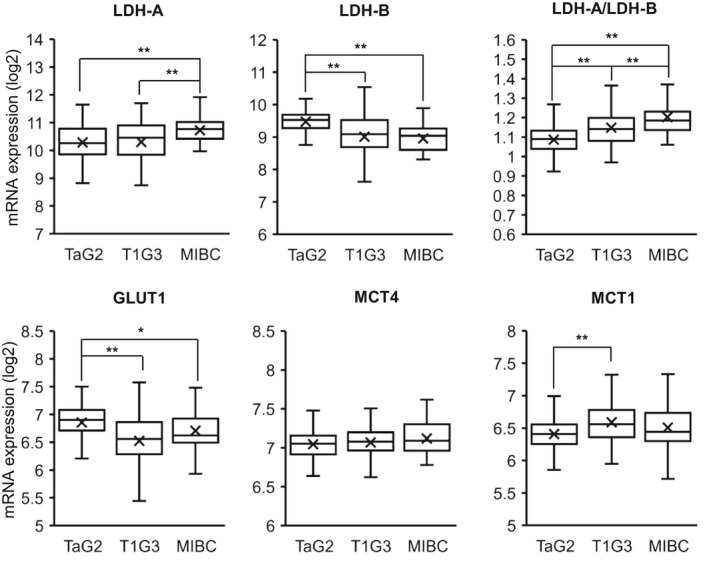
mRNA expression of putative targets associated with the Warburg effect in Ta, T1, and muscle‐invasive bladder cancer (MIBC) clinical specimens. mRNA expression analysis (log2) of the indicated targets associated with the Warburg effect (LDH‐A, GLUT1, MCT4) and intratumoral metabolic coupling (lactate import transporter MCT1) in 263 urothelial tumor specimens (125 Ta G2, 106 T1 G3, and 32 MIBC clinical specimens). LDH‐A/LDH‐B indicates ratio of LDH‐A expression relative to LDH‐B. Box and whisker plots showing the median, mean (x), and minimum and maximum expression following clustering by stage. *P < .05, **P < .01; Student's t‐test
4. DISCUSSION
Treatments and outcomes for both NMIBCs and MIBCs have changed little over the past few decades, and there is an urgent need for new therapeutic approaches. Recent studies have identified several distinct genomic subtypes of both NMIBC and MIBC based on their mutational and gene copy number signatures.25, 37 It is expected that this will facilitate the development of targeted therapies against the specific subtypes. A key finding from this study is that aerobic glycolysis (the Warburg effect) is a feature of both NMIBCs and MIBCs (Figures 1, 6, and 7) indicating opportunity to develop targeted therapies exploiting this for both NMIBC and MIBC.
The Warburg effect in NMIBC is consistent with the recent identification of a NMIBC genomic subtype that is characterized by loss of chromosome region 9q and tumor suppressor TSC1 and upregulated mTORC1 signaling, which would be predicted to stimulate aerobic glycolysis.37 Genetic alterations to a number of different tumor suppressor genes and proto‐oncogenes are known to stimulate aerobic glycolysis.15, 16, 17 This includes loss of p53 and TSC1 tumor suppressor functions and activating mutations in PIK3CA, RAS, and AKT, all of which are found in different bladder cancer subtypes. Consistent with this, aerobic glycolysis, as indicated by extracellular lactate release, was evident in all bladder cancer cell lines analyzed (Figure 1) with only RT112 cells (lowest rate of cellular lactate release, Figure 1) lacking a mutation in any of these genes (Table S2). It is likely that this metabolic switch occurs via different genetic routes in different cell lines, which may account for the wide variation in rates of extracellular lactate release observed across the panel, with different integrating promoting (and inhibitory) contributions by different combinations of genetic mutations. In support of this, cell line clustering based on mutation status of three key genes commonly mutated in bladder cancers, TP53, RAS, and FGFR3, failed to indicate any significant correlation with rates of lactate release. Interestingly, median and mean lactate release rates were higher in the mutant Ras cell cluster, and although this was not statistically significant, it indicates a positive trend that is consistent with mutant Ras promoting aerobic glycolysis in other cancers. It is also noteworthy that the two bladder cancer cell lines with the lowest rate of lactate release and largest ratio of oxygen consumed to lactate released (indicating increased OXPHOS relative to aerobic glycolysis), RT112 and RT4, harbor FGFR3‐TACC3 genetic fusions54 (Table S2). The FGFR3‐TACC3 oncogenic fusion has recently been reported to increase OXPHOS and mitochondrial biogenesis.55
It is a commonly held view that cancer cells undergo a metabolic switch from OXPHOS to aerobic glycolysis.10, 43 Importantly, metabolic analyses of bladder cancer cell lines here show that cellular lactate release rates and oxygen consumption rates do not inversely correlate (Figure 2). Indeed, there is some indication of a weak positive correlation (Figure 2C). This supports metabolic analyses of tumors in vivo which suggests the metabolic switch involves upregulation of both aerobic glycolysis and mitochondrial respiration.10
The bladder cancer cell line studies identify for the first time the glycolytic enzyme LDH‐A as a potential, and cancer‐selective, therapeutic target for bladder cancers with normal urothelial cells shown to be refractory to LDH‐A knockdown (Figure 4). Bladder cancer cells were shown to have little or no oxidative metabolic reserve to compensate for inhibition of aerobic glycolysis (Figure S6). In bladder cancer clinical specimens, release of extracellular lactate was observed indicating that aerobic glycolysis is a metabolic feature of bladder cancer tissue that could potentially be targeted (Figures 6 and 7). LDH‐A as well as other targets linked to the Warburg effect (MCT4, GLUT1) are expressed in both NMIBC and MIBC (Figures 6 and 7), but protein expression was heterogeneous in IHC sections. This intratumoral heterogeneity suggests that some tumor regions may be carrying out aerobic glycolysis, whereas others may not. This raises the possibility of metabolic symbiosis or coupling between cancer cells in different regions of the same tumor as was first proposed and elegantly demonstrated by Sonveaux and colleagues.53, 56, 57 In support of this in the context of bladder cancers, sections showed increased staining for LDH‐A, GLUT1, and MCT4 furthest from blood vessels, consistent with tumor hypoxia and increased dependency on aerobic glycolysis. In contrast, cells nearer blood vessels could potentially utilize lactate produced from hypoxic cells as a fuel for OXPHOS,53, 56, 57 and this may account for the relatively low rates of extracellular lactate release shown by the clinical specimens compared with the cell lines. The extracellular release of lactate, in addition to potentially being utilized as a fuel,53, 56, 57 could also potentially facilitate the migration of glycolytic or hypoxic bladder cancer cells toward blood vessels or a more favorable environment.50, 51, 52 In two inducible EMT models, EMT induction, an important step in cells becoming more migratory, was positively associated with increased extracellular lactate release (Figure 5; Figure S7). While further studies are needed to fully understand this, it raises the possibility that the targeting of aerobic glycolysis and decreased lactate release might also be beneficial as a barrier against bladder cancer cell migration and invasion.
In summary, this work identifies aerobic glycolysis as a targetable feature of both NMIBC and MIBC. Intratumoral heterogeneity in expression of potential targets exemplifies the importance of the TME as well as cancer cell genetics, with aerobic glycolysis inhibitors (eg, LDH‐A inhibitors) predicted to be most effective against hypoxic regions of tumors. Intratumoral heterogeneity and possible lactate shuttling from glycolytic cancer cells to fuel OXPHOS in other cancer cells indicates the likely need to use aerobic glycolysis inhibitors in combination with other metabolic inhibitors (eg, of OXPHOS) for effective therapeutic targeting of bladder cancers. However, the option of intravesical delivery to the bladder may enable effective metabolic inhibitor combinations unavailable to treat other cancer types that would otherwise be toxic with systemic administration.
DISCLOSURE
The authors declare that they have no conflict of interest.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Fig S7
Fig S8
Table S1
Table S2
Supplementary Material
ACKNOWLEDGEMENTS
This work was funded by a Yorkshire Cancer Research pump priming grant (BPP028) awarded to RMP and MK and University of Huddersfield funding to SJA. We thank Joanne Brown for collection of freshly resected urothelial carcinoma tissue.
