

Abstract
Background
At diagnosis, the majority of patients with intrahepatic cholangiocarcinoma (IHCC) present with advanced disease and a poor prognosis. Comprehensive genomic profiling (CGP) early in the disease course may increase access to targeted therapies and clinical trials; however, unresolved issues remain surrounding the optimal biopsy type to submit for CGP.
Patients and Methods
Mutational frequencies between primary tumor biopsies (Pbx), metastatic biopsies (Mbx), and liquid biopsies (Lbx) in 1,632 patients with IHCC were compared.
Results
Potentially actionable alterations were found in 52%, 34%, and 35% of patients in the Pbx, Mbx, and Lbx cohorts, respectively. In Pbx, Mbx, and Lbx, FGFR2 rearrangements were found in 9%, 6%, and 4%, and IDH1 mutations were identified in 16%, 5%, and 9% patients, respectively. Moreover, alterations in FGFR2 and IDH1 were significantly associated with distinct ancestries, including 2.1‐fold enrichment for FGFR2 rearrangements in patients with African ancestry and 1.5‐fold enrichment for IDH1 mutations in patients with admixed American (Hispanic) ancestry. Finally, the publication of biomarker‐driven clinical trials in IHCC correlated with changing CGP testing patterns. Significant correlations between patient characteristics and IHCC trial disclosures were observed, including a significant decrease from time between biopsy and CGP testing, and more frequent testing of primary versus metastatic samples.
Conclusion
Overall, because of the high likelihood of identifying actionable genomic alterations, CGP should be considered for the majority of patients with inoperable IHCC, and Lbx and Mbx can be considered as part of the diagnostic suite.
Implications for Practice
Comprehensive genomic profiling (CGP) should be considered for all patients with intrahepatic cholangiocarcinoma (IHCC) or suspected IHCC, as actionable alterations were commonly found in multiple genes and a wide variety of FGFR2 fusion partners were identified. The disclosure of IHCC trial data correlated with increased use of CGP, an encouraging trend that moves new therapeutic options forward for rare cancers with a rare biomarker. Although tissue from the primary lesion may identify actionable alterations at higher rates, CGP of a liquid biopsy or metastatic site can be considered, particularly if the primary tissue block is exhausted.
Keywords: Intrahepatic cholangiocarcinoma, Bile duct neoplasms, Comparative genomics, Liquid biopsy
Short abstract
Comprehensive genomic profiling (CGP) early in the disease course may increase access to targeted therapies and clinical trials; however, unresolved issues remain for the use of CGP for patients with intrahepatic cholangiocarcinoma. This article focuses on the frequency of specific genomic alterations and encouraging trends toward therapeutic options.
Introduction
Intrahepatic cholangiocarcinoma (IHCC) is the second most common primary liver cancer, accounting for 10%–20% of tumors, and has increased in incidence by 350% over the past 35 years [1, 2, 3]. At diagnosis, the majority of patients with IHCC present with poor prognosis, and thus comprehensive genomic profiling (CGP) early in the disease course is critical to increase the chances of access to targeted therapies and biomarker‐driven clinical trials [4]. Currently, the prognosis is approximately 3 years with resectable disease, 12–15 months if unresectable, and 9 months for patients with stage IV disease [5, 6, 7, 8].
Previous studies estimate that 35%–50% of patients with IHCC have potentially actionable genomic alterations [9, 10, 11, 12, 13, 14]. In smaller datasets, rearrangements and fusions involving the FGFR2 gene have been observed in 11%–17% of IHCC, and patients harboring these alterations have an improved prognosis but may be less likely to benefit from first‐line platinum chemotherapy [15, 16, 17, 18, 19, 20, 21]. Recent phase I and II trials of fibroblast growth factor receptor inhibitors (FGFRis) have demonstrated promising results in the second‐line setting, and pemigatinib was recently granted accelerated approval by the U.S. Food and Drug Administration (FDA) for the treatment of adults with previously treated, unresectable, locally advanced or metastatic cholangiocarcinoma with an FGFR2 fusion or other rearrangement as detected by an FDA‐approved test [22]. FGFR2 can be activated by gene fusions that leave its tyrosine kinase domain intact as well as truncating rearrangements that delete inhibitory motifs encoded by the last exon, and in the FIGHT‐202 trial, responses to pemigatinib were observed in patients with both gene fusions and nonfusion rearrangements [23, 24]. Additional genomic alterations with phase II or III efficacy data in IHCC include mutant IDH1, BRAF V600E, and HER2 amplifications or mutations [25, 26, 27]. Actionable biomarkers in IHCC as the result of pantumor FDA approvals include microsatellite instability (MSI; MSI‐high/deficient mismatch repair), tumor mutational burden (TMB) ≥10 mutations (mut)/Mb, and NTRK fusions. Other genomic alterations with mature data in other tumor types and evolving data in IHCC include alterations in BRCA1/2, IDH2, MET, PIK3CA, EGFR, and others [9, 10, 11, 12, 28]. Thus, it is increasingly recognized that multiple potentially actionable genomic alterations are commonly observed in IHCC.
Several unresolved issues must be addressed to optimize routine CGP in IHCC. First, limited data exist regarding which biopsy types are most appropriate for testing. Although primary tumor tissue‐based testing (Pbx) is assumed to be the gold standard, it is not clear whether metastatic tissue‐based testing (Mbx) or liquid biopsy–based testing (Lbx) detect genomic alterations as frequently as Pbx. Liquid biopsy followed by sequencing of circulating tumor DNA (ctDNA) allows for noninvasive mutational profiling, and recently published studies suggest that Lbx profiling can interrogate tumor heterogeneity in cholangiocarcinoma and other gastrointestinal cancers, as well as acquired resistance mutations to FGFRis [29, 30]. Additionally, clinical responses to FGFRis solely based on Lbx profiling have been reported [30, 31]. However, discordance between Pbx and Lbx in small datasets has been observed, which can be caused by a variety of factors including baiting differences between assays and usage preferences [32, 33]. In the real‐world setting, Lbx can provide genomic results quickly, a key issue for patients with poor prognoses, and can reduce the risk associated with obtaining a new biopsy. Thus, a detailed analysis comparing biopsy types can help weigh risk versus benefit of obtaining a new biopsy.
The primary objective of this study was to query a large database of 1,632 patients diagnosed with IHCC to determine whether genomic alterations differ in frequency between Pbx, Mbx, and Lbx. Secondary objectives were to determine whether key alterations in IHCC are enriched in specific ancestries and to assess whether a trend exists toward patients receiving CGP earlier in their disease course as a likely result of the publication of biomarker‐driven trials.
Methods
Approval for this study, including a waiver of informed consent and a Health Insurance Portability and Accountability Act waiver of authorization, was obtained from the Western Institutional Review Board (Protocol No. 20152817). Analysis was performed on an international cohort of patients from the Foundation Medicine (FMI) database, which consisted of samples submitted for routine clinical testing from 2013 to 2019. Diagnosis, biopsy site, and the date of specimen collection were extracted from test requisition forms and pathology reports. A hybrid capture‐based next‐generation sequencing assay was performed on patient samples in a Clinical Laboratory Improvement Amendments–certified, College of American Pathologists–accredited, New York State–approved laboratory (Foundation Medicine, Cambridge, MA). Tissue‐based testing assessed 318–327 cancer‐related genes, MSI status, and TMB [34]. For Lbx, 20‐mL peripheral blood samples were collected, plasma was isolated, ≥20 ng of DNA was extracted, and CGP was performed with the 62‐gene FoundationACT or the 70‐gene FoundationOne Liquid assay (Foundation Medicine, Cambridge, MA) [35]. Both tissue and liquid biopsy–based tests assessed base substitutions, short insertions/deletions, rearrangements/fusions, and copy number variations. Programmed death‐ligand 1 (PD‐L1) expression in tumor cells (tumor proportion score [TPS]) was measured by immunohistochemistry (Dako 22C3). Low positive was defined as ≥1% and <50% expression, and high positive was defined as ≥50% expression. TMB was determined on 0.8–1.11 Mb of sequenced DNA [36]. MSI was determined on 95–114 loci [37], and the calculation of MSI‐high frequency excluded cases in which MSI could not be determined.
Biopsy site comparative analysis was performed on 1,632 patients with IHCC from the FMI database. Differences in alteration frequencies were evaluated by Fisher's exact test with Bonferroni correction for multiple comparisons. The analysis of ancestry and CGP testing patterns was performed on 5,241 patients with IHCC. The biopsy site analysis cohort is smaller than ancestry and testing pattern cohorts because of date of analysis and information availability from pathology reports. For pattern analysis, time to result represents the duration of time between the specimen collection date and the generation of results from the FoundationOne analysis pipeline. Differences in age and time to result were determined using the Wilcoxon rank sum test.
Genomic ancestry was determined on 5,241 patients with IHCC based on previously published methods [38, 39, 40]. Briefly, >40,000 single nucleotide polymorphism (SNP) sites sequenced by CGP were identified, and a random forest classifier was trained on the 1,000 Genomes samples to identify ancestral populations (African [AFR], admixed American/Hispanic [AMR], East Asian [EAS], European [EUR], South Asian [SAS]) using genetic variation at the SNP sites. Genetic variation was defined by five features that captured allele‐count variation as determined by principal component analysis. This classifier was applied to CGP samples to assign them to one of the ancestral populations. To quantitate the genetic admixture in CGP samples, an unsupervised, maximum likelihood estimation approach was applied to SNPs in the 1,000 Genomes samples to learn a population structure for five ancestral signatures. The population structure was then projected onto the SNP alleles in CGP samples to quantitate a sample's genetic admixture.
Results
Biopsy Site Comparative Analysis
The comparison between Pbx, Mbx, and Lbx was performed on all patients from the IHCC cohort who had detailed biopsy information available and received FoundationOne testing (n = 1,632 patients). Mbx sites included lymph nodes (63), soft tissues (47), peritoneum (34), lung/pleura (27), omentum (15), bone (10), abdomen (7), gynecologic tract (5), brain (2), upper gastrointestinal (2), colon (2), bladder (1), and adrenal (1). Patient characteristics and genomic findings are summarized in Table 1 and Figure 1. The Pbx cohort of 1,048 patients was 51% female and had a median age of 65 years. FGFR2 rearrangements (including fusions) were present in 9% of Pbx patients and IDH1 mutations in 16%. Fifty‐eight unique FGFR2 fusion partners were identified (Fig. 2).
Table 1.
