

Abstract
pET expression plasmids are widely used in the biotechnology, biopharmaceutical, and basic research sectors for the production of recombinant proteins. Typically, they are used off-the-shelf because they support high production titers; however, we have identified two design flaws in many pET plasmids that limit their production capacity. We used modern methods of DNA assembly and directed evolution to identify improved designs for these modules and demonstrated that these designs support higher protein production yields. Herein, we present two PCR protocols for implementing the designs and increasing protein production from existing pET expression plasmids.
Graphic abstract:
A simple workflow for implementing novel designs in pET expression plasmids.
Keywords: pET, Plasmid, T7lac, Transcription initiation, Translation initiation region (TIR), Synthetic evolution, Bacterial cell factory, Recombinant protein
Background
The basic architecture of pET expression plasmids was established over three decades ago by integrating the φ10 promoter for the T7 RNA polymerase (T7p) and the Tφ transcription terminator (T7t) into the pBR322 backbone ( Rosenberg et al., 1987 ). This architecture enables efficient transcription of cloned coding sequences in bacterial strains harbouring an inducible copy of the DE3 phage fragment encoding the T7 RNA polymerase. The basic pET vector architecture was then elaborated on by including optional add-ons. For example, a Shine-Dalgarno (SD) sequence originating from the major capsid protein of T7 (gene 10 protein) was incorporated to enable efficient translation initiation ( Rosenberg et al., 1987 ), and the lac O1 operator sequence was cloned adjacent to the T7 promoter (T7lac) so that basal gene expression was repressed in the absence of an inducer (Dubendorf and Studier, 1991). Alternative antibiotic cassettes and purification, solubility, and secretion tags were also included. Currently, 103 different pET expression plasmids are available (Shilling et al. 2020). It is also possible to construct bespoke T7p-based expression plasmids using the modular platform for Standard European Vector Architecture (SEVA) (Silva- Rocha et al., 2013 ).
pET expression plasmids are currently used ‘off-the-shelf’ because they are known to support high titers of recombinant protein production - as much as 50% of the total cell protein after a few hours of induction ( Mierendorf et al., 1998 ). Titers can also be increased by screening induction conditions or testing bacterial hosts that are supplemented with tRNAs and folding catalysts or that modulate the expression of T7 RNA polymerase (Rosano and Ceccarelli, 2014; Rosano et al., 2019 ). However, not all recombinant proteins can be expressed at high titers and many fall out of experimental pipelines. For example, analysis of structural genomics pipelines indicated that more than 40% of soluble proteins cannot be produced in sufficient titers for downstream structural, biochemical, and biophysical studies (Walsh, 2015; Parret et al., 2016 ).
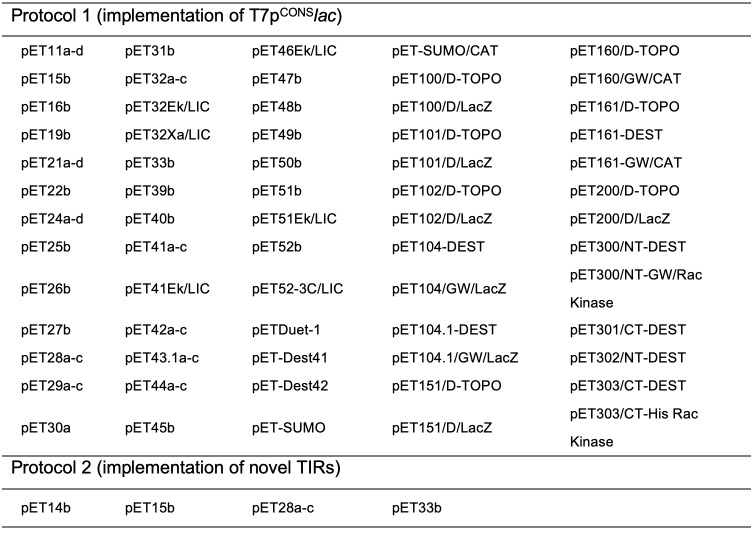
