

Summary
In Caenorhabditis elegans, targeted genome editing techniques are now routinely used to generate germline edits. The remarkable ease of C. elegans germline editing is attributed to the syncytial nature of the pachytene ovary which is easily accessed by microinjection. This protocol describes the step-by-step details and troubleshooting tips for the entire CRISPR-Cas genome editing procedure, including gRNA design and microinjection of ribonucleoprotein complexes, followed by screening and genotyping in C. elegans, to help accessing this powerful genetic animal system.
For complete details on the use and execution of this protocol, please refer to Ghanta and Mello (2020).
Subject areas: genetics, model organisms, molecular biology, CRISPR
Graphical abstract
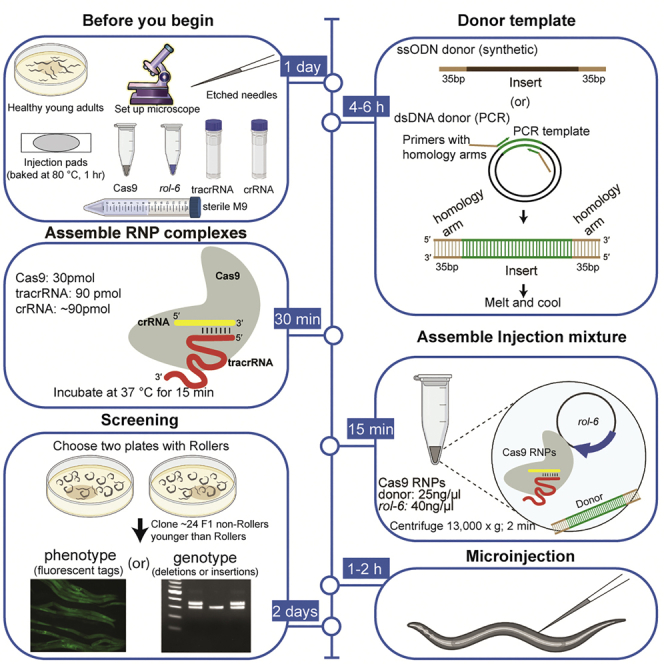
Highlights
-
•
Step-by-step microinjection protocol for CRISPR-Cas genome editing in C. elegans
-
•
Tips to troubleshoot microinjection and genome editing protocols
-
•
Melting dsDNA donor molecules potentiates precision genome editing
-
•
A single injected hermaphrodite can produce dozens of precisely edited progenies
In Caenorhabditis elegans, targeted genome editing techniques are now routinely used to generate germline edits. The remarkable ease of C. elegans germline editing is attributed to the syncytial nature of the pachytene ovary which is easily accessed by microinjection. This protocol describes the step-by-step details and troubleshooting tips for the entire CRISPR-Cas genome editing procedure, including gRNA design and microinjection of ribonucleoprotein complexes, followed by screening and genotyping in C. elegans, to help accessing this powerful genetic animal system.
Before you begin
Culturing worms
Timing: 3 days
Worms grown for microinjection should be healthy — fed well, contamination free and under controlled temperature. An approach that ensures ideal worms for injection is to culture broods from individual animals. Animals cultured by chunking Nematode Growth Medium (NGM) agar tend to vary significantly in their quality for the purpose of injections. If chunked worms must be used it is helpful to pick several L3 and L4 hermaphrodites onto a fresh plate one day prior to injection. Synchronized worms obtained by bleaching gravid adults can also be used, but synchrony limits the variety of stages available from the culture on a given day to a much narrower developmental window than is achieved by brood culture.
The choice of animals may also depend on the needle. A blunt or broken needle can still work for injections into older larger adults but will invariably kill young adults. When attempting the first injections for a new batch of animals, pay close attention to the size of the gonad cytoplasm. Recently starved adults may appear healthy otherwise but will have a shrunken core gonadal cytoplasm that provides a very poor target for microinjection. Unhealthy animals are a very common reason for unsuccessful injections. Refer to the WormBook chapter for detailed protocols on maintenance of C. elegans (Stiernagle, 2006).
Preparation of injection pads
Dried 2% agarose pads (also known as injection pads) are used to immobilize the worms during the microinjection procedure (Fire, 1986; Mello and Fire, 1995). If not prepared properly the pads can either dehydrate the worms too quickly and/or worms may not stick to them. High quality injection pads are critical to performing good injections.
-
1.
Prepare 2% (w/v) agarose solution in distilled water by briefly bringing to a boil in a microwave oven. Keep the solution as liquid and well mixed on a stir plate with medium heat (with a stir bar).
-
2.
Use a Pasteur pipette to place two drops (∼50 μL each) of hot agarose solution on a 24 x 60 mm glass coverslip and immediately drop another coverslip on top, forming an agarose sandwich, such that the coverslips are parallel to each other.
-
3.
After the flattened agarose solidifies (∼5 min) remove one coverslip by gently sliding.
-
4.
Place the coverslips with wet agarose facing up on Whatman paper cut to fit on an oven tray for baking. Array the coverslips singly so they do not stack or overlap.
-
5.
Repeat the agarose sandwich process until about 100 pads are arrayed on the Whatman paper for baking.
-
6.
Bake the agarose pads at 80°C for 1 h.
-
7.
Once dried, the agarose pads will not stick to each other can be transferred to a 10 cm petri dish and stored together indefinitely. In areas with high humidity, it may be necessary to use a sealed desiccated container, or to re-bake the pads periodically as needed. For convenience, correct side (agarose side) of the slide can be marked with an asymmetric letter before storing in the container.
Preparation of needles
Here we describe needle preparation method that we routinely use in the lab, other labs may use slightly different techniques. Injection needles are prepared by pulling and etching borosilicate glass capillaries (1.2 mm) with inner filaments. The inner glass filament provides capillary action and aids in easier back filling of the needles. Although pre-pulled needles (e.g., Eppendorf Femtotips or Tritech Research, MINJ-PP) can be purchased, they are unnecessary and cost-prohibitive in our opinion. Needles can be stored and used indefinitely (at least in the order of months) without losing their shape due to glass flow.
-
8.Pulling the capillaries: Needles suitable for penetrating the outer cuticle of adult worms must be both rigid and sharp. Such needles are easily prepared using an economical needle puller. There are three key adjustment parameters: the “heat” setting, a “gentle pull” setting, and a “strong pull” setting. During the gentle pull the needle gradually lengthens as the glass closest to the filament warms, upon reaching a predetermined length, this slow pull triggers a strong pull that draws the needle out and breaks the filament into two halves. If your device pulls from one end only, the end closer to the filament will have a longer taper, however both needles should be nearly identical at the tip and thus both halves should be saved for use.
-
a.Adjusting the needle puller is an empirical process that can be greatly aided by keeping a reference needle to use for comparison. Needles should taper to a sharp stiff point. These parameters are attained by adjusting the “heat” and “pull-strength” settings on the puller. In general, lower heat and greater pull strength give stiffer needles. Occasionally, the heating filament is bumped by the blank needle and becomes bent. Use extra care when loading or unloading the needles to avoid this. Examine the first few needles under a dissecting scope comparing their shapes to a reference needle kept for this purpose. The settings we use may not translate to other systems and are only approximate starting values (Heat: 97°C, magnetic sub:22 and magnetic main: 90 on Narishige PN-30 needle puller). These settings should be optimized for your system.
-
b.Once the settings are optimized, pull needles and monitor needle shape by eye and by periodically re-checking the needles under the dissecting scope. If the needles begin to deviate in shape or if the puller begins to delay during the slow pull phase or stops pulling altogether, the filament was likely bumped out of alignment. Carefully, re-align the filament, repeat the adjustments to the heat and pull strength, and then continue pulling until you have made enough needles to last for several weeks.
-
c.Load the pulled needles into a tray or box (with a lid) by propping them up in rows on Time Tape (rolled lengthwise with sticky side out) or along a thin line of rolled clay.
-
a.
-
9.Etching Needles: Etching the needles takes less than 1 min per needle and is worth the added time as it saves hours that will otherwise be lost attempting to brake open needles on the inverted microscope. To etch the needles, pressurize each needle one at a time by loading them onto the microinjection system set to 80 psi. This is easily done by sliding the Tygon tubing from the system’s compressed air source directly over the blunt end of the needle. While holding the needle in your hand, apply pressure (using the foot pedal from the injector system). Keep the needle pressurized throughout the test. If the needle tubing interface leaks (emits a hissing sound) it is necessary to seat the needle more firmly.
-
a.Pipette 50 μL of distilled water near the center of a 35 mm plastic petri dish (do not use a glass dish). Pipette a second drop of 15 μL water nearby about a half inch from the larger drop. Carefully, using a plastic pipette tip add 15 μL of a commercially sourced 48% HF solution to the 15 μL drop of water to obtain a drop containing 24% HF.
-
b.Bubble test: Place the petri dish containing the water drop and 24% HF droplets under the dissection scope and carefully, at low magnification, bring the pressurized needle close to the (larger) water drop, pause before touching. If the needle is badly broken, then before the needle touches the water drop you will see the water drop distort from air blown from the needle. Immediately discard such needles. Otherwise, while keeping the needle pressurized, insert the tip of the needle into the water drop. If the needle emits a slow stream of tiny air bubbles, immediately remove it from the water while keeping it pressurized (to prevent water from entering the needle). The needle can then be transferred directly to a final storage tray, with lid (marked as “bubbles in water”). These needles have coarse openings and though useful are not ideal, unless for example an injection solution contains particulates that are clogging finer tipped needles.
-
c.If the needle fails to bubble in water, transfer the needle (while keeping it pressurized) into the drop of HF solution. In about one or two seconds it should start to bubble in HF. Immediately transfer back to water, (again maintaining pressure at all times, including during the transfer). Usually, the needle will not bubble in water. If so, transfer it to a tray labeled “bubbles in HF.” These are ideal needles. If the needle produces a slow stream of bubbles in the second water test, it can be saved in the “bubbles in water” tray. A fast bubble stream in water, indicates a faulty needle that is over etched, and should be discarded. If this happens repeatedly add additional water to further dilute the HF solution.
-
d.Store the etched needles in a closed container on rolled tape or clay so that the needles are firmly secured.
-
a.
CRITICAL: Keep the needle pressurized throughout the entire process. If HF enters the needle, it will quickly over-etch and destroy the needle. If water is allowed to enter the needle it will leave residues when it evaporates and could also dilute your injection mixes.
Alternatives: Validating the needles in the bubble tests is a major time saver. When using pre-tested needles, a single needle can be loaded with each injection mix and will almost always yield excellent flow. If untested needles are used, or if a needle becomes clogged while in use, the needle can be opened by physically breaking the tip against the agarose pad or by touching it to the edge of a coverslip.
Note: After several needles are etched it may be necessary to trim a few mm off of the Tygon tubing to remove cracks that prevent a good seal. Etching can also be done by loading each needle into a needle holder that creates a seal with a screw lock and gaskets. However, we find this takes more time than using the simpler direct tubing interface.
Preparation of stock solutions
Note: proceed to either step 10 or 11 based on the Cas protein used in this protocol.
-
10.If using Cas9, prepare Cas9 RNPs
-
a.Cas9 protein- aliquot 0.5 μL (5 μg or 30 pmol) either in PCR tubes or 1.5 mL tubes and store at −80°C (avoid freeze/thaw cycles)
-
b.tracrRNA – 0.4 μg/μL (18 μM) in IDT nuclease free duplex buffer, store at −20°C (store 30 μL aliquots at −80°C for long term storage)
-
c.crRNA – 0.4 μg/μL (34 μM) in TE pH 7.5, store at −20°C (store 10 μL aliquots at −80°C for long term storage)
-
a.
-
11.If using Cas12a, prepare Cas12a RNPs
-
a.Cas12a protein- aliquot 0.5 μL (5 μg or 32 pmol) and store at −80°C (avoid freeze/thaw cycles)
-
b.Cas12a-crRNA – 40 μM in TE pH 7.5, store at −20°C (store 10 μL aliquots at −80°C for long term storage)
-
a.
-
12.
ssDNA oligo donor – 1 μg/μL in nuclease free water, store at −20°C
-
13.
PRF4::rol-6 (su1006) plasmid (midi prep): 500 ng/μL working solution in nuclease free water, store at −20°C.