Primary tissue vs. metastatic tissue vs. liquid biopsy patient characteristics and comparative genomics
| Characteristic | Pbx | Mbx | Lbxa |
|---|---|---|---|
| Patients, n | 1,048 | 216 | 364 |
| Male/female, % | 49/51 | 56/44 | 52/48 |
| Median age (range), yr | 65 (23–89+) | 64 (29–89+) | 66 (29–88) |
| Cases with an actionable alteration, % | 52 | 34 | 35a |
| Potentially targetable alterations, % | |||
| BRAF V600E | 6 | 2 | 3 |
| BRCA1/2 | <0.1/<0.1 | <0.1/<0.1 | 1.9/3.3 |
| ERBB2 (HER2) | |||
| Amplification | 6 | 4 | 4 |
| Mutation | 2 | 2 | 2 |
| FGFR2 rearrangement | 9 | 6 | 4 |
| IDH1/2 | 16/4 | 5/4 | 9/3 |
| KRAS G12 | <1 | 2 | 1 |
| PIK3CA | 4 | 4 | 4 |
| Currently untargetable alterations, % | |||
| TP53 | 32 | 35 | 40 |
| CDKN2A | 31 | 31 | 4b |
| CDKN2B | 23 | 24 | 1b |
| KRAS | 16 | 34 | 13 |
| MTAP | 16 | 16 | N/A |
| BAP1 | 15 | 11 | N/A |
| TERT | 8 | 4 | 6 |
| SMAD4 | 5 | 11 | N/A |
| MYC | 5 | 5 | 1 |
| Biomarkers and alterations linked to immunotherapy response | |||
| MSI‐high, % | 0.7 (n = 1,036) | 1 (n = 215) | 0 (n = 224) |
| PD‐L1 low positive, % | 15 (n = 345) | 18 (n = 66) | N/A |
| PD‐L1 high positive, % | 5 (n = 345) | 18 (n = 66) | N/A |
| TMB median, mut/Mb | 2.5 | 2.5 | N/A |
| TMB ≥10 mut/Mb, % | 4 | 4 | N/A |
| TMB ≥20 mut/Mb, % | 1 | 1 | N/A |
| ARID1A, % | 19 | 16 | N/A |
| PBRM1, % | 12 | 14 | N/A |
| STK11, % | 2 | 8 | 3 |
| MDM2, % | 4 | 7 | 2 |
| KEAP1, % | 1 | <1 | N/A |
The liquid biopsy gene panel was narrower than tissue testing and did not include TMB and genes listed as N/A.
Short variants only.
Abbreviations: Lbx, liquid biopsy; Mbx, metastatic biopsy; MSI, microsatellite instability; mut, mutation; Pbx, primary tumor biopsy; PD‐L1, programmed death‐ligand 1; TMB, tumor mutational burden.
Figure 1.

The genomic landscape of intrahepatic cholangiocarcinoma by biopsy type. (A): Pbx, (B): Mbx, (C): Lbx, and (D): Pbx vs. Mbx. Pbx was not compared with Lbx because of gene panel differences. Abbreviations: Lbx, liquid biopsy; Mbx, metastatic tissue; Pbx, primary tumor tissue.
Figure 2.
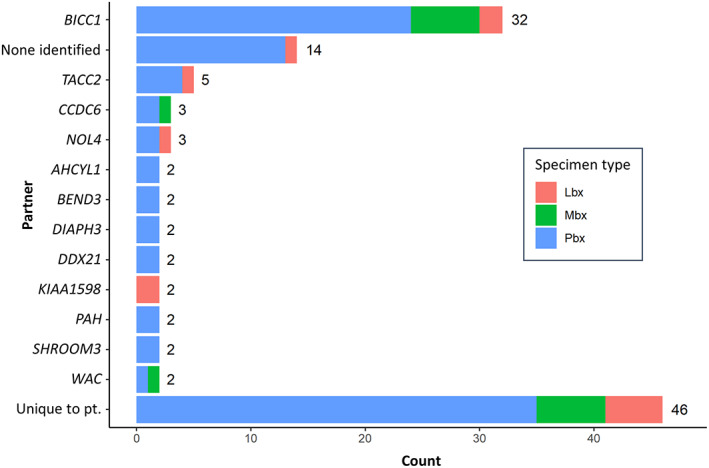
FGFR2 fusion partners in intrahepatic cholangiocarcinoma identified by comprehensive genomic profiling. Abbreviations: Lbx, liquid biopsy; Mbx, metastatic tissue; Pbx, primary tumor tissue; pt, patient.
European Society for Medical Oncology Scale for Clinical Actionability of molecular Targets framework was used to establish a list of actionable alterations for patients with IHCC [41]. A patient was defined as having a potentially actionable alteration if they had either of the following: (a) alterations with positive data in IHCC or basket trials (i.e., FGFR2 rearrangements, IDH1/2 mutations, HER2 copy number amplifications or mutations, BRAF mutations, MSI‐high, TMB ≥10 mut/Mb, and NTRK1‐3 fusions) or (b) alterations with positive phase III clinical data in other tumor types (i.e., BRCA1/2 pathogenic alterations, EGFR activating alterations, KRAS G12C mutations, MET exon 14 skipping alterations, PIK3CA mutations, and fusions in ALK, RET, and ROS1). Using this definition, the Pbx cohort was more likely to have a potentially actionable alteration than the Mbx cohort (52% vs. 34%, respectively; p = .0008). More specifically, IDH1 alterations were significantly less frequent in Mbx than Pbx (p < .001; Fig. 1D). IDH1 alterations and FGFR rearrangements were found in 16% and 9% of the 1,048 Pbx patients, respectively, and 5% and 6% of the 216 Mbx patients, respectively. However, KRAS mutation frequency was doubled in Mbx compared with Pbx and Lbx (p < .001). Immunotherapy biomarkers were detected at similar levels between tissue biopsy cohorts (Table 1). The frequency of TMB‐high (≥10 mut/Mb) was 4% in both Mbx and Pbx, and median TMB was also identical (2.5 mut/Mb). TMB was not assessed in the Lbx cohort. MSI‐high was rare in all cohorts, with <0.01%, 1%, and 0% of patients classified as MSI‐high in the Pbx, Mbx, and Lbx, respectively. Alterations in STK11 (p < .001) and SMAD4 (p = .0016) were more frequently identified in Mbx.
The analysis of Lbx samples revealed that despite more limited baiting for FGFR2, IDH1, and other genes on the older liquid biopsy assays studied, ctDNA‐based genomic profiling identified multiple genomic alterations in IHCC and was generally representative of the landscape observed in Pbx (Fig. 1). However, IDH1 and FGFR2 alterations were detected at a lower frequency in Lbx compared with Pbx (Table 1), possibly related to variable ctDNA shed. Thirty‐five percent of Lbx patients had actionable alterations compared with 52% and 34% in the Pbx and Mbx cohorts, respectively. Notably, alterations in genes associated with clonal hematopoiesis (CH), such as CHEK2, ATM, and TP53, were more prevalent in Lbx versus Pbx and Mbx (Fig. 1C), which is consistent with this known limitation of ctDNA genotyping. Overall, the biopsy site comparative analysis found differences between cohorts; however, the overall genomic landscape between Pbx, Mbx, and Lbx were very similar and high frequencies of actionability were observed.
Ancestry Analysis of FGFR2 Rearrangements and IDH1 Mutations
The worldwide epidemiology of cholangiocarcinoma shows significant geographic variations, with lower rates in Western countries versus Asia [5]. It is unclear whether genomic alterations relevant to IHCC correlate with ancestry. We determined genomic ancestry for each patient from >40,000 single nucleotide polymorphisms based on previously published methodology [38, 39, 40]. Ancestral populations were defined as AFR, AMR, EAS, EUR, and SAS. In IHCC tissue specimens, frequencies of FGFR2 rearrangements and IDH1 mutations were both significantly associated with ancestry (Fig. 3A). FGFR2 rearrangements were more frequently observed in AFR, followed in order by EUR, EAS, AMR, and SAS (16%, 8.9%, 8.9%, 6.5%, and 4.7%, respectively). In contrast, IDH1 mutations were most common in AMR, followed in order by EUR, SAS, EAS, and AFR (19.4%, 15%, 12%, 9.5%, and 7%, respectively). Relative to the general population, AFR was 2.1‐fold enriched for FGFR2 rearrangements (p = 2.3e−05) and AMR was 1.5‐fold enriched for IDH1 mutations (p = .0006). Because differences between Pbx and Mbx were observed in the general IHCC population, we compared ancestral groups in Pbx specimens only and observed similar trends (Fig. 3B). An analysis of hepatitis B virus (HBV) DNA sequences in tissue specimens revealed a significant association between HBV and EAS ancestry (odds ratio, 23.8; p < 10e−05), but significant correlations between HBV and FGFR2 rearrangements or IDH1 mutations were not observed.
Figure 3.

FGFR2 and IDH1 alteration frequency by ancestry for all tissue specimens (A) and Pbx only (B). +, positive.
Abbreviation: Pbx, primary tumor tissue.
Correlation Between Genomic Testing Patterns and Biomarker‐Driven Clinical Trial Publications
The disclosure date of IHCC targeted therapy clinical trials was determined via literature search and is summarized in Table 2. Publication of FGFRi trial data in IHCC began in 2018, including data that contributed to the FDA approval of pemigatinib. Thus, for statistical analyses, patients tested pre‐ versus post‐2018 were compared. We analyzed a cohort of 5,241 patients with IHCC who had received tissue‐based CGP testing and stratified patients by year of biopsy (Table 3). The median time between tissue collection and genomic testing in Pbx was significantly shorter in the post‐ versus pre‐2018 patient cohort (p = 1.68e‐11; Fig. 4A). In the post‐2018 cohort, Pbx samples were tested significantly more frequently compared with Mbx (p = .0006; Table 3) and the time from tissue collection to testing was more rapid compared with Mbx (p = 2.3e‐5; Fig. 4B). Interestingly, the median age of patients at time of testing was significantly older in the post‐2018 cohort (p = 5.82e‐21; Fig. 4C). Lbx testing began in 2016 and steadily increased each year with 21, 55, 119, and 169 patients tested in 2016 through 2019, respectively. Together, these findings suggest that the publication of targeted therapy trials in IHCC correlates with changes in genomic testing patterns favoring earlier genomic testing of a population that is more representative of the broader population with IHCC.
Table 2.
Initial disclosure dates of targeted therapy trials in intrahepatic cholangiocarcinoma
| Inhibitor | Target | Disclosure date | Design | Results | Reference |
|---|---|---|---|---|---|
| Infigratinib (QED) | FGFR2 | 1/2018 | PhII n = 61 | ORR 15%, PFS 5.8 mo | [41] |
| Futibatinib (Taiho) | FGFR2 | 6/2018 | PhI n = 45 | ORR 25% | [42] |
| Pemigatinib (Incyte) | FGFR2 | 10/2018 | PhII n = 47 | ORR 24%, PFS 6.8 mo | [43] |
| Dabrafenib + trametinib (Novartis) | BRAF | 1/2019 | PhII n = 33 | ORR 41%, PFS 7.2 mo | [44] |
| Derazantinib (Basilea) | FGFR2 | 1/2019 | PhI/II n = 29 |
ORR 21%, PFS 5.7 mo |
[45] |
| Debio 1347 (Debiopharm) | FGFR2 | 3/2019 | PhI n = 9 | ORR 22% | [46] |
| Neratinib (Puma) | HER2 | 7/2019 | PhII n = 19 | ORR 11%, PFS 1.8 mo | [27] |
| Erdafitinib (Janssen) | FGFR2 | 8/2019 | PhI n = 11 | ORR 27% | [47] |
| Ivosidenib (Agios) | IDH1 | 9/2019 | PhIII n = 185 | ORR 2.4%, PFS 2.7 mo | [48] |
Abbreviations: ORR, overall response rate; PFS, progression‐free survival; PhI, phase I; PhII, phase II; PhIII, phase III.
Table 3.
Patient characteristics by year of biopsy
| Characteristic | All cases | 2013 | 2014 | 2015 | 2016 | 2017 | 2018 | 2019 |
|---|---|---|---|---|---|---|---|---|
| Patients, n | 5,241 | 152 | 266 | 465 | 592 | 1,009 | 1,295 | 1,462 |
| Median age (range), yr | 63 (<1 to >89) | 59 (17–80) | 61 (17–87) | 60 (17–86) | 60 (16–87) | 62 (<1 to >89) | 64 (18–>89) | 65 (23–>89) |
| Female/male, % | 51/49 | 57/43 | 54/46 | 53/47 | 51/49 | 53/47 | 50/50 | 48/52 |
| Tissue source, n (%) | ||||||||
| Liver | 3,966 (76) | 112 (74) | 209 (79) | 347 (75) | 424 (72) | 748 (74) | 972 (75) | 1,154 (79) |
| Metastasis | 1,275 (24) | 40 (26) | 57 (21) | 118 (25) | 168 (28) | 261 (26) | 323 (25) | 308 (21) |
Figure 4.

