

Abstract
Long noncoding RNAs (lncRNAs) are a group of nonprotein coding transcripts that play a critical role in cancer progression. However, the role of lncRNA in metformin-induced inhibition of cell growth and its biological function in gastric cancer remain largely unknown. In this study, we identified an oncogenic lncRNA, Loc100506691, the expression of which was decreased in gastric cancer cells with metformin treatment. Moreover, Loc100506691 was significantly overexpressed in gastric cancer compared with adjacent normal tissues (p < 0.001), and high Loc100506691 expression was significantly correlated with poor survival of patients with gastric cancer. Additionally, Loc100506691 knockdown could significantly suppress gastric cancer cell growth in vitro, and ectopic Loc100506691 expression accelerated tumor growth in an in vivo mouse model. Analysis of the cell cycle revealed that Loc100506691 knockdown induced cell cycle arrest at the G2/M phase by impairing cell entry from the G2/M to G1 phase. Loc100506691 negatively regulated CHAC1 expression by modulating miR-26a-5p/miR-330-5p expression, and CHAC1 knockdown markedly attenuated Loc100506691 knockdown-induced gastric cancer cell growth and motility suppression. We concluded that anti-proliferative effects of metformin in gastric cancer may be partially caused by suppression of the Loc100506691-miR-26a-5p/miR-330-5p-CHAC1 axis.
Keywords: metformin, lncRNA, Loc100506691, gastric cancer, miRNA, CHAC1, miR-26a-5p, miR-330-5p
Graphical abstract
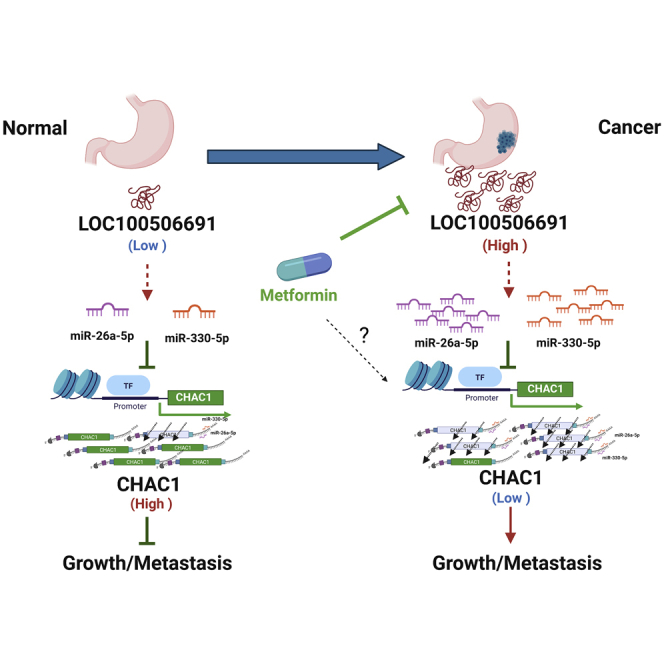
This new signaling pathway broadens our understanding of the mechanism and anticancer potential of metformin in the treatment of gastric cancer. Our findings showed anti-proliferative effects of metformin in gastric cancer may be caused by suppression of the novel Loc100506691-CHAC1 axis.
Introduction
Gastric cancer is one of most the common malignancies and is the third leading cause of cancer-related mortality worldwide.1,2 Early gastric cancer has no associated symptoms and is often diagnosed at an advanced stage. Despite efforts, not much improvement has been observed in surgical techniques, chemotherapy, and radiochemotherapy, and the prognosis of advanced gastric cancer remains poor.3 Therefore, further investigation of the molecular mechanisms of gastric cancer progression is crucial for developing an effective therapy.
Metformin is an antidiabetic drug for type2 diabetes with a safe tolerability profile. Retrospective studies have demonstrated that the antidiabetic drug metformin has a potential antitumor effect because a significant reduction in the risk of malignancy has been observed in patients with diabetes treated with metformin.4 Many studies have shown that metformin inhibits the tumor development of various human cancer cell types, such as those of lung,5 breast,6 colon,7 and pancreatic cancer.8 Furthermore, studies have revealed that metformin inhibits cellular proliferation, induces cell death, and causes partial cell cycle arrest in gastric cancer.9,10
Long noncoding RNAs (lncRNAs) are a group of nonprotein coding transcripts more than 200 bp in length.11,12 They have been demonstrated to participate in several biological functions by modulating complicated signaling pathways, such as those responsible for chromosome dosage compensation, imprinting, epigenetic regulation, cell cycle control, nuclear and cytoplasmic trafficking, transcription, translation, splicing, and cell differentiation.11,13,14 Several lncRNAs were observed to be deregulated in a wide variety of gastric cancers. lncRNA MALAT1 was observed to promote cell proliferation by recruiting SF2/ASF and be overexpressed in gastric cancer.15 HOTAIR was upregulated in gastric carcinoma tissues compared with adjacent normal gastric tissues,16 and the overexpression of HOTAIR in gastric cancer cells resulted in the enhancement of metastases of the liver in vivo.17,18 Overexpression of lncRNA H19 enhances cell proliferation and invasion of gastric cancer cells.19, 20, 21 Moreover, lncRNA CCAT1 is markedly increased in gastric carcinoma, promotes cell proliferation and migration, and is activated by c-Myc.22
Studies have demonstrated that metformin regulates the expression of two ncRNAs in cancer cells, namely HULC and H19. Thus, lncRNA HULC is overexpressed in hepatocellular carcinoma, and a decrease in its expression can be observed in hepatocellular carcinoma cells treated with metformin. In addition, the knockdown of HULC can inhibit cell growth and motility ability.23 The H19 is a potential oncogenic lncRNA in many types of human cancer.24, 25, 26 Furthermore, a report revealed a positive relationship between H19 expression and tumor cell motility ability, and metformin downregulates H19 in part by inducing DNA methylation.27 However, the antitumor role of lncRNA in patients with gastric cancer undergoing metformin treatment is also largely unknown.
In this study, we comprehensively profiled the expression of lncRNAs in gastric cancer cells after metformin treatment by adopting a microarray approach. We identified an oncogenic lncRNA, Loc100506691, which was significantly increased in patients with gastric cancer and could be suppressed after metformin treatment, suggesting that the antiproliferation effects of metformin in gastric cancer cells might result from the modulation of Loc100506691-miR-26a-5p/miR-330-5p-CHAC1 axis signaling.
Results
Metformin inhibited gastric cancer cell proliferation and invasion ability
Studies have revealed that metformin can inhibit cell proliferation in human cancer cells, including lung, breast, colon, and pancreatic cancer cells.5, 6, 7, 8 We evaluated the effects of metformin treatment in three human gastric cells (AGS, HR, and TSGH) by applying metformin treatment of various concentrations (0, 1, 5, and 10 mM) for 4 days. In Figures S1A–S1C, the growth of gastric cancer cells was significantly suppressed in a dose-dependent manner. The invasion ability was also suppressed in gastric cancer cells with metformin treatment (Figures S1D–S1F). The distribution of the cell cycle was examined in HR cells with metformin treatment. Our data indicated that HR cells with metformin treatment could significantly induce cell cycle arrest at the G2/M phase (Figures S1G and S1H). Expression levels of cell-cycle-related protein expression noticeably decreased after metformin treatment (Figures S1I and S1J). In summary, our results demonstrated that metformin could significantly inhibit gastric cancer growth by impairing cell cycle progression.
Identification of metformin-regulated lncRNAs in gastric cancer cells
To identify metformin-regulated lncRNAs, we performed transcriptome profiles of HR cells with and without metformin treatment by using a microarray approach. As indicated in Figures 1A and 1B, most of these genes were consistently expressed between the control group and the group that received metformin treatment. A total of 3,383 genes were downregulated in the fold change of <0.5 (i.e., 1,286 protein-coding genes and 2,097 lncRNAs), and 4,160 genes were upregulated in the fold change of >2 (i.e., 1,395 protein-coding genes and 2,765 lncRNAs) in HR cells after metformin treatment (Figures 1A and 1B). Pathway enrichment analysis revealed that these metformin-response genes were significantly enriched in the cell cycle, DNA replication, oocyte meiosis, cell division, and regulation of the cell-death-relative signaling pathway (Figure 1C; Figures S1K and S1L). These results are in strong agreement with our aforementioned data; that is, metformin treatment could suppress gastric cancer cell growth by impairing cell cycle progression.
Figure 1.