Note: Commercial Cas9 protein can be diluted 1:1 with 1× PBS and stored as 2.5 μg (0.5 μL) aliquots to reduce the total injection volume to 10 μL. We chose to reconstitute tracrRNA in Duplex buffer to provide salts to the Cas9 RNP complexes. We have not explored the long-term stability and cutting efficiencies of the injection mixtures prepared with tracrRNA reconstituted in other solvents such as TE or water. We have also not explored replacing TE or water with Duplex buffer in the injection mixture. Aliquots of Cas9 protein can be stored at −80°C for at least a year without compromising efficiencies. crRNAs and tracrRNA can be stored at −80°C for at least two years and at −20°C for at least a year without compromising efficiencies. Refer to previously published protocols if Cas9 and guide RNA plasmid-based approach is used (Dickinson et al., 2015; Schwartz and Jorgensen, 2016).
Guide RNA design
Depending on the GC content and the availability of Protospacer Adjacent Motif (PAM) sites either Cas9 (PAM: NGG; N is any DNA base) or Cas12a (Cpf1) (PAM: TTTV; V is A, C, or G) can be used as nuclease to introduce double strand breaks. In our experience Cas9 guide RNAs (gRNA) with low GC content perform poorly. It may be more efficient to use Cas12a (Ebbing et al., 2017; Ghanta and Mello, 2020) at those AT-rich target sites.
-
14.
To generate loss of function deletions, choose two gRNA target sites such that majority of the gene is deleted. To generate HDR mediated knock-in alleles, choose a PAM site that is as close as possible to the site of insertion. Bioinformatics tools such as CRISPOR (http://crispor.tefor.net/) (Concordet and Haeussler, 2018) or CHOPCHOP (https://chopchop.cbu.uib.no) (Labun et al., 2019) help in identifying gRNAs that have no off-target cleavage activity. When the choice of guide RNA is more restricted (e.g., to insert tags) these tools can be used to identify potential off-target sites that can be screened for later and crossed out of the strains. If using C. elegans strains other than N2, polymorphisms specific to your strains need to be considered for gRNA design and off-target assessment.
Templated precise editing
Although non-homologous end joining (NHEJ) has been shown to be the dominant type of repair mechanism in many cell types, this is not the case in the pachytene germline of C. elegans. When using properly prepared donor molecules, up to two thirds of the post injection progeny will exhibit homology-directed repair (Ghanta and Mello, 2020) (Ghanta et al., 2021). Depending on the length of the desired edit, one can choose to use short single stranded oligodeoxynuleotides (ssODN) (Arribere et al., 2014; Dokshin et al., 2018; Farboud et al., 2019; Paix et al., 2017; Prior et al., 2017; Zhao et al., 2014; Ward, 2015) or linear double stranded DNA (dsDNA) (Dokshin et al., 2018; Farboud et al., 2019; Ghanta and Mello, 2020; Paix et al., 2015; Paix et al., 2017; Vicencio et al., 2019) as donor templates. Refer to (Ghanta and Mello, 2020) (Ghanta et al., 2021) for HDR efficiencies achieved using the protocols described here.
-
15.
ssODN Donor Design
ssODNs up to 200 nt can be obtained commercially at reasonable prices. Therefore, any edit that is shorter than 130 bp can easily be obtained using a synthetic ssODN donor (35 nt homology arms) (Dokshin et al., 2018; Paix et al., 2017).
-
a.
To generate short inserts: To generate short inserts (<130 bp), add 5′ homology sequence (∼35 nt) in front of the tag (or mutations) and 3′ homology sequence (∼35 nt) at the end. To prevent recutting of the edited allele, introduce silent mutations into the PAM site or into the guide binding sequence in the donor molecule if it is not already split by the insert. Three or four silent mutations in the PAM proximal region should suffice and 35nt after the final silent mutation should be used as homology arm (Figure 1A).
Figure 1.

Donor design strategy
(A) If the site of insertion of a tag (knock-in) and the site of double strand break are not close to each other, disrupting the internal homology between these two sites increases the efficiency of precise repair. Sequence with disrupted homology should be considered as part of the insert and the homology arm should begin after the last silent mutation.
(B) Schematic design for a set of locus-specific homology arms (HA) with PCR primers which also serve as linkers between the tag and the protein of interest. For a given locus, the same set of oligos can be used to generate dsDNA donors to knock-in any tag. Plasmid containing the tag flanked by linkers is used as PCR template. Diagrams are not drawn to scale.
-
b.
To generate point mutations: Choose 35 nt sequence upstream and 35 nt sequence downstream of the DSB site as homology arms for the donor. Introduce the desired nucleotide changes in the donor and the PAM or guide binding sequences. Silent mutations can be introduced to create restriction sites to easily genotype precisely edited progeny of the injected animals.
-
c.
To generate large deletions: Use two guides that remove majority of the target locus (for example, to precisely remove a structural domain or an entire coding region). Order an ssODN that contains 35 nt of sequence upstream of the left DSB and 35 nt downstream of the right DSB to act as a donor. You may wish to incorporate a new gRNA binding site on this donor so that you can re-cut the locus for any downstream purposes. Deletions of 1 kb are routinely achieved with this strategy but it can also work for larger deletions. Donor can be omitted if precise deletion is not necessary.
-
16.
dsDNA Donor design and generation
Generate dsDNA donor templates using unmodified or 5′ Triethylene Glycol (TEG) modified primers. TEG modification is also known as spacer 9 (SP9). Melting the dsDNA donors is critical to achieve high HDR efficiencies (Ghanta and Mello, 2020). Using end-modified donors (and melting) further increases HDR efficiencies (Ghanta et al., 2021).-
a.Synthesize unmodified (or 5′ SP9 modified) PCR primers with standard desalting (IDT); 35 nt as homology arms and 20 nt complementary to the desired insert (e.g., gfp). SP9 modifications are available at 100 nmol scale from IDT.
-
b.Perform PCRs to amplify the insert (e.g., gfp) from a plasmid using High-Fidelity polymerase.
-
c.Perform agarose gel electrophoresis to ensure that a single specific band is obtained. If non-specific amplification is observed, set up a temperature gradient (50°C–64°C) and find the optimal annealing temperature.
-
d.Purification of donor DNA: Use one of the following methods depending on your experimental conditions.
-
i.Perform PCR purification using spin-columns (e.g., Qiaquick Minelute) and elute DNA in 15–20 μL of nuclease free water. Generally, column purification is sufficient, and you may proceed to step 18e. However, some primer (55 nt long) pairs produce long dimers that may contain the homology arms. Spin-columns may not be able to remove long (>80 bp) dimers completely. These short “dimer donors” out compete the full-length donors (e.g., gfp) and integrate heavily into the DSB site.Note: Dimers may or may not be visible on the agarose gel. It is helpful to use a primer analysis tool (e.g., OligoAnalyzer-IDT) to predict homo- or hetero-dimer formation.
-
ii.If primer-dimer formation is a concern, perform PCR clean-up using 0.6× SPRI paramagnetic beads (AMPure XP) to size-exclude shorter (<200 bp) products. For example, incubate 100 μL of PCR product with 60 μL of beads, wash the beads with 70% ethanol twice and elute with nuclease free water (refer to the manufacturer’s protocol for detailed procedure).
-
iii.Gel-extract the donor DNA if primer dimers are clearly visible on agarose gel. However, gel extracted DNA can be toxic due to the presence of guanidine hydrochloride (component of binding buffer) in the final elute. Strong absorbance at 230 nm on Nanodrop suggests presence of GuHCl in the elute. During the wash steps, incubating the spin-columns with wash buffer for 10 min before centrifugation helps in reducing salt contamination; repeat the washes 2 to 3 times. For best results, gel-extracted DNA should be further purified with 1× to 1.5× AMPure-XP beads (strongly recommended).
-
i.
-
e.Dilute a portion of the purified donor to 100 ng/μL and transfer about 5.5 μL (for 20 μL injection mixture) to a PCR strip tube and proceed to the melting step.
-
f.“Melt” the donor DNA using thermal cycler:
-
a.
| Temperature | Time |
|---|---|
| 95°C | 2 min |
| 85°C | 10 s |
| 75°C | 10 s |
| 65°C | 10 s |
| 55°C | 1 min |
| 45°C | 30 s |
| 35°C | 10 s |
| 25°C | 10 s |
| 4°C | hold |
Ramp down at 1°C/s at every step
Note: We store purified donors at −20°C and melt them right before adding to the injection mix. We have not explored storage and re-use of melted donors. We have only tested the editing efficiencies with donors melted at 50–150 ng/μL concentrations. Melting donors at concentrations that are too high or too low may negatively influence editing efficiencies.
Alternatives: If the goal is to insert a common tag such as FLAG, GFP, degron etc. then it is a good idea to order a set of homology arms with “universal” PCR primers which also serve as linkers between the tag and the protein of interest. A single set of gene-specific homology arm primers can then be used to insert any tag into your locus of interest (Figure 1B). It is almost always more cost effective to order universal primers, even if inserting a short tag such as FLAG could be achieved with ssODN donors. Any gene you decide to study will be tagged in multiple different ways during the course of your work, and universal primers will save both time and money. Plasmids containing a set of commonly used tags flanked by these linkers are available from Addgene (see key resources table).
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Hydrofluoric acid (HF) | Sigma-Aldrich | 339261 |
| Halocarbon oil- 700 CAS#9002-83-9 | Sigma-Aldrich | H8898 |
| Agarose | Genesee Scientific | 20-102GP |
| tracrRNA | IDT | Cat# 1072532 |
| TE 7.5 (10 mM Tris, 0.1 mM EDTA) | IDT | Cat#11-01-02-02 |
| Nuclease-Free Duplex Buffer (30 mM HEPES, pH 7.5; 100 mM potassium acetate) | IDT | Cat#11-01-03-01 |
| Polyethylene Glycol 8000 (PEG) | MP | #195445 |
| Q5 polymerase | NEB | Cat# M0491S |
| S. pyogenes Cas9 3NLS protein | IDT | Cat#1081058 |
| Cas12a protein | IDT | Cat#10001272 |
| Critical commercial assays | ||
| Gel Extraction Kit | QIAGEN-Qiaquick | #28706 |
| Experimental models: Organisms/Strains | ||
| C. elegans N2 strain | Caenorhabditis Genetics Center | N/A |
| Oligonucleotides | ||
| CRISPR-Cas9 crRNA, 2 nmol | IDT | N/A |
| A.s. Cas12a crRNA, 2 nmol | IDT | N/A |
| ssODN donors (ultramer) | IDT | N/A |
| GFP forward primer | This study | AGTAAAGGAGAAGAACTT |
| GFP reverse primer | This study | TTTGTATAGTTCATCCATGC |
| Universal linker forward | This study | TCCGGAGGGAGTGGA |
| Universal linker reverse | This study | AGAACCTCCGCCACC |
| Recombinant DNA | ||
| GFP-linker plasmid | Addgene | N/A |
| FLAG-TEV-degron-linker plasmid | Addgene | N/A |
| mCherry linker plasmid | Addgene | N/A |
| PRF4::rol-6 (su1006); now named pCCM958 | Addgene | N/A |
| Other | ||
| Glass capillaries | World Precision Instruments | #1B120F-4 |
| Cover slips 24 x 60 mm No.1 | Globe Scientific Inc | #1419-10 |
| Cover slips 22 x 22 mm No.1 | Globe Scientific Inc | #1404-10 |
| Mouth pipette -15 In Drummond aspirator tube assembly | Thermo Fisher Scientific | #2118010 |
| PCR purification columns | QIAGEN-Minelute | #28604 |
| Ampure XP beads | Beckman Coulter | Ref# A63880 |
| Tygon tubing E-3603 (ID:1/32 in; OD:3/32 in; Wall: 1/32 in) | Saint-Gobain | #00444 |
| Microloader Tips (Femtotips) | Eppendorf | 930001007 |
| Dissecting scope | Nikon | SMZ745 |
| Inverted microscope (DIAPHOT 200) | Nikon | Current successor: Eclipse Ti2 |
| Fluorescence dissecting microscope | Zeiss | Axio Zoom. V16 |
| Needle Puller Narishige PN-30 | Narishige | Current successor: PC-100 |
| Microinjector | Tritech Research, Inc | Analog MINJ-1 |
| Micromanipulator | Narishige | MN-151 |
| Thermal Cycler | Bio-Rad | 1851148 |
Materials and equipment
Reaction setup:
| Reagent | Amount |
|---|---|
| 5× Q5 Reaction Buffer | 10 μL |
| 10 mM dNTPs | 1 μL |
| 10 μM Forward Primer | 2.5 μL |
| 10 μM Reverse Primer | 2.5 μL |
| Q5 DNA Polymerase | 0.5 μL |
| Template Plasmid (5 ng) | 1.0 μL |
| Nuclease-Free Water | 33.5 μL |
| Total | 50 μL |
Thermocycling conditions:
| Step | Temperature | Time |
|---|---|---|
| Initial denaturation | 98°C | 1 min |
| Amplification (30 cycles) | 98°C | 10 s |
| 64°C | 20 s | |
| 72°C | 40 s | |
| Final extension | 72°C | 3 min |
| 4°C | Hold |
If necessary, find optimal annealing temperature by performing a thermal gradient (50°C–64°C).
