

Abstract
The exploration of three-dimensional chromatin interaction and organization provides insight into mechanisms underlying gene regulation, cell differentiation and disease development. Advances in chromosome conformation capture technologies, such as high-throughput chromosome conformation capture (Hi-C) and chromatin interaction analysis by paired-end tag (ChIA-PET), have enabled the exploration of chromatin interaction and organization. However, high-resolution Hi-C and ChIA-PET data are only available for a limited number of cell lines, and their acquisition is costly, time consuming, laborious and affected by theoretical limitations. Increasing evidence shows that DNA sequence and epigenomic features are informative predictors of regulatory interaction and chromatin architecture. Based on these features, numerous computational methods have been developed for the prediction of chromatin interaction and organization, whereas they are not extensively applied in biomedical study. A systematical study to summarize and evaluate such methods is still needed to facilitate their application. Here, we summarize 48 computational methods for the prediction of chromatin interaction and organization using sequence and epigenomic profiles, categorize them and compare their performance. Besides, we provide a comprehensive guideline for the selection of suitable methods to predict chromatin interaction and organization based on available data and biological question of interest.
Keywords: gene regulation, three-dimensional genome organization, DNA sequence and epigenomic features, methods evaluation
Introduction
Elucidation of the mechanisms underlying genome function is key for an understanding of how genetic molecules determine cell fates, and how the disruption of this function leads to disease. An understanding of the biological function of the genome requires investigation of two distinct aspects of human genome organization: dynamic chromatin interaction between regulatory elements and the higher-order three-dimensional (3D) chromatin organization in which 2 m DNA is fitted into a 6–10 μm diameter nucleus. Chromatin interaction among distant genomic elements participates in the initiation and regulation of gene transcription. Chromatin organization, in which chromatins organize themselves in different spatial structures, determines the interaction frequency between gene loci. Chromatin interaction and organization underlie different aspects of gene regulation, variation and evolution, highlighting the importance of continued effort to understand them. With the development of chromosome conformation capture technologies (e.g. chromosome conformation capture (3C) [1], circularized chromosome conformation capture [2], carbon-copy chromosome conformation capture [3] and high-throughput chromosome conformation capture (Hi-C) [4], multilevel hierarchical chromatin organizations have been explored (Figure 1). Chromosomes organize themselves into A and B compartments, which are associated with euchromatin and heterochromatin, respectively [4]. The compartments are composed of topologically associated domains (TADs), which can be further subdivided into chromatin loops, such as enhancer–promoter loops, which influence gene regulation [5, 6]. The loop extrusion (LE) model considers TAD formation to be mediated by an active extrusion mechanism in which ring-shaped extruding factors move along chromatin and squeeze it into loops when they encounter another extruding factor or two opposite-oriented boundary elements [7, 8]. The exploration of chromatin organization facilitates characterization of the spatial arrangement of genes and their regulatory elements in a manner that allows them to carry out their functions.
Figure 1 .
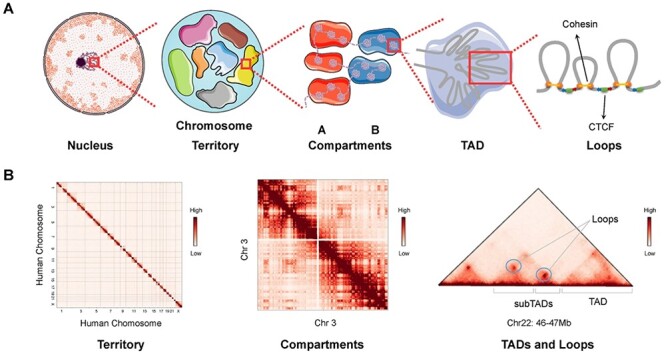
Hierarchical genome organization. (A) Multilevel 3D genome organizations. Chromosome territory, compartments, TADs and loops can be observed from left to right. Each chromosome territory is denoted by different colors. Compartment A and B are indicated by red and blue background, respectively. TADs are formed by LE. The ring-shaped cohesin squeeze the chromatin into loops and halt when they encounter the boundary elements CTCF in the specific orientation (indicated by the blue and red arrows) at two sites of cohesin ring. (B) Hi-C matrices of hierarchical chromatin organizations. Interchromosomal interaction between human chromosomes presents the organization of chromosome territories. Intrachromosomal interaction between chromosome 3 indicates the chromatin compartments. Matrix of a 1 Mb width subregion on chromosome 22 shows TADs, subTADs and loops, which are denoted as punctate signals. Hi-C matrices in panel B are generated from Rao’s (access code GSE63525) Hi-C data in GM12878 cell line.
Chromosome conformation capture techniques, such as Hi-C and chromatin interaction analysis by paired-end tag sequencing (ChIA-PET), have been used commonly to identify potential genome-wide interaction of regulatory elements and spatial chromatin organization [9, 10]. The resolution of Hi-C and ChIA-PET data directly impacts the effectiveness and accuracy of identification; high-resolution Hi-C data remain lacking for most tissues and cell lines, and their acquisition via substantial increases in sequencing depth remains expensive, time consuming and labor intensive [11]. The exponential data growth with increasing depth also brings new analytical challenges [12]. Comparatively, abundant sequence and epigenomic data for various tissues and cell types are publicly available in many databases, such as the Encyclopedia of DNA Elements (ENCODE) [13], Roadmap Epigenomic [14] and Gene Expression Omnibus [15] databases.
A growing body of evidence has shown that sequence features and epigenomic modification can serve as informative predictors for the identification of chromatin interaction and organization, due to their roles in transcriptional regulation and chromatin folding via the control of DNA accessibility and recruitment of specific proteins [16–18]. Besides, transcription factors (TFs) and their binding motifs underlie important aspects of transcription activity that facilitate the identification of chromatin interaction and organization [19]. Numerous computational methods have been developed to predict chromatin interaction and organization from sequence and epigenomic data, as alternatives to costly experimental approaches. However, the application of these approaches in biological research remains limited. The largest obstacle is the complexity of the algorithms, which cannot be understood by many biologists. In addition, the appropriate method is difficult to choose due to the diverse input data and software requirements. Furthermore, some computational methods are difficult to reproduce and apply in new research.
Here, we introduce 48 computational methods for the prediction of chromatin interaction and organization based on sequence and epigenomic profiles, classifying them based on their function, input features and categories and comparing their performance. We provide a comprehensive guideline and list several methods that can be applied easily with small sets of input data. Moreover, we introduce specific applications of these methods in biomedical research, which can serve as a reference to help researchers choose the appropriate methods for their studies. