
Figure 10: cartoon illustrating the mechanisms by which deficiency of Adam15 leads to exaggerated COPD in mice.
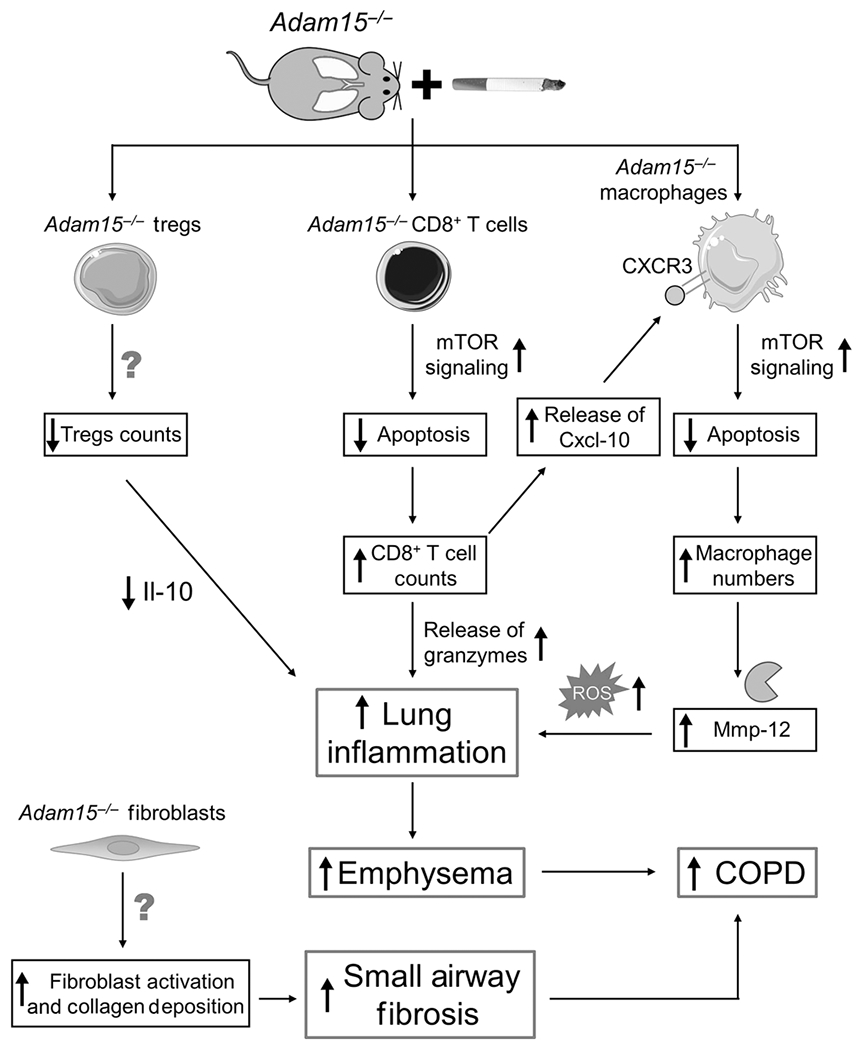
Exposing Adam15−/− mice to CS leads to a phlogistic pulmonary response characterized by increases in the number of innate and adaptive immune cells (alveolar macrophages [AMs], CD4+ lymphocytes, CD8+ lymphocytes and CD3-positive NKT cells) and reductions in leukocytes having anti-inflammatory activities (Foxp3+ T regulatory cells) in the lung, along with exaggerated emphysema development and small airway fibrosis. The results of studies of Adam15 bone marrow chimeras and Adam15−/− X Cd8−/− mice indicate that deficiency of Adam15 in leukocytes (especially CD8+ T cells) drives the increased emphysema development and small airway fibrosis observed in CS-exposed Adam15−/− mice. Likely, increased release of Mmp-12 and oxidants from the increased numbers of macrophages and the increased release of granzymes from the increased numbers of CD8+ T cells detected in the lungs of CS-exposed Adam15−/− mice contributed to their increased emphysema development. The exaggerated small airway fibrosis in CS-exposed Adam15−/− mice may be due to: 1) increased release of mediators from leukocytes and other cells that activate (myo)fibroblasts; and 2) Adam15 deficiency in (myo)fibroblasts increasing the survival and/or profibrotic activities of these cells in the small airways. Adam15 deficiency increases CS-induced pulmonary inflammation by several mechanisms. First, Adam15−/− CD8+ T cells (and macrophages) are protected from CS-induced activation of the mitochondrial apoptosis pathway and consequently accumulate in, and injure, the lung. Adam15 deficiency preserves mammalian target of rapamycin (mTOR) signaling in CD8+ T cells (and macrophages), leading to preserved expression of myeloid cell leukemia sequence-1 (Mcl-1) protein (an anti-apoptotic protein which binds to the mitochondrial membrane to prevent loss of mitochondrial membrane potential, mitochondrial release of cytochrome c and downstream caspase activation). The increased number of CD8+ T cells in lungs of Adam15−/− mice also promotes the survival of macrophages in the lung via Adam15−/− CD8+ T cells releasing increased quantities of Cxcl-10 (and possibly other ligands for Cxcr3) which bind to Cxcr3 on Adam15−/− macrophages to increase mTOR signaling in macrophages leading to increased macrophage survival and activation and release of Mmp-12, oxidants and other mediators that promote lung inflammation and destruction. Second, the accumulation of leukocytes leads to increased release of pro-inflammatory mediators that amplify pulmonary inflammation and destruction in Adam15−/− lungs. Third, lung levels of anti-inflammatory mediators, such as interleukin-10 (Il-10) are reduced in CS-exposed Adam15−/− lung, which may be due to the reduced numbers of Foxp3+ T regulatory cells (which are a source of Il-10) in their lungs. A reduced anti-inflammatory immune response likely contributes to the phlogistic environment that increases emphysema development and small airway fibrosis in CS-exposed Adam15−/− lungs.