While this protocol has focused on GFP knock-in with the microinjection equipment detailed in the key resources table, the general principles of the procedure described here can be used for inserts of comparable size using other equipment. GFP sequence is provided in Data S1.
Step-by-step method details
Note: If using Cas9, follow steps 1–8; if using Cas12a, follow steps 9–16.
Genome editing using Cas9 nuclease
Add components of the injection mixture to the tube containing Cas9 in the following order:
-
1.
Prepare Cas9 – 0.5 μL of 10 μg/μL stock (30 pmol)
-
2.
Add tracrRNA – 5 μL of 0.4 μg/μL stock (90 pmol)
-
3.
Add crRNA – 2.8 μL of 0.4 μg/μL stock (95 pmol) (if two guides are needed add 1.4 μL of each)
-
4.
Pipette the mixture gently several times and incubate at 37°C for 15 min.
-
5.Add ssODN donor – 2.2 μL of 1 μg/μL stock (or)
- Add melted dsDNA – 500 ng (final concentration: 25 ng/μL for ∼1 kb donors or 45 fmol/μL)
-
6.
Add PRF4::rol-6 (su1006) plasmid – 1.6 μL of 500 ng/μL solution
-
7.
Add nuclease free water to bring the final volume to 20 μL and pipette gently several times.
-
8.
To avoid needle clogging, centrifuge the mixture 13000 x g for 2 min, transfer about 17 μL of the mixture to a fresh tube and keep the tube on ice; proceed to loading the needles.
Genome editing using Cas12a nuclease
Add components of the injection mixture to the tube containing Cas12a in the following sequence:
-
9.
Cas12a – 0.5 μL of 10 μg/μL stock (32 pmol)
-
10.
Add Cas12a-crRNA – 2.5 μL of 40 μM stock (100 pmol)
-
11.
Add TE pH 7.5– 3.0 μL
-
12.
Pipette the mixture gently several times and incubate at 37°C for 15 min
-
13.Add ssODN donor – 2.2 μL of 1 μg/μL stock (or)
- Add melted dsDNA – 500 ng (final concentration: 25 ng/μL for ∼1 kb donors or 45 fmol/μL)
-
14.
Add PRF4::rol-6 (su1006) plasmid – 1.6 μL of 500 ng/μL stock
-
15.
Add nuclease free water to bring the final volume to 20 μL and pipette gently several times.
-
16.
To avoid needle clogging, centrifuge the mixture at 13000 x g for 2 min, transfer about 17 μL of the mixture to a fresh tube and keep the tube on ice; proceed to loading the needles.
Note:
Injection mixtures can be prepared at room temperature.
1:3 molar ratio of nuclease to guide RNA is used to saturate the nuclease. We have not explored the cutting efficiencies with other molar ratios.
Protein aggregation or precipitation is not an issue at these RNP concentrations.
TE is added in step 11 of Cas12a mixtures for easier pipetting. This step can be omitted by further reducing the concentration of crRNA stock.
Although we haven’t explored the optimal dose range for ssODNs, given the efficiencies obtained with dsDNA at 25 ng/μL, much lower doses of ssODN could be used (Paix et al., 2017).
Final injection mixture can be stored at 4°C and re-used for more than a year without compromising efficiencies. We have injected 16 months old mixes and confirmed that HDR efficiencies are high.
In our experience adding any double stranded DNA before RNP complex formation reduces HDR efficiency.
Loading needles and preparing the microscope
-
17.
Using microloader tips, load 1.5 μL of the injection mixture into an etched needle.
-
18.
Leave the needle pointing tip downward for several minutes to allow air bubbles to work their way out of the tip. This can be done easily by sticking the needle to double sided tape or clay on a vertical surface.
-
19.
Before mounting your needle onto the micro-manipulator make sure to align and adjust the microscope. To do this place a worm on an injection pad under halocarbon oil. Bring the worm into focus under the 10× objective, then close down the condenser iris and make sure the shadow of the iris is centered in the field of view and sharply in focus. Also adjust the Nomarski optics and ensure that you have the proper filters and polarizers in the light path. Consult your manual for appropriate steps to focus the condenser and adjust the microscope. These adjustments are essential and must be done properly or the injections will be difficult if not impossible.
-
20.
Insert the needle into the tube connected to the pressure regulator and lock it in place on the needle holder, then position the holder downward (∼15°) relative to the stage. Position the needle using the Z-axis adjustment such that it doesn’t touch the injection pad or the condenser when the microscope head is lowered.
-
21.
With the light on and the condenser iris closed down partially, position the needle using the coarse lateral adjustments so that the needle tip is illuminated in the center of the light. If your manipulator is mounted to the condenser arm, these adjustments can be made while the arm is tilted back away from the stage. Do not look through the eyepiece when making these coarse adjustments. Instead, look directly at the needle tip as you move the lateral and longitudinal adjustments on the manipulator. First, with the needle tip pushed past the center of the light path, move it forward or backward with the coarse adjustments until it is illuminated. You will see a spark of light when the needle is in the light path (close down the condenser iris to further pinpoint the needle, via this spark of light, in the center of the field of view). Next, move the needle laterally (to the right if using a right-handed system) until the spark of light disappears, and then move it back toward the left until it lights again. Now raise the needle up close to the condenser to ensure ample working distance when you lower the condenser arm. Assuming you successfully aligned your microscope light source as described in step 19 above, your needle tip is now in the center of the field of view and will only need to be lowered in order to bring it into focus when needed.
-
22.
Using the worm loaded earlier as a reference, focus on the worm using the 10× objective and then while watching the needle directly (not through the eyepiece) use the Z-axis adjustment to lower the needle until it touches the oil above the worm.
-
23.
Look through the eyepiece and slowly center and bring the tip of needle into focus using X-, Y- and Z-axis adjustments on the micromanipulator. Once the tip is in focus, do not change the positions of X- or Y- axis.
-
24.
Using 40× objective, examine the needle tip and apply pressure continuously for one or two seconds. One can judge the size of the opening (hence the quality of the needle) based on the diameter of the bubble produced in this interval. The bubble should be about the diameter of the gonad after one second. Adjust the pressure to the needle as needed. Pressures in the range of 40 to 80 psi are typically used during injection.