Transcription profiling was performed in HR cells with and without metformin treatment by adopting a microarray approach
(A) Genes with altered gene expression of HR treated with and without metformin, as identified by microarray analysis (fold change of ≥2 folds or ≤0.5 folds). (B) Pairwise sample comparisons of differentially expressed coding and noncoding genes, with red indicating significant upregulation (≥2 folds) and blue indicating significant downregulation (≤0.5 folds). (C) Differentially expressed genes were subjected to the Kyoto Encyclopedia of Genes and Genomes (KEGG) or Gene Ontology (GO) pathway enrichment analysis. (D) By combining transcriptome profiles of gastric cancer and corresponding adjacent normal tissue from two patients with gastric cancer, we precisely identified metformin-associated lncRNA candidates in gastric cancer. The heatmap reveals these lncRNA candidates’ expression in HR with metformin treatment and two gastric cancers. (E–G) Expression levels of lncRNA candidates (H19, RBMS3-AS3, and Loc100506691) were examined by real-time PCR in HR cells with metformin treatment at various doses (0, 1, 5, and 10 mM) for 4 days. (H–J) Expression levels of lncRNA candidates (H19, RBMS3-AS3, and Loc100506691) were examined by real-time PCR in HR cells with metformin treatment (10 mM) for various durations (0, 1, 2, 3, and 4 days) (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
To reduce the number of lncRNA candidates, we further performed transcriptome profiles of gastric cancer and the corresponding adjacent normal tissue from two patients with gastric cancer by using a microarray approach. We combined the microarray data and identified 7 putative oncogenic lncRNAs that could be suppressed by metformin treatment and 20 putative tumor-suppressive lncRNAs that could have their expression restored by metformin (Figure 1D). We also identified many protein-coding gene candidates by following the aforementioned criteria (Figure S2). Among these lncRNAs, H19, Loc100506691, and RBMS3-AS3 were selected for further validation using the real-time polymerase chain reaction (PCR) approach. In Figures 1E–1G, expression levels of H19, RBMS3, and Loc100506691 could be suppressed in HR cells with metformin treatment in a dose-dependent manner. In addition, our data indicated that expression levels of these lncRNAs were silenced significantly in HR cells following the application of 10 mM of metformin for more than 1 day (Figures 1H–1J). These results implied that metformin’s suppression of gastric cancer may occur partially by suppressing the expression of these lncRNAs. Among these lncRNAs, we selected the lncRNA Loc100506691 for further examination. We validated the metformin-induced suppression of Loc100506691 in AZ-521, NCI-N87, and TSGH cells, demonstrating that metformin could significantly suppress Loc100506691 expression in a dose- and time-dependent manner (Figures S3A–S3F).
High expression of Loc100506691 correlates with poor prognosis of gastric cancer
To clarify the role of Loc100506691 in gastric cancer, we assessed the expression levels of Loc100506691 in gastric cancer. We examined the expression levels of Loc100506691 in gastric cancer and determined that Loc100506691 expression significantly increased in gastric cancer tissues compared with normal stomach specimens from 42 patients (Figure 2A; p = 0.0001). Further examination of the public database revealed that expression levels of Loc100506691 significantly increased in gastric cancer cells compared with adjacent normal tissue. The analysis was conducted by examining Gene Expression Database of Normal and Tumor Tissues (GENT; p = 0.0002) and The Cancer Genome Atlas (TCGA) database (p = 0.0007) (Figures 2B and 2C). We further assessed the clinical impacts of Loc100506691 expression in patients with gastric cancer. In Table S1, expression levels of Loc100506691 had no correlation with clinical pathological features.
Figure 2.

Expression levels of Loc100506691 were analyzed in gastric cancer
(A) Expression levels of Loc100506691 were examined using the real-time PCR approach in gastric cancer cells and adjacent normal tissues from 42 patients with gastric cancer. (B) Expression levels of Loc100506691 were analyzed by RNA microarray analysis from the GENT database. (C) Expression levels of Loc100506691 were examined by analyzing the TCGA database. (D and E) The correlation between Loc100506691 expression and overall survival in patients with gastric cancer was analyzed according to a Kaplan-Meier plot; data were obtained from the GENT and TCGA databases, respectively. Patients with gastric cancer were separated into two groups representing high and low Loc100506691 expression on the basis of best cutoff value (GENT: 78 and TCGA: 0.184).
Kaplan-Meier survival curves obtained by analyzing the GENT database indicated that high expression levels of Loc100506691 were strongly associated with a shorter survival time compared with low expression levels in patients with gastric cancer (Figure 2D). Similar results in the TCGA database have demonstrated that high expression levels of Loc100506691 are significantly associated with poor survival curves in patients with gastric cancer (Figure 2E). Multivariate analysis revealed that high expression of Loc100506691 is significantly correlated with poor survival curves in patients with gastric cancer (p = 0.009) (Table S2). Our results indicated that Loc100506691 dysfunction might be involved in gastric cancer carcinogenesis.
Loc100506691 plays an oncogenic role in gastric cancer cell growth
We attempted to verify the biological role of Loc100506691. First, we identified the full length of the Loc100506691 sequence by performing 5′ and 3′ rapid amplification of cDNA ends (RACE). As indicated in Figures S4A and S4B, we identified three putative isoforms with different transcript lengths (Loc100506691-V1: 806 bp; Loc100506691-V2: 685bp; and Loc100506691-V3: 640 bp). The detailed sequence of these three isoforms was examined with the Sanger sequence (Table S3). Expression levels of Loc100506691 were as follows: high in TSGH and SUN-1 cells, medium in HR and AZ-521 cells, and low in AGS and NCI-N87 cells (Figure S4C). Expression levels of Loc100506691-V1 and -V2 were the most abundant compared with those of V3 (Figure S4D). Further examination of the localization of Loc100506691 revealed that it was expressed in the nucleus and especially the cytoplasm of gastric cancer cells (Figure S4E). In addition, expression levels of Loc100506691-V1 and -V2 were significantly increased in gastric cancer cells compared with adjacent normal tissues, and Loc100506691-V3 expression levels were not significantly increased (Figures S4F–S4H).
To examine the biological function of Loc100506691, we designed two small interfering RNAs (siRNAs) targeting sequences of Loc100506691 that could silence Loc100506691 expression. After siRNA transfection, the Loc100506691 expression levels were significantly decreased by 70% in TSGH cells and HR cells with siLoc100506691#1 (siLOC#1) transfection (Figures 3A and 3B). Our data indicated that Loc100506691 knockdown could significantly suppress cell proliferation, colony-formation ability and anchorage-dependent growth, invasion, and migration ability in HR and TSGH cells (Figures 3C–3J). Similarly, the results of the cell growth and motility assay revealed that both cell growth and motility were suppressed by another siRNA (siLoc100506691#2) in TSGH cells (Figures S5A–S5E). We further ectopically expressed Loc100506691-V1 and -V2 in HR cells. As displayed in Figures 4A and 4B, the expression levels of Loc100506691-V1 and -V2 were noticeably increased in HR cells after transfection with pLoc100506691-V1 and pLoc100506691-V2 expression vectors. The ectopic expression of Loc100506691-V1 and -V2 significantly accelerated gastric cancer cell proliferation (Figures 4C and 4D) and invasion ability (Figures 4E and 4F). Moreover, an in vivo mouse model also revealed that Loc100506691-V1 and -V2 expression could significantly increase tumor growth (Figures 4G–4J).
Figure 3.

Loc100506691 has an oncogenic role in the regulation of gastric cancer cell growth and motility
(A and B) Expression levels of Loc100506691 were examined in HR and TSGH after siLOC#1 transfection. (C and D) Cell proliferation of HR and TSGH with and without Loc100506691 knockdown was assessed. (E and F) Colony-formation ability in HR and TSGH with and without Loc100506691 knockdown was examined and quantified. (G and H) Anchorage-dependent growth of HR with and without Loc100506691 knockdown was examined and quantified. (I and J) Invasion ability of HR and TSGH with and without Loc100506691 knockdown was examined and quantified (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
Figure 4.