Comprehensive genomic profiling testing patterns over time. Time between specimen collection and testing in Pbx (A) and comparison between Pbx and Mbx (B). (C): Patient age at time of testing. Abbreviations: Mbx, metastatic tissue; Pbx, primary tumor tissue.
Discussion
This study compares the genomic landscape by biopsy site in a large cohort of patients with IHCC and provides key updates to mutational frequencies from previous smaller studies. Fifty‐two percent of patients with IHCC from the Pbx cohort had potentially actionable genomic alterations, including FGFR2 rearrangements and IDH1 mutations in 9% and 16% of patients, respectively. The definition of actionability incorporated recent clinical trial publications in IHCC and validated clinical evidence from other tumor types, which may explain the higher frequencies relative to previous studies, which ranged from 35% to 50% in IHCC [9, 10, 11, 12, 13, 14]. Previous studies in IHCC consistently reported higher frequencies of FGFR2 rearrangements, ranging from 11% to 17%, and have also found significant associations between FGFR2 rearrangements and younger age, female gender, and earlier‐stage disease [15, 16, 18, 20, 21]. Differences in patient characteristics likely contributed to differences in FGFR2 frequencies between studies, as we report that the median patient age and the proportion of men tested have both increased in recent years. As CGP expands to a broader population with IHCC, these revised frequencies should be noted.
The overall genomic landscape of IHCC is highly similar regardless of biopsy type (i.e., Pbx, Mbx, and Lbx), suggesting that all three types are appropriate for genomic testing if only one option is available or if other patient‐specific factors exist. However, there were statistically significant differences between these groups. Pbx patients had the highest probability of actionability versus Mbx or Lbx, including the highest percentage of FGFR2 rearrangements (9% vs. 6% vs. 4%, respectively). Differences in gene panels between tissue and liquid biopsy assays contributed to this difference and is a limitation of the present study. The Lbx cohort was profiled with older liquid biopsy assays that included narrower coverage of actionable genes such as FGFR2 and IDH1, and TMB and MSI status were not assessed. Newer liquid biopsy assays, such as FoundationOne Liquid CDx, provide broader coverage in genes relevant to IHCC versus the liquid biopsy assays used in this study (FoundationACT and FoundationOne Liquid) [50]. Future studies that control for differences between tissue and liquid gene panels may help strengthen conclusions. The lower frequencies of FGFR2 rearrangements in Lbx and Mbx observed may also be echoing previous reports of FGFR2 rearrangements significantly associating with earlier‐stage disease [16, 20], given that liquid and metastatic biopsies tend to be used more heavily in patients with more‐advanced disease. A limitation of the present study is that tumor size and stage data were unavailable and thus future studies that compare Pbx, Mbx, and Lbx while controlling for tumor stage may help further elucidate differences between cohorts. Previous reports in biliary tract cancers have observed liquid biopsy is more concordant with metastatic biopsies versus primary biopsies [33], which may have also contributed to differences in overall actionability between Lbx and Pbx. To further optimize biopsy type decision‐making, studies using broader liquid biopsy panels, patient‐matched concordance studies, and studies in patients with suspected acquired resistance to targeted therapies are needed. The Lbx cohort also contained a notably high frequency of BRCA1/2 alterations compared with both Pbx and Mbx, which requires further investigation. In the Lbx cohort, alterations in CHEK2, ATM, and TP53—genes that have been associated with CH [51]—were observed to be more prevalent than in the Pbx or Mbx cohorts. Additional studies are needed to clarify the degree of CH contribution to these findings.
Pathological misclassification of adenocarcinoma may have been a factor in the lower frequency of actionable alterations in Mbx. The Mbx cohort featured greater frequencies of KRAS alterations and lower IDH1 and FGFR2 alteration frequencies. As a pathological diagnosis of IHCC is often a diagnosis of exclusion, one potential contributing factor for these findings may be that the Mbx cohort may contain a significant number of non‐IHCC adenocarcinoma cases whose metastatic lesions were actually derived from other primary sites incorrectly assigned the diagnosis of IHCC (i.e., they represent carcinoma of unknown primary site cases). The Mbx findings share similarities with our previous study on the genomic landscape of extrahepatic cholangiocarcinoma (EHCC), which also revealed greater frequencies of KRAS alterations (43% of patients) and found no cases of FGFR2 rearrangements or IDH1 mutations (n = 99) [52]. The Mbx data set was not enriched in HER2 copy number amplifications, in contrast to previous reports of increased frequencies of HER2+ EHCC relative to IHCC [53]. The Mbx genomic findings highlight that treating patients based on pathological diagnosis of the Mbx without genomic results can potentially lead to suboptimal treatment.
The likelihood of patients testing positive for immunotherapy biomarkers was highly similar between the Pbx and Mbx cohorts. TMB ≥10 mut/Mb and median TMB levels were identical between the two cohorts. The Lbx cohort received CGP using assays that did not include TMB assessment. More recently developed liquid biopsy assays, such as FoundationOne Liquid CDx, include blood TMB (bTMB) [50], and additional studies that compare bTMB to tissue‐based TMB in IHCC will provide further insight into the concordance between Lbx and Pbx. MSI‐High was infrequent across specimen types but was higher in Mbx compared with Pbx and Lbx. PD‐L1 positive trended slightly higher in Mbx and was not evaluable via Lbx. Additional biomarker analyses of recently published immunotherapy trials relevant to IHCC, such as the phase II CA209‐538, may help refine the role of immunotherapy biomarkers [54].