Mounting worms
Worms move by swimming in a thin aqueous layer that surrounds their body surface. Agarose pads immobilize the animals by drying this layer of moisture, sticking the animals to the pad surface. If the pad is too thick it will quickly desiccate the animal, if too thin, the animal will crawl away. Even high-quality injection pads will dry the worms out after a few minutes, so do not mount more worms than you can easily inject in a 10-to-15-min interval. With experience mounting each worm should take less than one minute.
-
25.
Determine the upper side of the injection pad by scratching an edge of the dried agarose with a razor blade or other metal object. This scratch mark can also be used for breaking needles later if needed. Mark on the agarose side of the pad can also be used if the slide is marked with an asymmetric letter.
-
26.
If desired, you can mark the boundaries of the agarose with a thin marker on either surface of the coverslip. Since the dried agarose is virtually transparent marking the boundaries helps in placing the worms on the agarose.
-
27.
Use a Pasteur Pipette to transfer a small drop of halocarbon oil onto the injection pad away from the agarose (Figure 2). Use surface tension (not suction) on the pipette to move the oil to the injection pad. For working under the dissecting microscope place the injection pad on an inverted lid of petri dish for easier handling.
-
28.
Touch your worm pick to the surface of the halocarbon drop, and then use the viscous oil on the bottom of your pick to collect one or a few worms from your culture plate using the dissecting microscope. It is usually possible to choose worms that are off the bacterial lawn, but it may be necessary to move the worms briefly to an unseeded plate prior to picking them up with the halocarbon oil.
Alternatives: Excess bacteria can also be removed from the worms after transferring them to the large halocarbon oil droplet on the injection pad. After depositing the worms into the large oil droplet, flame the pick and return to the oil drop. First quench the pick in the oil away from the worms and then move the pick underneath the worms and then quickly pull the worms through the oil to remove bacteria. Once the desired number of animals are floating in the oil drop and free of bacteria, select the worms for injection by placing the pick under them in the oil and moving them by lifting them on the pick to a dry area of the pad. Depending on your abilities and your experimental needs you can transfer either one (recommended for beginners), or up to several animals onto your pad for the first round of injections.
-
29.
Touch the pick down to the surface and move it laterally to deposit the worms in a row one after another (Figure 2). They should float off the pick in the oil. Carefully orient the worms with vulva and row of eggs facing to the left (if a right-handed system is being used). Gently push the worm down with the worm-pick by rubbing the pick along the body of the animal. If the animal is mostly stuck, then move onto the next animal. Make sure all the animals remain stuck and properly oriented. If possible, keep the worms within about 2–3 mm of each other so that you can move from one to the next without difficulty while working on the inverted microscope (Figure 2). Finally, after the worms are stuck down dip your pick back into the large oil drop on the edge of the pad, then use your pick to deposit additional oil to fully cover the immobilized worms. This additional oil will prevent the worms from desiccating during the procedure.
Alternatives: If you accidently orient some worms incorrectly you can either reorient them by gently pushing the worm off the pad with your pick and re-sticking them, or you can inject the worms oriented one way and then flip the slide to inject the ones facing the other way.
Figure 2.

Mounting worms
Use halocarbon oil to mount worms onto the injection pad. Align all the pachytene zones (sites of injection) of all the gonad arms towards the needle. Diagrams are not drawn to scale.
Microinjection and Worm recovery
Microinjection into syncytium of hermaphrodite gonad (Mello et al., 1991; Stinchcomb et al., 1985) delivers Cas9-guideRNA complexes into dozens of germ nuclei simultaneously. Aim to inject the animals in the mid pachytene region. Injecting either in the distal or in the proximal end of the gonad is not ideal. Since the number of embryos produced by wildtype animals is limited by the availability of sperm, germ cells present in the distal region of the gonad arms do not get fertilized. Therefore, you should target the germ cells in the mid to late pachytene region. It should take approximately one minute to inject both gonad arms of each worm, and another minute or two to remove the injected animals to culture plates.
-
30.
Focus the objective (40×) onto the gonad (pachytene region) so that you can see two rows of nuclei (as shown in Figure 3A). This focal plane ensures that when inserted, the tip of the needle enters the core of the gonad and not above or below the somatic gonad. Do not focus on the honeycomb structure of the germ nuclei.
-
31.
Without changing the focus and while looking through the eyepiece, slowly lower the needle into the oil drop. By adjusting the position of the stage bring the cuticle (at the pachytene region) very close to the needle tip (Figure 3A).
-
32.
Gently tap the micromanipulator so that the needle penetrates the cuticle and the somatic gonad, and enters the core germplasm. Gently move the stage towards the needle to adjust the position of the tip in the gonad. Do not insert the needle too far into the gonad as it ruptures the cuticle.
-
33.
Press the foot pedal to inject the mixture into the germplasm and make sure that the injection mixture flows smoothly onto either side of the needle tip over a period of two seconds (Methods Video S1). You should see the fluid entering the proximal region of the gonad and pushing the oocytes towards the spermatheca (Methods Video S1 and Figure 3B). If the flow is excessive the worm or gonad can explode, reduce the pressure to achieve a lower flow rate. Inject into both arms of the gonad.
Figure 3.

Microinjection
(A) Using the 40× objective, focus on the two rows of germline nuclei as shown and insert the needle into the pachytene zone. Flow of the injection mixtures should reach mature oocytes in the proximal end of the germline as shown with yellow dotted line.
(B) Snapshots from Methods Video S1 are shown to illustrate the flow of the injection mixture. Germline is shown (1) before and (2) after injection. Arrows marks the region in the gonad until where the mixture has reached. 10× magnification is shown for illustration purposes, use 40× objective for the microinjection procedure.
-
34.
Raise the needle to remove the injection pad. Move the pad onto the inverted lid under the dissecting scope.
-
35.
Recover the injected worms using mouth pipette (pulled glass capillary attached to Drummond aspirator tube) and sterile M9 buffer or MPEG (0.5% PEG in M9). Occasionally, worms stick to the glass pipette during the recovery process, and they are less likely to stick when MPEG is used. Other buffers are not necessary. Avoid using plastic pipette tips to recover the injected worms because they will stick to the tips.
-
36.
If the injections differed in quality, for example if only one arm was injected, these differences should be noted, and the worms separated, NGM plates labeled according to injection quality. When all the worms are injected place each injected animal singly onto a fresh NGM plate and culture for about 3.5 days at room temperature (22°C–23°C) unless genetic or experimental conditions demand other temperature conditions.