Ectopic Loc100506691 expression accelerated gastric cancer cell growth
(A and B) Expression levels of Loc100506691-V1 and -V2 were respectively examined in HR after pLoc100506691-V1 and -V2 transfection. (C and D) The proliferation of HR with pLoc100506691-V1, -V2, or control vector expression was examined. (E and F) The invasion ability of HR in regard to pLoc100506691-V1, -V2, or control vector expression was examined. (G) The in vivo tumorigenic potential of Loc100506691-V1-overexpressing, Loc100506691-V2-overexpressing, and control HR cells was examined by subcutaneously injecting cancer cells into nude mice. (H–J) Weight and size of tumors were assessed (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
Loc100506691 knockdown suppresses gastric cancer cell proliferation by inducing cell cycle arrest at G2/M
To explore the mechanism of Loc100506691 that regulates gastric cancer cell growth, we examined cell cycle progression in gastric cancer with Loc100506691 knockdown. Cell cycle analysis demonstrated that knockdown of Loc100506691 could induce arrest at the G2/M phase in TSGH and HR cell lines (Figures S6A and S6B). Furthermore, expression levels of CDK1 were significantly decreased in TSGH with Loc100506691 knockdown compared with the control group, whereas those of cyclin A2, cyclin B1, CDK2, and CDK4 were significantly increased (Figures S6C and S6D). To elucidate the complex role of Loc100506691 in gastric cancer, we analyzed the dynamics of cell progression in TSGH cells with Loc100506691 knockdown. The cell cycles of TSGH cells with and without Loc100506691 knockdown were synchronized at the G2/M phase by using a nocodazole block for 14 h, and the cells were subsequently released and analyzed at 1, 2, 3, and 4 h. Our data indicated that TSGH cells with Loc100506691 knockdown might retard the progression of cells at the G2/M phase to the G0/G1 phase (Figure S6E). Thus, knockdown of Loc1005006691 could suppress cell growth by inducing cell cycle arrest at the G2/M phase by impairing cell cycle progression.
Identification of downstream targets of Loc100506691: a microarray approach
To understand the oncogenic function of Loc100506691 in gastric cancer progression, microarray profiles were conducted to identify Loc100506691-regulated genes in gastric cancer cells. We performed transcription profiling of HR cells with Loc100506691 overexpression and TSGH cells with Loc100506691 knockdown. In Figure 5A, expression levels of 14 genes, namely LITAF, APOLD1, ALPK2, COL9A3, EFEMP1, CYR61, SPEF1, SLC7A2, DLX2, RIN2, TOM1L2, KIAA0922, ID2, and NFIA, were increased in HR cells with Loc100506691 overexpression, whereas their expression levels were decreased in TSGH cells with Loc100506691 knockdown. The expression of four genes, namely ZNF567, LHX6, SERPINB8, and CHAC1, were downregulated in HR with Loc100506691 expression but upregulated in TSGH cells with Loc100506691 knockdown (Figure 5A). We confirmed the mRNA and protein levels of CHAC1 and revealed that expression levels of CHAC1 expression were upregulated in TSGH or HR cells with Loc100506691 knockdown but downregulated in HR with Loc100506691 expression (Figures 5B–5D). We also selected an additional four genes, namely DLX2, LITAF, NF1A, and SERPINB8, for further confirmation by real-time PCR. Consistent with the microarray data, the expression levels of DLX2, LITAF, and NF1A were coexpressed with Loc100506691, whereas those of SERPINB8 were opposite to Loc100506691 (Figures S7A–S7H). Our results indicated that these genes might be downstream genes involved in Loc100506691-regulated gastric cancer growth. Notably, the expression levels of CHAC1 could be upregulated in HR and TSGH cells through metformin treatment (Figures S2B, S8A, and S8B). These results suggested that metformin-induced CHAC1 upregulation may partially result from the silencing of Loc100506691 expression.
Figure 5.

Identification of putative downstream genes of Loc100506691 in gastric cancer by applying the microarray approach
(A) Differential expression genes were identified in HR with Loc100506691 overexpression and TSGH with Loc100506691 knockdown and compared with the corresponding control using a microarray approach. The heatmap presents putative downstream targets of Loc100506691in gastric cancer cells. (B) Expression levels of CHAC1 were examined by real-time PCR in TSGH and HR cells with Loc100506691 knockdown. (C) Expression levels of CHAC1 were assessed in HR cells with pLoc100506691-V1 or -V2 overexpression by applying the real-time PCR approach. (D) Expression levels of CHAC1 were examined by western blotting in gastric cancer cells with Loc100506691 knockdown or overexpression in TSGH and HR, respectively. Relative expression was further quantification (below panels). (E) The CHAC1 expression levels in human gastric cancer cells were examined through western blotting. Relative expression was further quantification (below panel). (F and G) After transfection for 24 h, CHAC1 expression levels were examined in TSGH with siCHAC1 using western blotting and real-time PCR. (H) Cell proliferation was examined in TSGH with CHAC1 knockdown. (I and J) The colony-formation ability of TSGH was investigated and quantified following siCHAC1 transfection. (K and L) The migration and invasion ability were assessed and quantified in TSGH with CHAC1 knockdown (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
CHAC1 is involved in gastric cancer growth and motility
The above results revealed that CHAC1 may be a downstream contributor to Loc100506691-regulated gastric cancer cell growth and motility. Therefore, we examined expression levels of CHAC1 in gastric cancer cells by using a real-time PCR approach and determined that the expression levels of CHAC1 were significantly decreased in gastric cancer cells compared with adjacent normal tissues (Figure S9A). Similar results were observed in the TCGA and GENT databases, namely that CHAC1 was significantly downregulated in gastric cancer cells (Figures S9B and S9C). Higher expression levels of CHAC1 were observed in SUN1 and TSGH cells compared with those in AGS, AZ521, HR, and N-87 cells (Figure 5E). We further assessed the biological function of CHAC1 in TSGH using a loss-of-function approach. As depicted in Figures 5F and 5G, the expression levels of CHAC1 were silenced in TSGH following siCHAC1 transfection for 48 h. Our data indicated that CHAC1 knockdown could significantly accelerate TSGH cell proliferation (Figure 5H), colony formation (Figures 5I and 5J), and cell motility ability (Figures 5K and 5L). Similar results were also observed in HR cells, indicating that CHAC1 knockdown significantly reduced HR cell growth and motility (Figure S10). To further verify that CHAC1 was a crucial target of Loc100506691, TSGH cells were simultaneously cotransfected with siCHAC1 and siLoc100506691, and both of their expression levels were examined using the real-time PCR approach (Figures 6A and 6B). Our data revealed that CHAC1 knockdown can functionally rescue the effects of the Loc100506691 knockdown-induced suppression of gastric cancer cell proliferation (Figure 6C), colony-formation ability (Figures 6D and 6E), migration, and invasion ability (Figures 6F–6H).
Figure 6.

CHAC1 knockdown rescues the suppression of gastric cancer cell growth and motility through Loc100506691 knockdown
TSGH cells were transfected with N.C, siLOC#1, siCHAC1, and siLOC#1 + siCHAC1 for 48 h. (A and B) The expression levels of Loc100506691 and CHAC1 in the different groups were examined using real-time PCR. (C–H) CHAC1 knockdown could functionally rescue the effect of the Loc100506691 knockdown-induced suppression of gastric cancer cell proliferation, colony formation, migration, and invasion ability (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
We evaluated the clinical impacts of Loc100506691 and CHAC1 in gastric cancer. The combined expression of Loc100506691 and CHAC1 was separated into four groups: Loc100506691 low-CHAC1 high, Loc100506691 low-CHAC1 low, Loc100506691 high-CHAC1 high, and Loc100506691 high-CHAC1 low. As indicated in Figure S11, the combination of high Loc100506691 expression levels and low CHAC1 expression levels was significantly associated with poor survival in patients with gastric cancer. Multivariate analysis revealed that Loc100506691 high-CHAC1 low expression was significantly correlated with a poor survival curve in patients with gastric cancer (Table S4).
Loc100506691 is involved in CHAC1 expression through the modulation of miR-26a-5p/miR-330-5p expression
To explore the detailed mechanism of Loc100506691 knockdown-accelerated CHAC1 expression in gastric cancer, we further clarified the relationship among Loc100506691, microRNA (miRNA), and CHAC1. We performed small RNA profiling of TSGH with Loc10050691 knockdown and control groups. As indicated in Figure 7A, we identified 32 miRNA candidates that had decreased TSGH with Loc100506691 knockdown. By using the micoRNA.org prediction tool, we determined that among these miRNAs, miR-26a-5p and miR-330-5p were potentially targeting the 3′ UTR of CHAC1 (Figure 7A). The expression levels of miR-26a-5p and miR-330-5p were markedly increased in TSGH following the transfection of mimics for 48 h (Figure 7B). Furthermore, ectopic expression of miR-26a-5p miR-330-5p revealed that the expression of CHAC1 could significantly suppress mRNA and protein levels (Figures 7C and 7D). A luciferase reporter assay revealed that both miR-26a-5p and miR-330-5p could significantly inhibit luciferase activity by directly targeting the 3′ UTR of CHAC1 (Figures 7E and 7F). When the binding sites of miR-26a-5p or miR-330-5p were respectively mutated, the suppression of luciferase activity was restored (Figure 7G). We further demonstrated that both CHAC1/miR-26a-5p and CHAC1/miR-330-5p were present in the Ago2 complex through an Ago2 protein precipitation assay (Figures 7H–7J). The results indicated that miR-26a-5p and miR-330-5p could silence CHAC1 expression by directly targeting its 3′ UTR.