Burns JE, Hurst CD, Knowles MA, Phillips RM, Allison SJ. The Warburg effect as a therapeutic target for bladder cancers and intratumoral heterogeneity in associated molecular targets. Cancer Sci. 2021;112:3822–3834. 10.1111/cas.15047
REFERENCES
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 2.Sternberg CN, Bellmunt J, Sonpavde G, et al. ICUD‐EAU International Consultation on Bladder Cancer 2012: chemotherapy for urothelial carcinoma‐neoadjuvant and adjuvant settings. Eur Urol. 2013;63:58‐66. [DOI] [PubMed] [Google Scholar]
- 3.Babjuk M, Böhle A, Burger M, et al. EAU guidelines on non‐muscle‐invasive urothelial carcinoma of the bladder: update 2016. Eur Urol. 2017;71:447‐461. [DOI] [PubMed] [Google Scholar]
- 4.Casadei C, Dizman N, Schepisi G, et al. Targeted therapies for advanced bladder cancer: new strategies with FGFR inhibitors. Ther Adv Med Oncol. 2019;11:1758835919890285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Kessel KE, Zuiverloon TC, Alberts AR, Boormans JL, Zwarthoff EC. Targeted therapies in bladder cancer: an overview of in vivo research. Nat Rev Urol. 2015;12:681‐694. [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 7.Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Golub D, Iyengar N, Dogra S, et al. Mutant isocitrate dehydrogenase inhibitors as targeted cancer therapeutics. Front Oncol. 2019;9:417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tennant DA, Duran RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010;10:267‐277. [DOI] [PubMed] [Google Scholar]
- 10.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2:e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luengo A, Gui DY, Vander Heiden MG. Targeting metabolism for cancer therapy. Cell Chem Biol. 2017;24:1161‐1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov. 2013;12:829‐846. [DOI] [PubMed] [Google Scholar]
- 13.Griffiths HBS, Williams C, King SJ, Allison SJ. Nicotinamide adenine dinucleotide (NAD+): essential redox metabolite, co‐substrate and an anti‐cancer and anti‐ageing therapeutic target. Biochem Soc Trans. 2020;48:733‐744. [DOI] [PubMed] [Google Scholar]
- 14.Vander Heiden MG, DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell. 2017;168:657‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dang CV, Semenza GL. Oncogenic alterations of metabolism. Trends Biochem Sci. 1999;24:68‐72. [DOI] [PubMed] [Google Scholar]
- 16.Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9:691‐700. [DOI] [PubMed] [Google Scholar]
- 17.Nagarajan A, Malvi P, Wajapeyee N. Oncogene‐directed alterations in cancer cell metabolism. Trends Cancer. 2016;2:365‐377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolpaw AJ, Dang CV. Exploiting metabolic vulnerabilities of cancer with precision and accuracy. Trends Cell Biol. 2018;28:201‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fantin VR, St‐Pierre J, Leder P. Attenuation of LDH‐A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425‐434. [DOI] [PubMed] [Google Scholar]
- 20.Le A, Cooper CR, Gouw AM, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci USA. 2010;107:2037‐2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patra K, Wang QI, Bhaskar P, et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell. 2013;24:213‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allison SJ, Knight JRP, Granchi C, et al. Identification of LDH‐A as a therapeutic target for cancer cell killing via (i) p53/NAD(H)‐dependent and (ii) p53‐independent pathways. Oncogenesis. 2014;3:e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonnet S, Archer SL, Allalunis‐Turner J, et al. A mitochondria‐K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37‐51. [DOI] [PubMed] [Google Scholar]
- 24.Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic‐targeting therapy for cancer. Br J Cancer. 2008;99:989‐994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robertson AG, Kim J, Al‐Ahmadie H, et al. Comprehensive molecular characterization of muscle‐invasive bladder cancer. Cell. 2017;171:540‐556 e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sarkar S, Julicher KP, Burger MS, et al. Different combinations of genetic/epigenetic alterations inactivate the p53 and pRb pathways in invasive human bladder cancers. Cancer Res. 2000;60:3862‐3871. [PubMed] [Google Scholar]
- 27.Yeager TR, DeVries S, Jarrard DF, et al. Overcoming cellular senescence in human cancer pathogenesis. Genes Dev. 1998;12:163‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Faveri LE, Hurst CD, Platt FM, et al. Putative tumour suppressor gene necdin is hypermethylated and mutated in human cancer. Br J Cancer. 2013;108:1368‐1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Earl J, Rico D, Carrillo‐de‐Santa‐Pau E, et al. The UBC‐40 Urothelial Bladder Cancer cell line index: a genomic resource for functional studies. BMC Genom. 2015;16:403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chapman EJ, Hurst CD, Pitt E, Chambers P, Aveyard JS, Knowles MA. Expression of hTERT immortalises normal human urothelial cells without inactivation of the p16/Rb pathway. Oncogene. 2006;25:5037‐5045. [DOI] [PubMed] [Google Scholar]