In a recent study, we identified a design flaw in the T7lac module ( Shilling et al., 2020 ). This module was originally engineered by fusing T7p to the lac O1 operator sequence in the early generation pET plasmids (Dubendorf and Studier, 1991). T7p is typically 23 nucleotides long and sits −17 to +6 relative to the messenger RNA (mRNA) start site (Figure 1) ( Dunn et al., 1983 ). However, it was truncated by four nucleotides when lac O1 was fused, as the architects used an StuI restriction site within T7p. In our previous study, we used an overlap PCR approach to insert the four truncated nucleotides into T7lac in the most commonly used pET expression plasmid, pET28a(+). We subsequently demonstrated that this design ( T7pCONSlac ) increased the production titers of recombinant proteins ( Shilling et al., 2020 ). Herein, we present a protocol for incorporating the T7pCONSlac design in vectors with an existing T7lac module (Protocol 1). This protocol is directly applicable to 88 different pET plasmids (Table 1), as well as the +LacIq-PT7/LacO (SEVA#4E) module of the pSEVA platform (Silva- Rocha et al., 2013 ). The remaining 15 pET vectors encode T7pCONS and do not include the lac O1 operator sequence.
Figure 1. Comparison of T7 promoters.

The T7 promoter in pET28a(+) and 87 other pET plasmids is a truncated variant fused to the lac operator. Protocol 1 uses an overlap PCR approach to insert four nucleotides into the T7 promoter, thus restoring the consensus sequence and increasing production titers. Figure adapted from Shilling et al. (2020) .
Table 1. pET plasmids that will benefit from each protocol.
|
Our recent study also identified a design flaw in the translation initiation region (TIR) of pET28a(+). This module is a stretch of 30 nucleotides that is recognised by the 30S subunit of the ribosome during translation initiation (i.e., the first ribosomal footprint). The sequence determines the efficiency of translation initiation, the rate-limiting step in protein synthesis (McCarthy and Gualerzi, 1990; Laursen et al., 2005 ; Milón and Rodnina, 2012), and significantly affects the production titers of recombinant proteins ( Mirzadeh et al., 2015 , 2016 and 2020; Shilling et al., 2020 ). The sequence is usually comprised of: (1) a Shine-Dalgarno sequence complementary to the 16S rRNA subunit; (2) an AUG start codon that is situated 5-9 nucleotides downstream; and (3) the first 5 codons of the coding sequence (Shine and Dalgarno, 1975; Chen et al., 1994 ; Osterman et al., 2013 ). A large body of work indicates that the TIR works most effectively when it is largely free of mRNA structures, which promotes accessibility of the 30S subunit ( Kudla et al., 2009 ; Plotkin and Kudla, 2011; Bentele et al., 2013 ; Goodman et al., 2013 ). In most pET expression plasmids, this region is comprised of the SD sequence and a seven-nucleotide spacer region from the major capsid protein of T7, and the first five codons of the coding sequence. However, there is no indication that this region has been optimised in any pET expression plasmid, which we considered a design flaw. We therefore carried out a directed evolution approach on the TIR in pET28a(+), which encodes an N-terminal poly-histidine tag and a thrombin protease cleavage site (Figure 2). We identified two TIRs that work more efficiently than the existing TIR, as judged by the fact that they increase protein production titers ( Shilling et al., 2020 ). Herein, we present a protocol for incorporating the improved TIRs (Protocol 2), which is directly applicable to four of the most widely used pET plasmids (Table 1). Utilising the optimisation strategy, we noted improvements to sfGFP expression levels, starting from a low of 0.8 mg/ml to a high of 97 mg/ml, without affecting protein quality ( Shilling et al., 2020 ). In instances where protein expression was already determined to be high, the addition of optimised TIRs described in this protocol did not always result in an increase in protein yield (unpublished data).