Figure 7.

Loc100506691 was involved in suppressing CHAC1 expression by modulating miR-26a-5p and miR-330-5p expression
(A) Identification of downstream miRNA candidates of Loc100506691 in gastric cancer cells by using next-generation sequences. Venn diagrams reveal that miR-26a-5p and miR-330-5p were regulated by Loc100506691 and were identified using microRNA.org as having potential to target the 3ʹ UTR of CHAC1. (B) Expression levels of miR-26a-5p and miR-330-5p were assessed in gastric cancer cells with miR-26a-5p or miR-330-5p mimic transfection, respectively. (C and D) Ectopic expression of miR-26a-5p or miR-330-5p significantly inhibited and suppressed the endogenous mRNA and protein levels of CHAC1 in TSGH cells. (E) Luciferase constructs: The miR-26a-5p (left panel) or miR-330-5p (right panel) targeting sequence in the 3′ UTR sequence of CHAC1 is presented in the upper panel, and the mutant of its seed region is presented in the color red. (F and G) Relative luciferase activity of the reporter with wild-type and mutant 3ʹ UTR of CHAC1 was determined in TSGH cells cotransfected with miR-26a-5p (left panels), miR-330-5p mimic (right panels), and the scramble control, respectively. Firefly luciferase activity was the normalization control. (H–J) TSGH cells were used to perform Ago2 protein precipitation, and the expression levels of miR-26a-5p, miR-330-5p, and CHAC1 were examined using real-time PCR (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
In summary, our results revealed a novel mechanism; that is, Loc100506691-CHAC1 expression may play a key coordination role in metformin-induced tumor growth suppression. Furthermore, the oncogenic Loc100501191-CHAC1 axis may itself be regulated by the expression of miRNAs, including miR-26a-5p and miR-330-5p (Figure 8). Our findings provided valuable insight into the novel oncogenic role of Loc100506691, which can serve as a useful prognostic biomarker and therapeutic target in patients with gastric cancer.
Figure 8.

The proposed model for the mechanistic metformin suppression of gastric cancer cell growth through modulation of the Loc100506691-microRNAs-CHAC1 axis
Discussion
Studies have demonstrated that metformin can act as therapeutic drug for human cancer by regulating cancer cell biological functions, including proliferation, colony formation, apoptosis, autophagy, and motility.8,10,28, 29, 30 We demonstrated that metformin could inhibit proliferation, colony formation, and invasion in gastric cancer cells. Notably, our data indicated that metformin suppressed gastric cancer cells by inducing cell cycle arrest at the G2/M phase. Consistent with our findings, Bost et al.31 reported that metformin induced arrest in cells in the G2/M or S phase in other cell types. In addition, metformin treatment was reported to enhance radio-sensitization through decreased CDK1-cyclin B activity and reduced Rad51 expression and lead to cell cycle G2/M arrest and impairment of homologous recombination repair.32
Metformin is a drug with the potential to suppress human cancer growth and metastasis through activation of AMPK by inhibiting the mTOR signaling pathway and stimulating the p53/p21 axis.33 Studies have reported that metformin could suppress gastric cancer cell growth and metastasis by regulating critical expressions, such as those of PTEN, MMP7, Calml3, NFκB, Vimentin, β-catenin, E-cadherin, VEGF, PARP, PI3K, AKT, PKM2, and COX.34, 35, 36, 37 We assumed that gene expression dysfunction could be restored by metformin treatment. Therefore, we selected gene candidates based on the expression profiles of the HR cell with or without metformin treatment and those of gastric cancer tissues from two patients. According to this hypothesis, we could more precisely identify critical metformin-regulated genes involved in gastric tumorigenesis. Thus, we identified many deregulated protein-coding genes in gastric cancer cells, the expression of which could be restored by metformin treatment (Figure S2). Vav3 expression could be induced under a high-glucose culture condition and was significantly suppressed after metformin treatment.38 Martin at el.39 also reported that VEGF-A could be significantly upregulated in A375 melanoma cells in vivo. Angiopoietin-like protein-4 is a multifunctional protein involved in lipid regulation, energy metabolism, angiogenesis, and inflammation. Moreover, angiopoietin-like protein-4 mRNA expression levels increased in diabetic mice, but treatment with metformin reversed this effect.40
The most encouraging finding of our study was the identification of several metformin-regulated lncRNA candidates, including H19, RBMS-AS3, and Loc100506691. Studies have reported that metformin suppresses human cancer growth and metastasis by modulating the expression of lncRNAs, including SNHG7, MALAT1, HULC, UCA1, NR_027710, ENSMUS00000138573, and H19.27,41, 42, 43, 44, 45, 46 Among them, H19 has a well-known oncogenic role in cancer tumorigenesis, which also results in decreased expression levels of H19 in several human cancer cells after metformin treatment. Li et al.42 reported that H19 participates in metformin-mediated inhibition of gastric cancer cell invasion by modulating the AMPK-MMP9 axis. This is consistent with our finding that H19 expression levels could be decreased in gastric cancer after metformin treatment (Figures 1E and 1H).
In this study, we, for the first time, identified that the Loc100506691-CHAC1 axis plays a key role in the metformin-induced suppression of growth and invasion in gastric cancer cells. CHAC1 is an apoptotic mediator that functions by modulating the Notch3 pathway.47,48 Our studies are the first to report a significant reduction in CHAC1 expression in gastric cancer cells and the ability of CHAC1 knockdown to promote gastric cancer cell growth and invasion ability. In addition to CHAC1, several downstream genes of Loc100506691 were identified and may contribute to the metformin-induced inhibition of gastric cancer growth via the Loc100506691-related signaling pathway. Among them, CYR61 expression significantly accelerated gastric cancer cell invasion ability, and high-expression CYR61 levels were strongly associated with metastasis of gastric cancer.49,50 High DLX2 expression is highly correlated with poor prognosis and is involved in cancer stemness, radio-resistance, and drug resistance in human cancers, including gastric cancer.51, 52, 53 Our previous study reported that EFEMP1 plays an oncogenic role in melanoma cell invasion and growth.28 Han et al.54 also reported that high expression levels of EFEMP1 were significantly correlated with the muscle invasion of bladder cancer. In this study, we also determined that Loc100506691 negatively regulated the miR-590-3p, which could bind at the 3′ UTR of EFEMP1 (data not provided). Therefore, Loc100506691 promoted gastric cancer growth and invasion ability partially by modulating miR-590-3p-EFEMP1 axis activation.
lncRNAs can be localized in either the cytoplasm or the nucleus, depending on their biological function. Loc100506691 was expressed in the cytoplasm of gastric cancer cells, indicating its involvement in promoting gastric cancer growth and metastasis through lncRNA-miRNA or lncRNA-protein interactions to silence tumor suppressors or upregulate oncogene expression. Our findings revealed that Loc100506691 positively regulated miR-26a-5p and miR-330-5p expression and that both miRNAs participated in the Loc100506691 suppression of CHAC1 expression by directly targeting the 3′ UTR of CHAC1. Studies have reported that both miR-26a-5p and miR-330-5p can play oncomiR roles in the promotion of cancer cell proliferation and migration.55, 56, 57, 58 Until now, the regulatory mechanisms of miR-26a-5p and miR-330-3p expression were largely unknown. Study revealed that TGFβ can induce miR-26a expression through the direct binding of Smad 4 at the promoter region of miR-26a.59 Wang et al.60 also demonstrated that C/EBPα regulates the transcriptional expression of miR-26a by directly binding to its promoter region. In turn, the EZH2-mediated H3K27 trimethylation within the promoter region of miR-26a resulted in the dramatic repression of its expression in hepatocellular carcinoma cells.61 Through pathway enrichment analysis, we observed that Loc100506691 was significantly involved in the Wnt signaling pathway, and C/EBPα may be a downstream target gene of this signaling pathway.62 Taken together, we suggested that Loc100506691 regulation of miR-26a-5p might be through its modulation of the Wnt/C/EBPα signaling pathway. However, the detailed mechanism of Loc100506691 involved in miR-26a-5p and miR-330-3p expression in gastric cancer remains unclear and a focus of our future study.