Although FGFR2 rearrangements and IDH1 mutations were both commonly observed in all ancestry groups, significant variation was observed. The AFR ancestry group was significantly enriched for FGFR2 rearrangements, and the AMR ancestry group was significantly enriched for IDH1 mutations. Differences in genomic alteration frequencies between ancestry groups may help reconcile differences in alteration frequencies between this study and previous smaller studies in IHCC. Causal factors behind the observed variation between ancestry groups remain unknown and could be explored in future studies. Previous studies have identified differences in genomic alterations, including IDH1 mutations, between HBV+ and HBV– patients with IHCC of EAS ancestry [55, 56]. Although a significant association between HBV DNA in patient specimens and EAS ancestry was observed in this data set, significant correlations between HBV and FGFR2 or IDH1 alterations were not observed.
By segmenting patients by year of biopsy, it was found that the disclosure of FGFRi and other biomarker‐driven trial data in IHCC beginning in 2018 significantly correlated with changes in genomic testing patterns. The number of patients with IHCC receiving CGP has rapidly grown in the last few years, with growth observed in both tissue and liquid biopsy–based testing. Additionally, patients in the post‐2018 cohort had specimens submitted for CGP testing more rapidly, and these specimens were more likely to be primary tumor versus metastatic. The post‐2018 cohort was significantly older, which more closely aligns with the average age of diagnosis in Western countries [2] and may also explain why the frequency of FGFR2 rearrangements is lower in this study compared with previous findings. Taken together, these data suggest that prior to FDA approval of the first targeted therapy in IHCC, CGP and precision oncology are potentially being considered earlier in the disease course in patients with IHCC. This encouraging trend may continue to help provide biomarker informed therapeutic options for patients with rare cancers.
We anticipate that when targeted therapies, such as FGFRis, are ultimately approved in the first‐line setting for patients with IHCC, CGP has the potential to become the standard of care to identify these patients earlier in their disease course. Additionally, given the potential misclassification between IHCC and EHCC in Mbx samples, these patients with unknown or uncertain primaries could be considered for inclusion in biomarker‐driven trials. The current trends in testing patterns and actionability of findings suggest that IHCC is evolving into a leading example of a rare tumor type benefiting from advances in precision oncology and personalized medicine.
Conclusion
In patients with IHCC or suspected IHCC, actionable alterations in multiple genes were commonly found and a wide variety of FGFR2 fusion partners were identified, demonstrating that CGP should be considered for all these patients. Actionable alterations may be identified at higher rates using tissue from the primary tumor; however, a liquid biopsy or metastatic site can be considered, particularly if the primary tissue block is exhausted. This study found that the disclosure of IHCC trial data correlated with increased use of CGP, which is an encouraging trend, suggesting new therapeutic options are moving to the forefront for patients with rare cancers and rare biomarkers.
Author Contributions
Conception/design: Mason A. Israel, Natalie Danziger, Kimberly A. McGregor, Karthikeyan Murugesan, Ethan S. Sokol, Jeffrey S. Ross
Data analysis and interpretation: Mason A. Israel, Natalie Danziger, Kimberly A. McGregor, Karthikeyan Murugesan, Ole Gjoerup, Ethan S. Sokol, Hanna Tukachinsky, Razelle Kurzrock, Shumei Kato, Jason K. Sicklick, Halla S. Nimeiri, Geoffrey R. Oxnard, Jeffrey S. Ross
Manuscript writing: Ole Gjoerup
Final approval of manuscript: Mason A. Israel, Natalie Danziger, Kimberly A. McGregor, Karthikeyan Murugesan, Ole Gjoerup, Ethan S. Sokol, Hanna Tukachinsky, Razelle Kurzrock, Shumei Kato, Jason K. Sicklick, Halla S. Nimeiri, Geoffrey R. Oxnard, Jeffrey S. Ross
Disclosures
Mason A. Israel: Foundation Medicine (E, OI); Natalie Danziger: Foundation Medicine (E, OI); Kimberly A. McGregor: Foundation Medicine (E, OI); Karthikeyan Murugesan: Foundation Medicine (E, OI); Ole Gjoerup: Foundation Medicine (E, OI); Ethan S. Sokol: Foundation Medicine (E, OI); Hanna Tukachinsky: Foundation Medicine (E, OI); Razelle Kurzrock: IDbyDNA, CureMatch, Soluventis (OI), Gaido Health, Loxo Oncology, XBiotech, Actuate Therapeutics, Roche, Neomed Pharmaceutical, Soluventis, Pfizer (C/A), Roche (H), Incyte, Genentech, Merck Serono, Pfizer, Sequenom, Foundation Medicine, Guardant Health, Grifols, Konica Minolta, Debiopharm, Boehringer Ingelheim, OmniSeq (RF—inst), CureMatch, CureMetrix (Other—Board member); Shumei Kato: Foundation Medicine (C/A), Roche (H); Jason K. Sicklick: Novartis, Amgen, Foundation Medicine (RF), Grand Rounds, Loxo Oncology, Deciphera (C/A), Roche, Deciphera (H); Halla S. Nimeiri: Foundation Medicine (E, OI); Geoffrey R. Oxnard: Foundation Medicine (E, OI); Jeffrey S. Ross: Foundation Medicine (E, OI).
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Acknowledgments
Medical writing assistance in the form of copyediting, editorial assistance, and production assistance, provided by Sarah Nordquist, Ph.D., and Claire Stedden, Ph.D., of Health Interactions Inc, was furnished by Foundation Medicine Inc.