Figure 2. Features of the pET28a(+) plasmid and position of oligonucleotides used in the two PCR protocols.

The pET28a plasmid includes the φ10 (T7) promoter and the lac operator, as well as the translation initiation region (TIR) encompassing the Shine-Dalgarno (SD) sequence, a spacer, and the first five codons of the open reading frame. Protocol 1 oligonucleotides (blue) incorporate four nucleotides within the T7 promoter at +3-6 relative to the mRNA transcriptional start site. Protocol 2 oligonucleotides (purple) incorporate nucleotide mutations in the TIR, which increases protein production titers.
Materials and Reagents
Petri dishes (VWR, catalog number: 391-0440)
Plate culture spreaders (VWR, catalog number: 612-1561)
Inoculation loops (VWR, catalog number: 612-9352)
50-ml conical tubes (VWR, catalog number: 525-0402)
Chemically competent E. coli MC1061
Q5 polymerase (NEB, catalog number: M0491L, storage: -20°C)
DpnI (NEB, catalog number: R0176L, storage: -20°C)
dNTPs (Thermo Scientific, catalog number: R0181, storage: -20°C)
Oligonucleotides (Eurofins Genomics)
Agarose (Sigma-Aldrich, catalog number: A9539)
O’GeneRuler DNA Ladder (Thermo Scientific, catalog number: SM1163)
DNA miniprep kit (OmegaTek, catalog number: D6943-02)
Yeast extract (Oxoid, catalog number: LP0021)
Tryptone (Oxoid, catalog number: LP0042)
NaCl (VWR, catalog number: ICNA0219473805)
Agar (VWR, catalog number: 20767.298)
Kanamycin sulfate (VWR, catalog number: 0408-EU-25G, storage: 4°C)
Ampicillin sodium salt (Sigma, catalog number: A0166-25G, storage: 4°C)
LB medium (see Recipes)
Antibiotic stock solutions (see Recipes)
LB agar (see Recipes)
50× TAE buffer (see Recipes)
DNA loading buffer (see Recipes)
Equipment
Thermocycler (Techne, catalog number: 5PRIME/02)
DNA mini horizontal submarine unit (Hoefer, catalog number: HE33)
Electrophoresis power supply (GE Healthcare, catalog number: EPS601)
Gel Imager Azure C200 (Azure Biosystems, catalog number: AC2001)
Thermomixer Comfort (Eppendorf, catalog number: 5355000.011)
New Brunswick Incubator (New Brunswick, catalog number: M1282-0012)
Benchtop centrifuge (Eppendorf, model: 5417C)
NanoDrop Spectrophotometer (Thermo Scientific, catalog number: ND-2000)
Procedure
-
Check whether the protocols are relevant to the pET plasmid of interest by cross-referencing Tables 1 and 2. If so, order the required oligonucleotide set.
Protocol 1 will correct a design flaw in the T7lac module, converting it to T7pCONSlac. This protocol only applies to the pET plasmids listed in Table 1.
Protocol 2 will replace the standard TIR with one of two optimised TIRs. This protocol only applies to pET expression plasmids encoding an N-terminal poly-histidine tag, such as those listed in Table 1.
Order the oligonucleotide sets required for implementing the T7CONSlac promoter (Protocol 1) and/or the TIR (Protocol 2). The oligonucleotides have complementarity to the plasmid template at their 3’ ends and to each other at their 5’ ends. The sequences are provided in Table 2.