Metformin could act as a therapeutic drug to suppress cancer growth and metastasis through the modulation of several signaling pathways, including the PI3K/AKT, mTOR-, AMPK-, and Wnt-signaling pathways.63 In this study, we, for first time, identified an oncogenic lncRNA, Loc100506691, that accelerates gastric cancer growth and invasion ability through the modulation of miR-26a-5p-CHAC1 and miR-330-5p-CHAC1 signaling. This new signaling pathway broadens our understanding of the mechanism and anticancer potential of metformin in the treatment of gastric cancer. Furthermore, we determined that an abnormal Loc100506691–CHAC1 axis acts as a poor prognostic biomarker for patients with gastric cancer.
Conclusions
Our results suggest that metformin has potential as a therapeutic drug in gastric cancer, is controlled through a Loc100506691-associated signaling pathway, and plays a critical role in the growth and motility of gastric cancer cells by modulating the miR-26a-5p/miR-330-5p-CHAC1 axis pathway.
Materials and methods
Cells of six gastric cancers, AGS, AZ-521, HR, NCI-N87, SNU-1, and TSHG, obtained from ATCC (Manassas, VA, USA) were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) (HyClone, Thermo Scientific, UT, USA), 2 mM glutamine, 10 μg/mL streptomycin, and 100 U/mL penicillin. Gastric cancer cells were treated at various concentrations (1, 5, and 10 mM) of metformin for 4 days, and cell growth was monitored for 3 consecutive days.
Extraction of RNA and microarray analysis
The total RNA of HR cells with and without metformin treatment, TSGH cells with and without siLoc100506691 knockdown, HR cells with and without Loc100506691-V2 overexpression, and two gastric cancer tissues and their corresponding adjacent normal tissues were extracted using a TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The detailed process was described in our previous study.28 Total RNA was subjected to profiling by Agilent SurePrint G3 Human V2 GE 8 × 60K Microarray (Agilent Technologies, Santa Clara, CA, USA). The detailed process and data analyses were performed as described in our previous study.64
Clinical samples
Patients with gastric cancer underwent surgery at the Department of Surgery, Taipei Tzu Chi Hospital. Gastric cancer tissue and the corresponding adjacent normal samples were obtained from 48 patients and had been collected for processing in the hospital’s biobank. This study was approved by the Institutional Review Board of Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, Taiwan (No. 09-XD-038).
Cell proliferation assay
Cells (1,000/well) were seeded into a 96-well plate, and cell growth was assessed using a CellTiter-Glo One Solution Assay (Promega Corporation, Madison, WI, USA) at various times (0, 1, 2, 3, and 4 days). All experiments were performed in triplicate. The detailed process and data analyses were performed as described in our previous study.64
Expression data from TCGA and GENT
The transcriptome expression profiles of gastric cancer were downloaded from the TCGA data portal (https://www.cancer.gov/tcga/). The expression profiles of 375 gastric cancer tissues and 32 adjacent normal tissues were obtained from the TCGA data portal. The detailed clinical information of these patients was also downloaded from the TCGA database. The microarray expression data of gastric cancer cells were obtained from the GENT database (http://gent2.appex.kr/gent2/). The total expression data of 311 gastric cancer and 57 adjacent normal tissues were collected and analyzed.
Full-length RNA ligase-mediated RACE (RLM-RACE)
To identify the sequence of Loc100506691, the RNA ligase-mediated RACE was performed using the GeneRacer kit (Invitrogen, Thermo Fisher Scientific) according to the manufacturer’s instructions. PCR products were gel-purified and cloned into a pCR4 TOPO vector (Invitrogen, Thermo Fisher Scientific) for sequencing. Three colonies were selected, and plasmid DNA was extracted using miniprep (QIAprep Spin Miniprep Kit, QIAGEN). Next, the sequence of individual clones was verified using an autosequencer.
Gene expression by real-time PCR
Total RNA was reverse transcribed into cDNA by using SuperScript III and random primers (Invitrogen, Thermo Fisher Scientific). Real-time PCR was performed using the SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA) on an Applied Biosystems 7900HT Real-Time System. The relative quantity of the target gene was computed for each sample using the ΔΔCt method by comparing the mean Ct of the gene to the mean Ct of the housekeeping gene S26. The sequences of all primers used are presented in Table S5.
Colony formation assay
The 4,000 cells per well were plated into a 6-well plate and incubated at 37°C for 2 weeks. Cell culture plates containing colonies were fixed with 3.7% formaldehyde for 10 min, and colonies were stained with 0.2% crystal violet solution in 10% ethanol for 3 h. Wells were rinsed with H2O after air-drying. The crystal violet staining of cells from each well was solubilized using 2 mL of 10% acetic acid, and the absorbance (optical density) of the solution was measured on a spectrophotometer at a wavelength of 620 nm.
Cell cycle analysis by image flow cytometry assay
We collected 1 × 106 cells and added 70% ethanol to the fixative, which was then left to sit for 3 h. Next, the cells were stained with 4′,6-diamidino-2-phenylindole (ChemoMetec, Gydevang, Lillerød, Denmark) and detected using NucleoView NC-3000 software (ChemoMetec, Gydevang, Lillerød, Denmark).
Cell synchronization
TSGH cells were seeded in 6-cm dish and transfected as described. Then, cells were synchronized at G2/M using a nocodazole block at a final concentration of 100 ng/mL for 14 h after 10 mM siRNA transfection for 24 h. Cells were then washed with 1× phosphate-buffered saline (PBS) and further cultured in normal cell maintenance media, in which they began the cell cycle. The progression of the cycle of cells was assessed hourly using flow cytometry from 0 to 4 h. Cell cycle profiles were analyzed with NucleoView NC-3000 software to determine the percentage of cells in G1, S, and G2/M.
Migration and invasion assay
Cells were assessed for their migration and invasion ability in vitro by using a Transwell assay in accordance with our previous study.65 In summary, gastric cancer cells with siLoc100506691, siCHAC1, or scramble control transfection were suspended in 2% FBS and seeded into the upper chamber of the Transwells (Falcon, Corning, NJ, USA). A Matrigel coating (BD Biosciences, Bedford, MA, USA) was used for the invasion assay but not employed for the migration assay. Cells were subsequently incubated on a low chamber with 10% FBS for 24 h or 48 h. Noninvading cells were removed using a cotton swab. Invading cells on the undersurface of the Transwells were fixed with a 10% formaldehyde solution. Cells were stained with a crystal violet solution, and the number of gastric cancer cells was calculated by counting three fields under a phase-contrast microscope. All experiments were performed in triplicate.
RNA interference
siRNA for Loc100506691 (siRNA#1: sense: 5′-CCAACAUAUGAUGCUAGAATT-3′ and antisense: 5′-UUCUAGCAUCAUAUGUUGGTT-3′; siRNA#2: sense: 5′-CUCUUUCUCGAGUAGUCCCTT-3′ and antisense: 5′-GGGACUACUCGAGAAAGAGTT-3′), for CHAC1(SI05142375, QIAGEN, Hilden, Germany) and negative control (sense: 5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense: 5′-ACGUGACACGUUCGGAGAATT-3′) siRNA were purchased from Gendiscovery. The HR and TSGH cells were cultured in a 60-mm cell culture dish for 24 h at 37°C. Transfection of duplex siRNAs (50 nmol/L) was conducted using Lipofectamine RNAiMAX (Invitrogen, Thermo Fisher Scientific), as previously described.65
Small RNA transcriptome extraction through next-generation sequencing
RNA was extracted from two samples, namely TSGH cells with Loc100506691knockdown and TSGH control cells, by using a TRIZOL reagent (Invitrogen, Carlsbad, CA, USA). The small RNA library preparation was performed as described in our previous study.66 The sequencing was performed using the MiSeq V2 reagent kit (150 cycles; Illumina, San Diego, CA, USA). The sequencing data were analyzed using our developed tool.67
Stem-loop reverse-transcription PCR
The detailed process was performed as described in our previous study.68 Expression levels of miR-26a-5p or miR-330-5p were normalized to those of U6 small RNA (ΔCt = target miRNA Ct-U6 Ct). The sequences of all primers used are presented in Table S5.