Screening and genotyping
If both arms of the hermaphrodite gonad are injected, a good injection should yield 20–40 F1 Rollers. Screening strategy depends on the type of edit generated.
-
37.About 72 h post injection, score the number of F1 Rollers and choose two plates with the highest number. The Rollers provide an important trouble-shooting metric (see below) and should always be included whenever possible.
-
a.For indels (without donor template) or ssODN donors, choose two P0 plates that segregate the highest number of F1 Rollers; pick 12–24 young adult F1 Rollers onto separate plates and allow them to have progeny prior to genotyping by PCR.
-
b.For long dsDNA donors screening should be performed differently depending on the quality of the injections and the type of edit desired. If excellent bilateral injections were performed resulting in greater than 20 Roller progeny per injected animal, then choose two plates that segregate the highest number of F1 Rollers. If a fluorescent protein tag was inserted a quick look under the compound fluorescence microscope (or a dissecting Fluorescence microscope if the fluorescence is expected to be bright) will allow you to gauge how many F1 progeny to pick. To check under the compound microscope (preferred for genes whose expression levels are low or unknown) place ∼50 gravid adult F1 progeny younger than the rollers onto 2% wet agarose pads on a microscope slide (do not use dry injection pads) and immobilize with levamisole, then push the worms into proximity with the pick, gently place a cover slip on the top and check for fluorescent signal. If you wish you can memorize the location of positive animals and while working on a dissecting scope push the cover slip aside (do not flick it open) to directly recover the fluorescent positive animals. If PCR genotyping is the desired approach for detection, choose about 24 non-Rollers that are younger than the Rollers and place them onto separate NGM plates. Younger animals among Rollers can also be picked. For inexperienced injectors, we recommend using 5′ end-modified dsDNA donors and picking F1 Rollers.
-
a.
-
38.
Genotype the F1 adults directly after allowing them to lay progeny. Alternatively, to avoid PCR false positives due to mosaicism, lyse several F2s from each plate and genotype. At least one of the genotyping primers should lie outside of the homology arms to avoid amplification from transiently retained donor molecules.
Note: Strains obtained by genome editing should be back crossed to eliminate off-target indels. However, off-target indels on the same chromosome and close to the target site may be linked to the target site. Therefore, generating at least two independent alleles using different gRNAs is recommended.
Expected outcomes
This protocol is used to generate precise genome edits in C. elegans germline. Loss of function alleles, point mutants and fluorescent tags can be generated with high efficiencies. A single injected animal can produce precision edits in among as many as two thirds of progeny from the post-injection cohort (Ghanta and Mello, 2020). These advances increase the utility of C. elegans for understanding the fundamentals of animal biology and for investigating the physiology of alleles implicated in disease.
Limitations
Obtaining high knock-in efficiencies of large inserts (for example, 3 kb or more) is still a challenge. Future improvements in protocols and development of new strategies are required to address this limitation.
Troubleshooting
Problem 1
Worms mounted onto injection pads dry immediately (step 29)
Potential solution
Ensure that the agarose solution is made with water. Beginners may mistakenly use DNA gel running buffers instead of water.
Ensure that the pads are thoroughly dried by baking and that they are not too thick
Animals that are too young tend to dry much faster
Ensure that the animals have never starved and are well fed
Problem 2
Worms don’t stick to the injection pads (step 29)
Potential solution
Injection pads may not be dry. Re-bake the pads at 80°C for an hour
Worms may be covered with excess amounts of bacteria. Make sure that bacteria are not transferred onto the injection pad
Problem 3
Needles clog during injections (step 33)
Potential solution
Dust particles in the injection mixture clog the needles. Make sure that the mixtures are spun down at high speed for at least 2 min prior to loading into the needles
High concentrations of protein could clog the needles. Ensure that the correct concentrations are used in the injection mixture
Needle opening may be too small: gently touch the tip of the needle to the agarose to break it open
Problem 4
Needle opening is either too wide or too narrow, or tip is too flexible (bends when pushed against cuticle (step 32)).
Potential solution
Adjust the settings of the needle puller to obtain tapered stiff needles
Adjust the contact time of the needles in HF
Problem 5
Low survival rate of the injected animals (step 35)
Potential solution
Choose young adults for injection
Blunt needles with wide openings could severely damage the cuticle
Injected worms should be recovered with care using M9 buffer
Problem 6
Small brood sizes after injection (step 37)
Potential solution
Small brood sizes are observed if the target locus of the guide RNA is essential for viability
Make sure that the donor DNA is free of contaminants such as high salts and that the concentrations of all the injection mixture components are optimal
Use young adults for injections. Old animals do not yield many progeny
Do not inject excess amounts of the mixture. If flow is excessive, reduce the pressure to lower the flow rate
Problem 7
No Rollers (step 37)
Potential solution
Check the integrity of the rol-6 marker plasmid on agarose gel. If degradation is observed, re-extract plasmid DNA (midi-prep).
Use appropriate concentrations of the Cas9 RNPs, donor DNA and rol-6 plasmid as suggested in the protocol. Excess amounts of Cas9 RNPs or DNA can be toxic.
For beginners, practicing microinjection procedure by injecting Prf-4: rol-6 plasmid alone can be helpful. More than 40 F1 Rollers can be obtained from each injected animal.
If loss of function alleles of the target locus impair gamete or embryo viability, the number of F1 Rollers produced will be dramatically low.
Problem 8
No indels (step 38)
Potential solution
AT rich Cas9 guide RNAs can be inefficient. If the target sequence is AT rich, switch to Cas12a based editing.
Use appropriate concentrations of Cas9 and crRNAs as suggested in the protocol. We recommend using a final concentration of 1.5 μM Cas9 RNPs in the mixture. Increasing the concentration to 3 μM does not increase cutting efficiency (Dokshin et al., 2018).
Problem 9
Indels at homologous locus (step 38)
Potential solution
If an identical target site exists in another locus, majority of the F1 progeny may have indels at both the loci. Use a different guide to avoid indels at the homologous locus.
If the guide choice is limited, reduce Cas9 RNP concentration by five-fold. Overall indel efficiencies may drop but single mutants can be easily isolated.