-
PCR setup and program for Protocol 1
-
Combine the following reagents (Final reaction volume 25 μl):
5 μl Q5 reaction buffer
5 μl GC enhancer solution (optional: included with the Q5 polymerase kit)
1 μl dNTPs from a 10 mM stock (0.4 mM final concentration)
1.25 μl primer 1 from a 10 μM stock (0.5 μM final concentration)
1.25 μl primer 2 from a 10 μM stock (0.5 μM final concentration)
0.5 μl template plasmid from a 2 ng/μl stock (0.04 ng final concentration)
0.25 μl Q5 polymerase
10.75 μl sterile ultrapure water
-
Set up a PCR program with the following parameters:
Hold at 95°C for 5 min
-
First cycle with 5 repeats
95°C for 30 s
48°C for 30 s
72°C for 3.5 min
-
Second cycle with 20 repeats
95°C for 30 s
60°C for 30 s
72°C for 3.5 min
Infinite hold at 10°C
-
-
PCR setup and program for Protocol 2
-
Combine the following reagents (TIR-1 or TIR-2) (Final reaction volume 25 μl):
5 μl Q5 reaction buffer
5 μl GC enhancer solution (optional)
1 μl dNTPs from a 10 mM stock (0.4 mM final concentration)
1.25 μl primer 3 (TIR-1) or 4 from a 10 μM stock (TIR-2) (0.5 μM final concentration)
1.25 μl primer 5 from a 10 μM stock (0.5 μM final concentration)
0.5 μl template plasmid from a 2 ng/μl stock (2 ng final concentration)
0.25 μl Q5 polymerase
10.75 μl sterile ultrapure water
-
Set up a PCR program with the following parameters:
Hold at 95°C for 5 min
-
First cycle with 5 repeats
95°C for 30 s
48°C for 30 s
72°C for 3.5 min
-
Second cycle with 20 repeats
95°C for 30 s
60°C for 30 s
72°C for 3.5 min
Infinite hold at 10°C
-
-
Check the PCR product by agarose gel electrophoresis
Cast a 1% (w/v) agarose gel with 1× TAE buffer and an appropriate gel stain.
Mix 3 μl PCR reaction with a suitable DNA loading buffer.
Load the samples and perform electrophoresis at a constant 100 V for 30 min.
Visualise the gel on a suitable imaging workstation (such as the Azure 200, Azure Biosystems). An example of an expected PCR product is shown in Figure 3.
-
Perform DpnI treatment of the PCR product
-
Combine the following in a 200-μl PCR tube:
4 μl PCR product (Optional: Perform PCR clean-up of the sample prior to DpnI treatment)
0.5 μl Cutsmart buffer (included in the DpnI kit)
0.5 μl DpnI
Incubate the samples at 37°C for 1 h in a thermocycler (such as the Techne Large-Format Gradient Thermal Cycler).
Optional: Heat inactivate DpnI at 80°C for 15 min.
-
-
Transform MC1061 with the DpnI-treated PCR product
Add 5 μl DpnI-treated sample to 50 μl chemically competent E. coli such as MC1061 or an alternative strain capable of in vivo DNA assembly.
Incubate on ice for 30 min.
Heat shock at 42°C for 1 min.
Incubate on ice for 2 min.
Add 150 μl LB medium.
Incubate at 37°C for 30 min with shaking at 900 rpm using a thermomixer (such as the Eppendorf Thermomixer R).
Plate 100 μl cell culture onto LB-agar containing a suitable antibiotic.
Allow the plate to dry in an aseptic environment.
Incubate the plate face-down overnight at 37°C.
-
Pick colonies and sequence
Pick two individually separated colonies and inoculate 10 ml LB media with a suitable antibiotic in a 50-ml conical tube.
Incubate the inoculated cultures at 37°C overnight with shaking at 180 rpm.
Harvest the cultures by centrifugation at 3,220 × g for 10 min.
Extract the plasmids using a miniprep kit such as the E.Z.N.A DNA mini kit from Omega Bio-Tek.
Measure the DNA concentration using a spectrophotometer such as the NanoDrop 2000.
-
Send the purified plasmids for sequencing.
Note: Use appropriate primers that are a minimum of 60 bp from the site of mutagenesis.
Confirm insertion of the new modified DNA sequence by comparison with the expected sequence using a standalone program such as SnapGene (download from https://www.snapgene.com/) or an online browser-based program such as Benchling (access from https://www.benchling.com/).