Ectopic expression of miRNAs
Gastric cancer cells were transfected with 10-nM miR-26a-5p, miRNA-330-5p mimics or a scramble control (GenDiscovery Biotechnology, Taiwan) by using the Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, CA, USA). Following 24 h of transfection, the expression levels of miRNAs were assessed using stem-loop quantitative real-time PCR.
miRNA target candidates and luciferase reporter assay
We identified a total of 15 miRNA candidates for binding the 3′ UTR of CHAC1 by employing the microRNA.org prediction tool.69 The 3′ UTR sequences and seed region mutant of CHAC1 were cloned into a pMIR-REPROT vector (AM5795, Thermo Scientific). Subsequently, the pMIR-REPROT-CHAC1 or pMIR-REPROT-CHAC1(mutant) vector was cotransfected with or without the miR-26a-5p or miR-330-5p expression vector into a TSGH cell line by using Lipofectamine 2000 (Invitrogen, Thermo Fisher Scientific). After 24 h of transfection, cell lysates were used to measure luciferase activity with the Dual-Glo Luciferase Reporter Assay System (Promega, Madison, WI, USA).
RNA immunoprecipitation chip assay
In this study, an RNA immunoprecipitation chip (RIP) assay was performed to further investigate the interaction of CHAC1 and miR-26a-5p or miR-330-5p using an RNA-binding protein immunoprecipitation kit (Millipore, Billerica, MA, USA). TSGH cells were transfected with miR-26a-5p mimics, miR-330-5p mimics, or Negative control (N.C.) for 48 h and then lysed with RIP buffer. The cell lysate was supplemented with Dynabeads M-280 Streptavidin (Invitrogen, Waltham, MA, USA) conjugated with an anti-Ago2 antibody or anti-IgG. Finally, Ago2-bounding RNAs were extracted using the TRIzol reagent, and the levels of purified RNAs were determined using quantitative real-time PCR.
Western blotting
The detailed process of western blotting was as described in our previous study.65 The primary antibodies used in this study were as follows: CHAC1 antibody (1:1,000, GTX120775, GeneTex, Irvine, CA, USA), anti-primary antibodies of actin, and cell cycle-related protein (for details, see our previous study).64
Xenograft tumor growth assay
Animal experiments were approved by the Kaohsiung Veterans Hospital Laboratory Animal Center and Use Committee. Nude mice (aged 4 weeks) were used; 2 × 106 HR cells with Loc100506691-V1 or -V2 stable expression or control cells were suspended in PBS and implanted in the backs of nude mice. The mice were monitored, and tumor volume was evaluated as V (in mm3) = largest length × 0.52 × (shortest length)2 each week for 4 weeks. At 4 weeks after implantation, the mice were sacrificed, and tumor size and weight were assessed.
Statistical analysis
Expression levels of Loc100506691 and CHAC1 in gastric cancer, obtained from the TCGA and GENT databases, were analyzed using Student’s t tests. Cumulative survival curves were estimated using the Kaplan-Meier method, and the comparison of survival curves was performed using the log-rank test. The difference was considered significant when p < 0.05. Expression levels of Loc100506691 or CHAC1 in paired gastric cancer cells were analyzed using a paired t test. The cell function assay comprised cell proliferation, colony formation, and soft agar assay, and invasion/migration experiments were performed in triplicate. Histograms presented the mean values, and the error bars indicated the standard deviation. These data were analyzed using Student t tests.
Acknowledgments
The authors appreciated the assistance of Biobank of Taipei Tzu Chi Hospital for the processing of clinical specimens. The authors would also like to thank the Core Laboratory in the Department of Research, Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, for their technical support and facilities. This study was supported by grants from the Ministry of Science Technology (MOST 106-2320-B-075B-004 and MOST 107-2320-B-003-MY2), Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation (TCRD-TPE-109-66 and TCRD-TPE-109-70), and Yen Tjing Ling Medical Foundation (CI-107-19 and CI-108-14).
Author contributions
H.H.T. executed this study and drafted the manuscript. Y.Z.C. and Y.T.T. performed the biological function assay in gastric cancer cell. N.H.C., Y.C.C., and C.C.W. provided assistance to collect clinical samples and analyzed this study. L.F.L. provided assistance with manuscript drafting. C.Y.Y. performed xenograft tumor growth assay. Y.F.Y. and M.L.K. assisted in manuscript proofreading. K.W.T. supervised the study and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.08.006.
Supplemental information
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2015. CA Cancer J. Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J., Soerjomataram I., Dikshit R., Eser S., Mathers C., Rebelo M., Parkin D.M., Forman D., Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 3.Power D.G., Kelsen D.P., Shah M.A. Advanced gastric cancer--slow but steady progress. Cancer Treat. Rev. 2010;36:384–392. doi: 10.1016/j.ctrv.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 4.Pollak M. Metformin and other biguanides in oncology: advancing the research agenda. Cancer Prev. Res. (Phila.) 2010;3:1060–1065. doi: 10.1158/1940-6207.CAPR-10-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo Q., Liu Z., Jiang L., Liu M., Ma J., Yang C., Han L., Nan K., Liang X. Metformin inhibits growth of human non-small cell lung cancer cells via liver kinase B-1-independent activation of adenosine monophosphate-activated protein kinase. Mol. Med. Rep. 2016;13:2590–2596. doi: 10.3892/mmr.2016.4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.El-Haggar S.M., El-Shitany N.A., Mostafa M.F., El-Bassiouny N.A. Metformin may protect nondiabetic breast cancer women from metastasis. Clin. Exp. Metastasis. 2016;33:339–357. doi: 10.1007/s10585-016-9782-1. [DOI] [PubMed] [Google Scholar]
- 7.Zaafar D.K., Zaitone S.A., Moustafa Y.M. Role of metformin in suppressing 1,2-dimethylhydrazine-induced colon cancer in diabetic and non-diabetic mice: effect on tumor angiogenesis and cell proliferation. PLoS ONE. 2014;9:e100562. doi: 10.1371/journal.pone.0100562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kato K., Iwama H., Yamashita T., Kobayashi K., Fujihara S., Fujimori T., Kamada H., Kobara H., Masaki T. The anti-diabetic drug metformin inhibits pancreatic cancer cell proliferation in vitro and in vivo: Study of the microRNAs associated with the antitumor effect of metformin. Oncol. Rep. 2016;35:1582–1592. doi: 10.3892/or.2015.4496. [DOI] [PubMed] [Google Scholar]
- 9.Han G., Gong H., Wang Y., Guo S., Liu K. AMPK/mTOR-mediated inhibition of survivin partly contributes to metformin-induced apoptosis in human gastric cancer cell. Cancer Biol. Ther. 2015;16:77–87. doi: 10.4161/15384047.2014.987021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kato K., Gong J., Iwama H., Kitanaka A., Tani J., Miyoshi H., Nomura K., Mimura S., Kobayashi M., Aritomo Y. The antidiabetic drug metformin inhibits gastric cancer cell proliferation in vitro and in vivo. Mol. Cancer Ther. 2012;11:549–560. doi: 10.1158/1535-7163.MCT-11-0594. [DOI] [PubMed] [Google Scholar]
- 11.Daniel C., Lagergren J., Öhman M. RNA editing of non-coding RNA and its role in gene regulation. Biochimie. 2015;117:22–27. doi: 10.1016/j.biochi.2015.05.020. [DOI] [PubMed] [Google Scholar]
