
Fig. 1 |. Workflow for single-cell analysis in immuno-oncology.
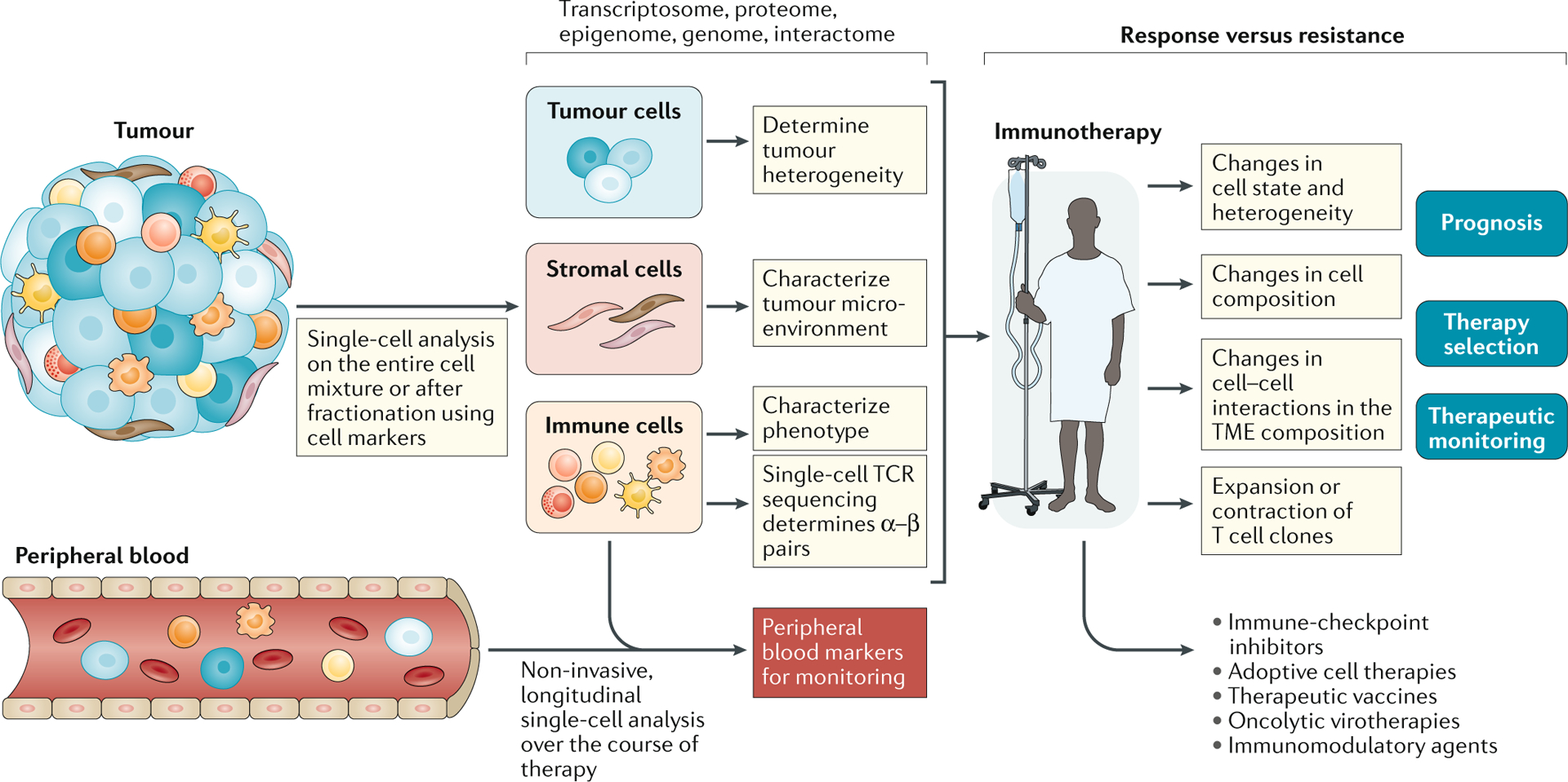
The tumour microenvironment (TME) is often complex and contains myriad cell types and states that can be disaggregated, with varying degrees of success, into a single-cell suspension. Cells can then be processed without further manipulation, or specific cell subsets can be isolated for downstream analysis. Single-cell analysis — most commonly by single-cell RNA sequencing (scRNA-seq) — enables the assessment of tumour, immune and/or stromal cells to yield high-dimensional information on tumour heterogeneity and an atlas of the immune and/or stromal microenvironment in relation to clinical stage, disease subtype and tumour location. Sampling before and after immunotherapy, especially if patients can be stratified based on their response, facilitates a detailed dissection of the mechanisms underlying response and resistance. The ability to track immune cell populations, specifically T cells through paired T cell receptor (TCR) α and β subunit sequencing, enables the detailed characterization of T cell clones involved in mediating response. These findings can be applied to a wider selection of patients, with the goals of improving prognostication and optimal treatment selection in the face of an ever-expanding range of therapeutic options, and enabling therapeutic monitoring through the identification of cellular populations or markers that can be monitored using high-throughput conventional technologies. Findings from within the TME might also be linked to changes in the peripheral circulation, which are more amenable to regular monitoring.