- 31.Tomlinson DC, Baxter EW, Loadman PM, Hull MA, Knowles MA. FGFR1‐induced epithelial to mesenchymal transition through MAPK/PLCgamma/COX‐2‐mediated mechanisms. PLoS One. 2012;7:e38972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Polson ES, Kuchler VB, Abbosh C, et al. KHS101 disrupts energy metabolism in human glioblastoma cells and reduces tumor growth in mice. Sci Transl Med. 2018;10:eaar2718. [DOI] [PubMed] [Google Scholar]
- 33.Allison SJ, Milner J. RNA interference by single‐ and double‐stranded siRNA With a DNA extension containing a 3' nuclease‐resistant mini‐hairpin structure. Mol Ther Nucleic Acids. 2014;2:e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Knight JR, Allison SJ, Milner J. Active regulator of SIRT1 is required for cancer cell survival but not for SIRT1 activity. Open Biol. 2013;3:130130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fan J, Hitosugi T, Chung T‐W, et al. Tyrosine phosphorylation of lactate dehydrogenase A is important for NADH/NAD(+) redox homeostasis in cancer cells. Mol Cell Biol. 2011;31:4938‐4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Allison SJ, Milner J. SIRT3 is pro‐apoptotic and participates in distinct basal apoptotic pathways. Cell Cycle. 2007;6:2669‐2677. [DOI] [PubMed] [Google Scholar]
- 37.Hurst CD, Alder O, Platt FM, et al. Genomic subtypes of non‐invasive bladder cancer with distinct metabolic profile and female gender bias in KDM6A mutation frequency. Cancer Cell. 2017;32:701‐715.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim JW, Dang CV. Cancer's molecular sweet tooth and the Warburg effect. Cancer Res. 2006;66:8927‐8930. [DOI] [PubMed] [Google Scholar]
- 39.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029‐1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeBerardinis RJ, Mancuso A, Daikhin E, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA. 2007;104:19345‐19350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.di Martino E, Tomlinson DC, Knowles MA. A decade of FGF receptor research in bladder cancer: past, present, and future challenges. Adv Urol. 2012;2012:429213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.di Martino E, Tomlinson DC, Williams SV, Knowles MA. A place for precision medicine in bladder cancer: targeting the FGFRs. Future Oncol. 2016;12:2243‐2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev. 2008;18:54‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matoba S, Kang JG, Patino WD, et al. p53 regulates mitochondrial respiration. Science. 2006;312:1650‐1653. [DOI] [PubMed] [Google Scholar]
- 45.Heinz S, Freyberger A, Lawrenz B, Schladt L, Schmuck G, Ellinger‐Ziegelbauer H. Mechanistic investigations of the mitochondrial complex I inhibitor rotenone in the context of pharmacological and safety evaluation. Sci Rep. 2017;7:45465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Markert CL, Shaklee JB, Whitt GS. Evolution of a gene. Multiple genes for LDH isozymes provide a model of the evolution of gene structure, function and regulation. Science. 1975;189:102‐114. [DOI] [PubMed] [Google Scholar]
- 47.Read JA, Winter VJ, Eszes CM, Sessions RB, Brady RL. Structural basis for altered activity of M‐ and H‐isozyme forms of human lactate dehydrogenase. Proteins. 2001;43:175‐185. [DOI] [PubMed] [Google Scholar]
- 48.Cheng A, Zhang P, Wang BO, et al. Aurora‐A mediated phosphorylation of LDHB promotes glycolysis and tumor progression by relieving the substrate‐inhibition effect. Nat Commun. 2019;10:5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stacpoole PW. Therapeutic targeting of the pyruvate dehydrogenase complex/pyruvate dehydrogenase kinase (PDC/PDK) axis in cancer. J Natl Cancer Inst. 2017;109:djx071. [DOI] [PubMed] [Google Scholar]
- 50.Kato Y, Ozawa S, Miyamoto C, et al. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013;13:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suzuki A, Maeda T, Baba Y, Shimamura K, Kato Y. Acidic extracellular pH promotes epithelial mesenchymal transition in Lewis lung carcinoma model. Cancer Cell Int. 2014;14:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bonuccelli G, Tsirigos A, Whitaker‐Menezes D, et al. Ketones and lactate "fuel" tumor growth and metastasis: evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle. 2010;9:3506‐3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest. 2013;123:3685‐3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Knowles MA. FGFR3—a central player in bladder cancer pathogenesis? Bladder Cancer. 2020;6:403‐423. [Google Scholar]
- 55.Frattini V, Pagnotta SM, Tala , et al. A metabolic function of FGFR3‐TACC3 gene fusions in cancer. Nature. 2018;553:222‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sonveaux P, Végran F, Schroeder T, et al. Targeting lactate‐fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118:3930‐3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Semenza GL. Tumor metabolism: cancer cells give and take lactate. J Clin Invest. 2008;118:3835‐3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Fig S7
Fig S8
Table S1
Table S2
Supplementary Material