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
References
- 1.Rizvi S, Borad MJ. The rise of the FGFR inhibitor in advanced biliary cancer: The next cover of time magazine? J Gastrointest Oncol 2016;7:789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghouri YA, Mian I, Blechacz B. Cancer review: Cholangiocarcinoma. J Carcinog 2015;14:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mukkamalla SKR, Naseri HM, Kim BM et al. Trends in incidence and factors affecting survival of patients with cholangiocarcinoma in the United States. J Natl Compr Canc Netw 2018;16:370–376. [DOI] [PubMed] [Google Scholar]
- 4.Sicklick JK, Fanta PT, Shimabukuro K et al. Genomics of gallbladder cancer: The case for biomarker‐driven clinical trial design. Cancer Metastasis Rev 2016;35:263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bridgewater J, Galle PR, Khan SA et al. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J Hepatol 2014;60:1268–1289. [DOI] [PubMed] [Google Scholar]
- 6.Ray CE Jr, Edwards A, Smith MT et al. Metaanalysis of survival, complications, and imaging response following chemotherapy‐based transarterial therapy in patients with unresectable intrahepatic cholangiocarcinoma. J Vasc Interv Radiol 2013;24:1218–1226. [DOI] [PubMed] [Google Scholar]
- 7.Valle J, Wasan H, Palmer DH et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 2010;362:1273–1281. [DOI] [PubMed] [Google Scholar]
- 8.Meng ZW, Pan W, Hong HJ et al. Macroscopic types of intrahepatic cholangiocarcinoma and the eighth edition of AJCC/UICC TNM staging system. Oncotarget 2017;8:101165–101174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Javle MM, Murugesan K, Shroff RT et al. Profiling of 3,634 cholangiocarcinomas (CCA) to identify genomic alterations (GA), tumor mutational burden (TMB), and genomic loss of heterozygosity (gLOH). J Clin Oncol 2019;37(suppl 15):4087a. [Google Scholar]
- 10.Silverman IM, Murugesan K, Lihou CF et al. Comprehensive genomic profiling in FIGHT‐202 reveals the landscape of actionable alterations in advanced cholangiocarcinoma. J Clin Oncol 2019;37(suppl 15):4080a. [Google Scholar]
- 11.Sia D, Losic B, Moeini A et al. Massive parallel sequencing uncovers actionable FGFR2‐PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat Commun 2015;6:6087. [DOI] [PubMed] [Google Scholar]
- 12.Ross JS, Wang K, Gay L et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next‐generation sequencing. The Oncologist 2014;19:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lowery MA, Ptashkin R, Jordan E et al. Comprehensive molecular profiling of intrahepatic and extrahepatic cholangiocarcinomas: Potential targets for intervention. Clin Cancer Res 2018;24:4154–4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chun YS, Javle M. Systemic and adjuvant therapies for intrahepatic cholangiocarcinoma. Cancer Control 2017;24:1073274817729241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Churi CR, Shroff R, Wang Y et al. Mutation profiling in cholangiocarcinoma: Prognostic and therapeutic implications. PLoS One 2014;9:e115383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Javle M, Bekaii‐Saab T, Jain A et al. Biliary cancer: Utility of next‐generation sequencing for clinical management. Cancer 2016;122:3838–3847. [DOI] [PubMed] [Google Scholar]
- 17.Almquist DR, Javle M, Ciombor KK et al. FGFR2 fusions and its effect of patient (pt) outcomes in intrahepatic cholangiocarcinoma (iCCA). Ann Oncol 2019;30(suppl 5):v279. [Google Scholar]
- 18.Graham RP, Barr Fritcher EG, Pestova E et al. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum Pathol 2014;45:1630–1638. [DOI] [PubMed] [Google Scholar]
- 19.Goyal L, Lamarca A, Strickler JH et al. The natural history of fibroblast growth factor receptor (FGFR)‐altered cholangiocarcinoma (CCA). J Clin Oncol 2020;38(suppl 15):e16686. [Google Scholar]
- 20.Jain A, Borad MJ, Kelley RK et al. Cholangiocarcinoma with FGFR genetic aberrations: A unique clinical phenotype. JCO Precis Oncol 2018;(2):1–12. [DOI] [PubMed] [Google Scholar]
- 21.Arai Y, Totoki Y, Hosoda F et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014;59:1427–1434. [DOI] [PubMed] [Google Scholar]
- 22.Hoy SM. Pemigatinib: First approval. Drugs 2020;80:923–929. [DOI] [PubMed] [Google Scholar]
- 23.Hollebecque A, Silverman I, Owens S et al. Comprehensive genomic profiling and clinical outcomes in patients (pts) with fibroblast growth factor receptor rearrangement‐positive (FGFR2+) cholangiocarcinoma (CCA) treated with pemigatinib in the fight‐202 trial. Ann Oncol 2019;30(suppl 5):v276. [Google Scholar]
- 24.Li F, Peiris MN, Donoghue DJ. Functions of FGFR2 corrupted by translocations in intrahepatic cholangiocarcinoma. Cytokine Growth Factor Rev 2020;52:56–67. [DOI] [PubMed] [Google Scholar]
- 25.Abou‐Alfa GK, Macarulla T, Javle MM et al. Ivosidenib in IDH1‐mutant, chemotherapy‐refractory cholangiocarcinoma (ClarIDHy): A multicentre, randomised, double‐blind, placebo‐controlled, phase 3 study. Lancet Oncol 2020;21:796–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subbiah V, Lassen U, Élez E et al. Dabrafenib plus trametinib in patients with BRAFV600E‐mutated biliary tract cancer (ROAR): A phase 2, open‐label, single‐arm, multicentre basket trial. Lancet Oncol. 2020;21:1234–1243. [DOI] [PubMed] [Google Scholar]
- 27.Harding J, Cleary J, Shapiro G et al. Treating HER2‐mutant advanced biliary tract cancer with neratinib: Benefits of HER2‐directed targeted therapy in the phase 2 SUMMIT basket trial. Ann Oncol 2019;30(suppl 4):iv127. [Google Scholar]