- 12.Cheetham S.W., Gruhl F., Mattick J.S., Dinger M.E. Long noncoding RNAs and the genetics of cancer. Br. J. Cancer. 2013;108:2419–2425. doi: 10.1038/bjc.2013.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rinn J.L., Chang H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prensner J.R., Chinnaiyan A.M. The emergence of lncRNAs in cancer biology. Cancer Discov. 2011;1:391–407. doi: 10.1158/2159-8290.CD-11-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhuang M., Gao W., Xu J., Wang P., Shu Y. The long non-coding RNA H19-derived miR-675 modulates human gastric cancer cell proliferation by targeting tumor suppressor RUNX1. Biochem. Biophys. Res. Commun. 2014;448:315–322. doi: 10.1016/j.bbrc.2013.12.126. [DOI] [PubMed] [Google Scholar]
- 16.Hajjari M., Behmanesh M., Sadeghizadeh M., Zeinoddini M. Up-regulation of HOTAIR long non-coding RNA in human gastric adenocarcinoma tissues. Med. Oncol. 2013;30:670. doi: 10.1007/s12032-013-0670-0. [DOI] [PubMed] [Google Scholar]
- 17.Lee N.K., Lee J.H., Park C.H., Yu D., Lee Y.C., Cheong J.-H., Noh S.H., Lee S.K. Long non-coding RNA HOTAIR promotes carcinogenesis and invasion of gastric adenocarcinoma. Biochem. Biophys. Res. Commun. 2014;451:171–178. doi: 10.1016/j.bbrc.2014.07.067. [DOI] [PubMed] [Google Scholar]
- 18.Niinuma T., Suzuki H., Nojima M., Nosho K., Yamamoto H., Takamaru H., Yamamoto E., Maruyama R., Nobuoka T., Miyazaki Y. Upregulation of miR-196a and HOTAIR drive malignant character in gastrointestinal stromal tumors. Cancer Res. 2012;72:1126–1136. doi: 10.1158/0008-5472.CAN-11-1803. [DOI] [PubMed] [Google Scholar]
- 19.Zhang E.-B., Han L., Yin D.-D., Kong R., De W., Chen J. c-Myc-induced, long, noncoding H19 affects cell proliferation and predicts a poor prognosis in patients with gastric cancer. Med. Oncol. 2014;31:914. doi: 10.1007/s12032-014-0914-7. [DOI] [PubMed] [Google Scholar]
- 20.Li H., Yu B., Li J., Su L., Yan M., Zhu Z., Liu B. Overexpression of lncRNA H19 enhances carcinogenesis and metastasis of gastric cancer. Oncotarget. 2014;5:2318–2329. doi: 10.18632/oncotarget.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang F., Bi J., Xue X., Zheng L., Zhi K., Hua J., Fang G. Up-regulated long non-coding RNA H19 contributes to proliferation of gastric cancer cells. FEBS J. 2012;279:3159–3165. doi: 10.1111/j.1742-4658.2012.08694.x. [DOI] [PubMed] [Google Scholar]
- 22.Yang F., Xue X., Bi J., Zheng L., Zhi K., Gu Y., Fang G. Long noncoding RNA CCAT1, which could be activated by c-Myc, promotes the progression of gastric carcinoma. J. Cancer Res. Clin. Oncol. 2013;139:437–445. doi: 10.1007/s00432-012-1324-x. [DOI] [PubMed] [Google Scholar]
- 23.Gandhy S.U., Imanirad P., Jin U.-H., Nair V., Hedrick E., Cheng Y., Corton J.C., Kim K., Safe S. Specificity protein (Sp) transcription factors and metformin regulate expression of the long non-coding RNA HULC. Oncotarget. 2015;6:26359–26372. doi: 10.18632/oncotarget.4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo M., Li Z., Wang W., Zeng Y., Liu Z., Qiu J. Long non-coding RNA H19 increases bladder cancer metastasis by associating with EZH2 and inhibiting E-cadherin expression. Cancer Lett. 2013;333:213–221. doi: 10.1016/j.canlet.2013.01.033. [DOI] [PubMed] [Google Scholar]
- 25.Tsang W.P., Kwok T.T. Riboregulator H19 induction of MDR1-associated drug resistance in human hepatocellular carcinoma cells. Oncogene. 2007;26:4877–4881. doi: 10.1038/sj.onc.1210266. [DOI] [PubMed] [Google Scholar]
- 26.Lottin S., Adriaenssens E., Dupressoir T., Berteaux N., Montpellier C., Coll J., Dugimont T., Curgy J.J. Overexpression of an ectopic H19 gene enhances the tumorigenic properties of breast cancer cells. Carcinogenesis. 2002;23:1885–1895. doi: 10.1093/carcin/23.11.1885. [DOI] [PubMed] [Google Scholar]
- 27.Yan L., Zhou J., Gao Y., Ghazal S., Lu L., Bellone S., Yang Y., Liu N., Zhao X., Santin A.D. Regulation of tumor cell migration and invasion by the H19/let-7 axis is antagonized by metformin-induced DNA methylation. Oncogene. 2015;34:3076–3084. doi: 10.1038/onc.2014.236. [DOI] [PubMed] [Google Scholar]
- 28.Tseng H.W., Li S.C., Tsai K.W. Metformin Treatment Suppresses Melanoma Cell Growth and Motility Through Modulation of microRNA Expression. Cancers (Basel) 2019;11:E209. doi: 10.3390/cancers11020209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feng Y., Ke C., Tang Q., Dong H., Zheng X., Lin W., Ke J., Huang J., Yeung S.-C., Zhang H. Metformin promotes autophagy and apoptosis in esophageal squamous cell carcinoma by downregulating Stat3 signaling. Cell Death Dis. 2014;5:e1088. doi: 10.1038/cddis.2014.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu N., Gu C., Gu H., Hu H., Han Y., Li Q. Metformin induces apoptosis of lung cancer cells through activating JNK/p38 MAPK pathway and GADD153. Neoplasma. 2011;58:482–490. doi: 10.4149/neo_2011_06_482. [DOI] [PubMed] [Google Scholar]
- 31.Bost F., Sahra I.B., Le Marchand-Brustel Y., Tanti J.-F. Metformin and cancer therapy. Curr. Opin. Oncol. 2012;24:103–108. doi: 10.1097/CCO.0b013e32834d8155. [DOI] [PubMed] [Google Scholar]
- 32.Wang Z., Lai S.-T., Ma N.-Y., Deng Y., Liu Y., Wei D.-P., Zhao J.-D., Jiang G.-L. Radiosensitization of metformin in pancreatic cancer cells via abrogating the G2 checkpoint and inhibiting DNA damage repair. Cancer Lett. 2015;369:192–201. doi: 10.1016/j.canlet.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 33.Pulito C., Sanli T., Rana P., Muti P., Blandino G., Strano S. Metformin: On Ongoing Journey across Diabetes, Cancer Therapy and Prevention. Metabolites. 2013;3:1051–1075. doi: 10.3390/metabo3041051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen G., Yu C., Tang Z., Liu S., An F., Zhu J., Wu Q., Cao J., Zhan Q., Zhang S. Metformin suppresses gastric cancer progression through calmodulin-like protein 3 secreted from tumor-associated fibroblasts. Oncol. Rep. 2019;41:405–414. doi: 10.3892/or.2018.6783. [DOI] [PubMed] [Google Scholar]
- 35.Sekino N., Kano M., Matsumoto Y., Sakata H., Murakami K., Toyozumi T., Otsuka R., Yokoyama M., Shiraishi T., Okada K. The Antitumor Effects of Metformin on Gastric Cancer In Vitro and on Peritoneal Metastasis. Anticancer Res. 2018;38:6263–6269. doi: 10.21873/anticanres.12982. [DOI] [PubMed] [Google Scholar]
- 36.Valaee S., Yaghoobi M.M., Shamsara M. Metformin inhibits gastric cancer cells metastatic traits through suppression of epithelial-mesenchymal transition in a glucose-independent manner. PLoS ONE. 2017;12:e0174486. doi: 10.1371/journal.pone.0174486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen G., Feng W., Zhang S., Bian K., Yang Y., Fang C., Chen M., Yang J., Zou X. Metformin inhibits gastric cancer via the inhibition of HIF1α/PKM2 signaling. Am. J. Cancer Res. 2015;5:1423–1434. [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Z., Shao Y., Tan L., Shi H., Chen S., Guo J. Clinical significance of the low expression of FER1L4 in gastric cancer patients. Tumour Biol. 2014;35:9613–9617. doi: 10.1007/s13277-014-2259-4. [DOI] [PubMed] [Google Scholar]
- 39.Martin M.J., Hayward R., Viros A., Marais R. Metformin accelerates the growth of BRAF V600E-driven melanoma by upregulating VEGF-A. Cancer Discov. 2012;2:344–355. doi: 10.1158/2159-8290.CD-11-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mizutani N., Ozaki N., Seino Y., Fukami A., Sakamoto E., Fukuyama T., Sugimura Y., Nagasaki H., Arima H., Oiso Y. Reduction of insulin signaling upregulates angiopoietin-like protein 4 through elevated free fatty acids in diabetic mice. Exp. Clin. Endocrinol. Diabetes. 2012;120:139–144. doi: 10.1055/s-0031-1291258. [DOI] [PubMed] [Google Scholar]
- 41.Wu P., Tang Y., Fang X., Xie C., Zeng J., Wang W., Zhao S. Metformin Suppresses Hypopharyngeal Cancer Growth by Epigenetically Silencing Long Non-coding RNA SNHG7 in FaDu Cells. Front. Pharmacol. 2019;10:143. doi: 10.3389/fphar.2019.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li P., Tong L., Song Y., Sun J., Shi J., Wu Z., Diao Y., Li Y., Wang Z. Long noncoding RNA H19 participates in metformin-mediated inhibition of gastric cancer cell invasion. J. Cell. Physiol. 2019;234:4515–4527. doi: 10.1002/jcp.27269. [DOI] [PubMed] [Google Scholar]