Table 2. Oligonucleotide sequences for correction of design flaws.
| Primer set | Number | Sense | Primer sequence (5’-3’) |
| Protocol 1 | 1 | Forward | CTATAGGGAGAGGAATTGTGAGCGGATAAC |
| 2 | Reverse | CCTCTCCCTATAGTGAGTCGTATTAATTTCGCGG | |
| Protocol 2 | 3 | Forward | AACTTTAAGAAGGAGAGTTATCATGGGTAGCAGCCATCATCATCATCATCA |
| 4 | Forward | AACTTTAAGAAGGAGAGCAGCTATGCAGCTTAGCCATCATCATCATCATCA | |
| 5 | Reverse | CTCCTTCTTAAAGTTAAACAAAATTATTTCTAGAGGGGAATTGTTATC |
Figure 3. Example of an expected PCR product for protocol 2.

A 1% agarose gel in TAE buffer. Lane 1: O’GeneRuler 1-kb Plus DNA Ladder (Thermo Scientific). Lane 2: PCR product using primers 3 and 5. Lane 3: PCR product using primers 4 and 5.
Recipes
-
LB medium
10 g/L NaCl
10 g/L tryptone
5 g/L yeast extract
Dissolve in 1 L ultrapure water and sterilise by autoclaving
-
Antibiotic stock solutions
Weigh the required quantity of antibiotics; 50 mg/ml kanamycin or 100 mg/ml ampicillin.
Add ultrapure water to the desired volume and dissolve by vortexing.
Under aseptic conditions, filter sterilise the antibiotic solution into the appropriate volumes.
Use directly or store at -20°C.
-
LB agar
LB medium plus 15 g/L agar
Dissolve in 1 L ultrapure water and sterilise by autoclaving.
Allow the media to cool to ~50°C.
Add the appropriate antibiotic and pour 20-ml volumes into 9.5-cm Petri dishes.
-
Tris acetic acid (TAE) (50×)
242.2 g/L Tris base (2 M)
57.1 ml/L acetic acid (1 M)
18.6 g/L EDTA (50 mM)
-
6× DNA loading buffer
60% v/v glycerol
20 mM Tris pH 8.0
60 mM EDTA
0.03% bromophenol blue
Acknowledgments
This work was supported by the Swedish Research Council (2017-00704) and the Carl Tryggers Stiftelse. The original research leading to these protocols is described in Shilling et al. (2020); we would like to acknowledge all co-authors from that publication. We also thank James Cumming and Diana Khananisho for testing the protocols.
Competing interests
The protocols and designs described herein are free from intellectual property. However, the synthetic evolution process used to identify TIR-1 and TIR-2 is patent-protected (PCT/SE2015/051343; European Patent no. 3234146). These patents are the property of CloneOpt AB, of which P.J.S. is a former employee and D.O.D. is a shareholder.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
References
- 1.Bentele K., Saffert P., Rauscher R., Ignatova Z. and Blüthgen N.(2013). Efficient translation initiation dictates codon usage at gene start. Mol Syst Biol 9: 675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen H., Bjerknes M., Kumar R. and Jay E.(1994). Determination of the optimal aligned spacing between the Shine-Dalgarno sequence and the translation initiation codon of Escherichia coli mRNAs. Nucleic Acids Res 22(23): 4953-4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dubendorf J.W. and Studier F.W.(1991). Controlling basal expression in an inducible T7 expression system by blocking the target T7 promoter with lac repressor. J Mol Biol 219(1): 45-59. [DOI] [PubMed] [Google Scholar]
- 4.Dunn J.J., Studier F.W. and Gottesman M.(1983). Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J Mol Biol 166(4): 477-535. [DOI] [PubMed] [Google Scholar]
- 5.Goodman D.B., Church G.M. and Kosuri S.(2013). Causes and effects of N-terminal codon bias in bacterial genes. Science 342(6157):475-479. [DOI] [PubMed] [Google Scholar]