- 28.Stein EM, DiNardo CD, Pollyea DA et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017;130:722–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goyal L, Saha SK, Liu LY et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion–positive cholangiocarcinoma. Cancer Discov 2017;7:252–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parikh AR, Leshchiner I, Elagina L et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat Med 2019;25:1415–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kasi PM. Favorable outcomes in FGFR fusion‐positive cholangiocarcinomas and evolution on treatment noted on circulating tumor DNA liquid biopsies. Case Rep Oncol 2020;13:941–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ettrich TJ, Schwerdel D, Dolnik A et al. Genotyping of circulating tumor DNA in cholangiocarcinoma reveals diagnostic and prognostic information. Sci Rep 2019;9:13261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okamura R, Kurzrock R, Mallory RJ et al. Comprehensive genomic landscape and precision therapeutic approach in biliary tract cancers. Int J Cancer 2021;148:702–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frampton GM, Fichtenholtz A, Otto GA et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31:1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clark TA, Chung JH, Kennedy M et al. Analytical validation of a hybrid capture‐based next‐generation sequencing clinical assay for genomic profiling of cell‐free circulating tumor DNA. J Mol Diagn 2018;20:686–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chalmers ZR, Connelly CF, Fabrizio D et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 2017;9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trabucco SE, Gowen K, Maund SL et al. A novel next‐generation sequencing approach to detecting microsatellite instability and pan‐tumor characterization of 1000 microsatellite instability–high cases in 67,000 patient samples. J Mol Diag 2019;21:1053–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Newberg J, Connelly C, Frampton G. Abstract 1599: Determining patient ancestry based on targeted tumor comprehensive genomic profiling. Cancer Res 2019;79(suppl 13):1599a. [Google Scholar]
- 39.Carrot‐Zhang J, Chambwe N, Damrauer JS et al. Comprehensive analysis of genetic ancestry and its molecular correlates in cancer. Cancer Cell 2020;37:639–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alexander DH, Novembre J, Lange K. Fast model‐based estimation of ancestry in unrelated individuals. Genome Res 2009;19:1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mateo J, Chakravarty D, Dienstmann R et al. A framework to rank genomic alterations as targets for cancer precision medicine: The ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann Oncol 2018;29:1895–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Javle M, Lowery M, Shroff RT et al. Phase II study of BGJ398 in patients with FGFR‐altered advanced cholangiocarcinoma. J Clin Oncol 2018;36:276–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meric‐Bernstam F, Arkenau H, Tran B et al. Efficacy of TAS‐120, an irreversible fibroblast growth factor receptor (FGFR) inhibitor, in cholangiocarcinoma patients with FGFR pathway alterations who were previously treated with chemotherapy and other FGFR inhibitors. Ann Oncol 2018;29(suppl 5):v100. [Google Scholar]
- 44.Hollebecque A, Borad M, Sahai V et al. Interim results of fight‐202, a phase II, open‐label, multicenter study of INCB054828 in patients (pts) with previously treated advanced/metastatic or surgically unresectable cholangiocarcinoma (CCA) with/without fibroblast growth factor (FGF)/FGF receptor (FGFR) genetic alterations. Ann Oncol 2018;29(suppl 8):viii258a. [Google Scholar]
- 45.Wainberg ZA, Lassen UN, Elez E et al. Efficacy and safety of dabrafenib (D) and trametinib (T) in patients (pts) with BRAF V600E–mutated biliary tract cancer (BTC): A cohort of the ROAR basket trial. J Clin Oncol 2019;37(suppl 4):187a. [Google Scholar]
- 46.Mazzaferro V, El‐Rayes BF, Busset MDD et al. Derazantinib (ARQ 087) in advanced or inoperable FGFR2 gene fusion‐positive intrahepatic cholangiocarcinoma. Br J Cancer 2019;120:165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ng MCH, Goyal L, Bang YJ et al. AB065. P‐36. Debio 1347 in patients with cholangiocarcinoma harboring an FGFR gene alteration: Preliminary results. Hepatobiliary Surg Nutr 2019;8(suppl 1):AB065. [Google Scholar]
- 48.Bahleda R, Italiano A, Hierro C et al. Multicenter phase I study of erdafitinib (JNJ‐42756493), oral pan‐fibroblast growth factor receptor inhibitor, in patients with advanced or refractory solid tumors. Clin Cancer Res 2019;25:4888–4897. [DOI] [PubMed] [Google Scholar]
- 49.Abou‐Alfa GK, Macarulla Mercade T, Javle M et al. ClarIDHy: A global, phase III, randomized, double‐blind study of ivosidenib (IVO) vs placebo in patients with advanced cholangiocarcinoma (CC) with an isocitrate dehydrogenase 1 (IDH1) mutation. Ann Oncol 2019;30(suppl 5):v872–v873a. [Google Scholar]
- 50.Woodhouse R, Li M, Hughes J et al. Clinical and analytical validation of FoundationOne Liquid CDx, a novel 324‐Gene cfDNA‐based comprehensive genomic profiling assay for cancers of solid tumor origin. PLoS One 2020;15:e0237802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Razavi P, Li BT, Brown DN et al. High‐intensity sequencing reveals the sources of plasma circulating cell‐free DNA variants. Nat Med 2019;25:1928–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee H, Wang K, Johnson A et al. Comprehensive genomic profiling of extrahepatic cholangiocarcinoma reveals a long tail of therapeutic targets. J Clin Pathol 2016;69:403–408. [DOI] [PubMed] [Google Scholar]
- 53.Galdy S, Lamarca A, McNamara MG et al. HER2/HER3 pathway in biliary tract malignancies; systematic review and meta‐analysis: A potential therapeutic target? Cancer Metastasis Rev 2017;36:141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klein O, Kee D, Nagrial A et al. Evaluation of combination nivolumab and ipilimumab immunotherapy in patients with advanced biliary tract cancers: Subgroup analysis of a phase 2 nonrandomized clinical trial. JAMA Oncol 2020;6:1405–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wardell CP, Fujita M, Yamada T et al. Genomic characterization of biliary tract cancers identifies driver genes and predisposing mutations. J Hepatol 2018;68:959–969. [DOI] [PubMed] [Google Scholar]
- 56.Fujimoto A, Furuta M, Shiraishi Y et al. Whole‐genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nat Commun 2015;6:6120. [DOI] [PubMed] [Google Scholar]