- 43.Xia C., Liang S., He Z., Zhu X., Chen R., Chen J. Metformin, a first-line drug for type 2 diabetes mellitus, disrupts the MALAT1/miR-142-3p sponge to decrease invasion and migration in cervical cancer cells. Eur. J. Pharmacol. 2018;830:59–67. doi: 10.1016/j.ejphar.2018.04.027. [DOI] [PubMed] [Google Scholar]
- 44.Jiang Z., Liu H. Metformin inhibits tumorigenesis in HBV-induced hepatocellular carcinoma by suppressing HULC overexpression caused by HBX. J. Cell. Biochem. 2018;119:4482–4495. doi: 10.1002/jcb.26555. [DOI] [PubMed] [Google Scholar]
- 45.Li T., Sun X., Jiang X. UCA1 involved in the metformin-regulated bladder cancer cell proliferation and glycolysis. Tumour Biol. 2017;39 doi: 10.1177/1010428317710823. 1010428317710823. [DOI] [PubMed] [Google Scholar]
- 46.Wang Y., Tang H., Ji X., Zhang Y., Xu W., Yang X., Deng R., Liu Y., Li F., Wang X., Zhou L. Expression profile analysis of long non-coding RNAs involved in the metformin-inhibited gluconeogenesis of primary mouse hepatocytes. Int. J. Mol. Med. 2018;41:302–310. doi: 10.3892/ijmm.2017.3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Joo N.E., Ritchie K., Kamarajan P., Miao D., Kapila Y.L. Nisin, an apoptogenic bacteriocin and food preservative, attenuates HNSCC tumorigenesis via CHAC1. Cancer Med. 2012;1:295–305. doi: 10.1002/cam4.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen P.H., Shen W.L., Shih C.M., Ho K.H., Cheng C.H., Lin C.W., Lee C.C., Liu A.J., Chen K.C. The CHAC1-inhibited Notch3 pathway is involved in temozolomide-induced glioma cytotoxicity. Neuropharmacology. 2017;116:300–314. doi: 10.1016/j.neuropharm.2016.12.011. [DOI] [PubMed] [Google Scholar]
- 49.Wei J., Yu G., Shao G., Sun A., Chen M., Yang W., Lin Q. CYR61 (CCN1) is a metastatic biomarker of gastric cardia adenocarcinoma. Oncotarget. 2016;7:31067–31078. doi: 10.18632/oncotarget.8845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yan J., Yang B., Lin S., Xing R., Lu Y. Downregulation of miR-142-5p promotes tumor metastasis through directly regulating CYR61 expression in gastric cancer. Gastric Cancer. 2019;22:302–313. doi: 10.1007/s10120-018-0872-4. [DOI] [PubMed] [Google Scholar]
- 51.Tang P., Huang H., Chang J., Zhao G.F., Lu M.L., Wang Y. Increased expression of DLX2 correlates with advanced stage of gastric adenocarcinoma. World J. Gastroenterol. 2013;19:2697–2703. doi: 10.3748/wjg.v19.i17.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi Y.J., Baek G.Y., Park H.R., Jo S.K., Jung U. Smad2/3-Regulated Expression of DLX2 Is Associated with Radiation-Induced Epithelial-Mesenchymal Transition and Radioresistance of A549 and MDA-MB-231 Human Cancer Cell Lines. PLoS ONE. 2016;11:e0147343. doi: 10.1371/journal.pone.0147343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yilmaz M., Maass D., Tiwari N., Waldmeier L., Schmidt P., Lehembre F., Christofori G. Transcription factor Dlx2 protects from TGFβ-induced cell-cycle arrest and apoptosis. EMBO J. 2011;30:4489–4499. doi: 10.1038/emboj.2011.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Han A.L., Veeneman B.A., El-Sawy L., Day K.C., Day M.L., Tomlins S.A., Keller E.T. Fibulin-3 promotes muscle-invasive bladder cancer. Oncogene. 2017;36:5243–5251. doi: 10.1038/onc.2017.149. [DOI] [PubMed] [Google Scholar]
- 55.Xiao S., Yang M., Yang H., Chang R., Fang F., Yang L. miR-330-5p targets SPRY2 to promote hepatocellular carcinoma progression via MAPK/ERK signaling. Oncogenesis. 2018;7:90. doi: 10.1038/s41389-018-0097-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen T., Yang Z., Liu C., Wang L., Yang J., Chen L., Li W. Circ_0078767 suppresses non-small-cell lung cancer by protecting RASSF1A expression via sponging miR-330-3p. Cell Prolif. 2019;52:e12548. doi: 10.1111/cpr.12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cui L., Nai M., Zhang K., Li L., Li R. lncRNA WT1-AS inhibits the aggressiveness of cervical cancer cell via regulating p53 expression via sponging miR-330-5p. Cancer Manag. Res. 2019;11:651–667. doi: 10.2147/CMAR.S176525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ding K., Wu Z., Wang N., Wang X., Wang Y., Qian P., Meng G., Tan S. MiR-26a performs converse roles in proliferation and metastasis of different gastric cancer cells via regulating of PTEN expression. Pathol. Res. Pract. 2017;213:467–475. doi: 10.1016/j.prp.2017.01.026. [DOI] [PubMed] [Google Scholar]
- 59.Chen X., Xu H., Ding L., Lou G., Liu Y., Yao Y., Chen L., Huang W., Fu X. Identification of miR-26a as a target gene of bile acid receptor GPBAR-1/TGR5. PLoS ONE. 2015;10:e0131294. doi: 10.1371/journal.pone.0131294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Z., Xie Q., Yu Z., Zhou H., Huang Y., Bi X. A regulatory loop containing miR-26a, GSK3β and C/EBPα regulates the osteogenesis of human adipose-derived mesenchymal stem cells. Sci. Rep. 2005;5:15280. doi: 10.1038/srep15280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhuang C., Wang P., Huang D., Xu L., Wang X., Wang L., Hu L. A double-negative feedback loop between EZH2 and miR-26a regulates tumor cell growth in hepatocellular carcinoma. Int. J. Oncol. 2016;48:1195–1204. doi: 10.3892/ijo.2016.3336. [DOI] [PubMed] [Google Scholar]
- 62.Bennett C.N., Ross S.E., Longo K.A., Bajnok L., Hemati N., Johnson K.W., Harrison S.D., MacDougald O.A. Regulation of Wnt signaling during adipogenesis. J. Biol. Chem. 2002;277:30998–31004. doi: 10.1074/jbc.M204527200. [DOI] [PubMed] [Google Scholar]
- 63.Courtois S., Lehours P., Bessède E. The therapeutic potential of metformin in gastric cancer. Gastric Cancer. 2019;22:653–662. doi: 10.1007/s10120-019-00952-w. [DOI] [PubMed] [Google Scholar]
- 64.Tsai K.W., Lo Y.H., Liu H., Yeh C.Y., Chen Y.Z., Hsu C.W., Chen W.S., Wang J.H. Linc00659, a long noncoding RNA, acts as novel oncogene in regulating cancer cell growth in colorectal cancer. Mol. Cancer. 2018;17:72. doi: 10.1186/s12943-018-0821-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tsai K.W., Leung C.M., Lo Y.H., Chen T.W., Chan W.C., Yu S.Y., Tu Y.T., Lam H.C., Li S.C., Ger L.P. Arm Selection Preference of MicroRNA-193a Varies in Breast Cancer. Sci. Rep. 2016;6:28176. doi: 10.1038/srep28176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tseng H.H., Tseng Y.K., You J.J., Kang B.H., Wang T.H., Yang C.M., Chen H.C., Liou H.H., Liu P.F., Ger L.P., Tsai K.W. Next-generation Sequencing for microRNA Profiling: MicroRNA-21-3p Promotes Oral Cancer Metastasis. Anticancer Res. 2017;37:1059–1066. doi: 10.21873/anticanres.11417. [DOI] [PubMed] [Google Scholar]
- 67.Pan C.T., Tsai K.W., Hung T.M., Lin W.C., Pan C.Y., Yu H.R., Li S.C. miRSeq: a user-friendly standalone toolkit for sequencing quality evaluation and miRNA profiling. BioMed Res. Int. 2014;2014:462135. doi: 10.1155/2014/462135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu W.S., Chan S.H., Chang H.T., Li G.C., Tu Y.T., Tseng H.H., Fu T.Y., Chang H.Y., Liou H.H., Ger L.P., Tsai K.W. Isocitrate dehydrogenase 1-snail axis dysfunction significantly correlates with breast cancer prognosis and regulates cell invasion ability. Breast Cancer Res. 2018;20:25. doi: 10.1186/s13058-018-0953-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Betel D., Wilson M., Gabow A., Marks D.S., Sander C. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36:D149–D153. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.