- 6.Kudla G., Murray A.W., Tollervey D. and Plotkin J.B.(2009). Coding-sequence determinants of gene expression in Escherichia coli . Science 324(5924): 255-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laursen B.S., Sørensen H.P., Mortensen K.K. and Sperling-Petersen H.U.(2005). Initiation of Protein Synthesis in Bacteria. Microbiol Mol Biol Rev 69(1):101-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCarthy J.E. and Gualerzi C.(1990). Translational control of prokaryotic gene expression. Trends Genet 6(3):78-85. [DOI] [PubMed] [Google Scholar]
- 9.Mierendorf R.C., Morris B.B., Hammer B. and Novy R.E.(1998). Expression and Purification of Recombinant Proteins Using the pET System. Methods Mol Med 13: 257-292. [DOI] [PubMed] [Google Scholar]
- 10.Milón P. and Rodnina M.V.(2012). Kinetic control of translation initiation in bacteria. Crit Rev Biochem Mol Biol 47(4): 334-348. [DOI] [PubMed] [Google Scholar]
- 11.Mirzadeh K., Martínez V., Toddo S., Guntur S., Herrgård M.J. and Elofsson A., et al.(2015). Enhanced protein production in Escherichia coli by optimization of cloning scars at the vector-coding sequence junction . ACS Synth Biol 4(9): 959-965. [DOI] [PubMed] [Google Scholar]
- 12.Mirzadeh K., Shilling P.J., Elfageih R., Cumming A.J. and Cui. H.L. Rennig. M. et al. (2020). Increased production of periplasmic proteins in Escherichia coli by directed evolution of the translation initiation region . Microb Cell Fact 19(1): 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mirzadeh K., Toddo S., Nørholm M.H.H. and Daley D.O.(2016). Codon optimizing for increased membrane protein production: A minimalist approach. Methods Mol Biol 1432: 53-61. [DOI] [PubMed] [Google Scholar]
- 14.Osterman I.A., Evfratov S.A., Sergiev P.V. and Dontsova O.A.(2013). Comparison of mRNA features affecting translation initiation and reinitiation. Nucleic Acids Res 41(1): 474-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parret A.H., Besir H. and Meijers R.(2016). Critical reflections on synthetic gene design for recombinant protein expression. Curr Opin Struct Biol 38:155-162. [DOI] [PubMed] [Google Scholar]
- 16.Plotkin J.B. and Kudla G.(2011). Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet 12(1): 32-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosano G.L. and Ceccarelli E.A.(2014). Recombinant protein expression in Escherichia coli: advances and challenges . Front Microbiol 5:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosano G.L., Morales E.S. and Ceccarelli E.A.(2019). New tools for recombinant protein production in Escherichia coli: A 5-year update . Protein Sci 28(8):1412-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenberg A.H., Lade B.N., Dao-shan C., Lin S.W., Dunn J.J., Studier F.W.(1987). Vectors for selective expression of cloned DNAs by T7 RNA polymerase. Gene 56(1): 125-135. [DOI] [PubMed] [Google Scholar]
- 20.Shilling P.J., Mirzadeh K., Cumming A.J., Widesheim M., Köck Z. and Daley D.O.(2020). Improved designs for pET expression plasmids increase protein production yield in Escherichia coli . Commun Biol 3(1): 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shine J. and Dalgarno L.(1975). Determinant of cistron specificity in bacterial ribosomes. Nature 254(5495): 34-38. [DOI] [PubMed] [Google Scholar]
- 22.Silva-Rocha R., Martínez-García E., Calles B., Chavarría M., Arce-Rodríguez A. and A. las Heras, et al.(2013). The Standard European Vector Architecture(SEVA): a coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Research 41(D1): D666-D675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Walsh, G. Large-Scale Protein Production. In: Proteins: Biochemistry and Biotechnology, Second Edition. 141-176.