Problem 10
No GFP insertions (steps 37 and 38)
Potential solution
Make sure that the dsDNA donor is melted before adding to the Cas9 RNP mixture
Donor DNA should be free of any primer dimer or smaller DNA species that can potentially inhibit HDR efficiencies
It is difficult to obtain HDR insertions with inefficient guide RNA. Genotype and sanger sequence the target locus in about 12-24 Rollers and ensure that cutting efficiency of the guide is high. Alternatively, TIDE analysis on pooled F1 genomic DNA can be performed (Dokshin et al., 2018) (Brinkman et al., 2014). Also, rule out partial insertion of the donor (e.g., due to primer dimer insertion).
If the expression pattern of the protein is unknown it may be helpful to perform fluorescence-based screens with animals of mixed stages including older embryos on the plates.
If the initial screening was performed by fluorescence microscopy, genotype the locus by PCR and sanger sequence the target site. Low expression levels may be the reason for “No GFP”.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Craig C. Mello (craig.mello@umassmed.edu).
Materials availability
All materials are available upon reasonable request.
Acknowledgments
We thank the members of the Mello lab and the worm community for sharing their observations and difficulties which helped in making the protocols better. This work was funded by NIH grant R37GM058800 to C.C.M. C.C.M is a Howard Hughes Medical Investigator. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
Author contributions
K.S.G. and C.C.M. wrote the manuscript. K.S.G., T.I., and C.C.M. performed the experiments. C.C.M. supervised the study.
Declaration of interests
Some of the findings described here are part of the patent applications filed by the University of Massachusetts Medical School with K.S.G and C.C.M. as co-inventors. C.C.M. is a scientific founder and advisory board member of CRISPR therapeutics.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100748.
Supplemental information
Data S1. GFP sequence
Flow of injection mixture is shown. Foot pedal was briefly pressed for three times to push the injection mixture towards mature oocytes in the proximal gonad. Video was recorded with 10× objective for illustration purposes, use 40× objective for the microinjection procedure.
Data and code availability
This study did not generate or analyze any data sets
References
- Arribere J.A., Bell R.T., Fu B.X., Artiles K.L., Hartman P.S., Fire A.Z. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics. 2014;198:837–846. doi: 10.1534/genetics.114.169730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman E.K., Chen T., Amendola M., van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concordet J.P., Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018;46:W242–W245. doi: 10.1093/nar/gky354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson D.J., Pani A.M., Heppert J.K., Higgins C.D., Goldstein B. Streamlined genome engineering with a self-excising drug selection cassette. Genetics. 2015;200:1035–1049. doi: 10.1534/genetics.115.178335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokshin G.A., Ghanta K.S., Piscopo K.M., Mello C.C. Robust genome editing with short single-stranded and long, partially single-stranded DNA donors in Caenorhabditis elegans. Genetics. 2018;210:781–787. doi: 10.1534/genetics.118.301532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebbing A., Shang P., Geijsen N., Korswagen H. Extending the CRISPR toolbox for C. elegans: Cpf1 as an alternative gene editing system for AT-rich sequences. MicroPubl Biol. 2017;2017 doi: 10.17912/W2237D. 10.17912/W2237D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farboud B., Severson A.F., Meyer B.J. Strategies for efficient genome editing using CRISPR-Cas9. Genetics. 2019;211:431–457. doi: 10.1534/genetics.118.301775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A. Integrative transformation of Caenorhabditis elegans. EMBO J. 1986;5:2673–2680. doi: 10.1002/j.1460-2075.1986.tb04550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanta K.S., Chen Z., Mir A., Dokshin G.A., Krishnamurthy P.M., Yoon Y., Gallant J., Xu P., Zhang X.-O., Ozturk A. 5′ modifications improve potency and efficacy of DNA donors for precision genome editing. bioRxiv. 2021:354480. doi: 10.7554/eLife.72216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanta K.S., Mello C.C. Melting dsDNA donor molecules greatly improves precision genome editing in Caenorhabditis elegans. Genetics. 2020;216:643–650. doi: 10.1534/genetics.120.303564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labun K., Montague T.G., Krause M., Torres Cleuren Y.N., Tjeldnes H., Valen E. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019;47:W171–W174. doi: 10.1093/nar/gkz365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello C., Fire A. DNA transformation. Methods Cell Biol. 1995;48:451–482. [PubMed] [Google Scholar]
- Mello C.C., Kramer J.M., Stinchcomb D., Ambros V. Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paix A., Folkmann A., Rasoloson D., Seydoux G. High efficiency, homology-directed genome editing in Caenorhabditis elegans using CRISPR-Cas9 ribonucleoprotein complexes. Genetics. 2015;201:47–54. doi: 10.1534/genetics.115.179382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paix A., Folkmann A., Seydoux G. Precision genome editing using CRISPR-Cas9 and linear repair templates in C. elegans. Methods. 2017;121-122:86–93. doi: 10.1016/j.ymeth.2017.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior H., Jawad A.K., MacConnachie L., Beg A.A. Highly efficient, rapid and Co-CRISPR-independent genome editing in Caenorhabditis elegans. G3 (Bethesda) 2017;7:3693–3698. doi: 10.1534/g3.117.300216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M.L., Jorgensen E.M. SapTrap, a toolkit for high-throughput CRISPR/Cas9 gene modification in Caenorhabditis elegans. Genetics. 2016;202:1277–1288. doi: 10.1534/genetics.115.184275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiernagle T. WormBook. 2006. Maintenance of C. elegans; pp. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinchcomb D.T., Shaw J.E., Carr S.H., Hirsh D. Extrachromosomal DNA transformation of Caenorhabditis elegans. Mol. Cell Biol. 1985;5:3484–3496. doi: 10.1128/mcb.5.12.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicencio J., Martinez-Fernandez C., Serrat X., Ceron J. Efficient generation of endogenous fluorescent reporters by nested CRISPR in Caenorhabditis elegans. Genetics. 2019;211:1143–1154. doi: 10.1534/genetics.119.301965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward J.D. Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics. 2015;199:363–377. doi: 10.1534/genetics.114.172361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P., Zhang Z., Ke H., Yue Y., Xue D. Oligonucleotide-based targeted gene editing in C. elegans via the CRISPR/Cas9 system. Cell Res. 2014;24:247–250. doi: 10.1038/cr.2014.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. GFP sequence
Flow of injection mixture is shown. Foot pedal was briefly pressed for three times to push the injection mixture towards mature oocytes in the proximal gonad. Video was recorded with 10× objective for illustration purposes, use 40× objective for the microinjection procedure.
Data Availability Statement
This study did not generate or analyze any data sets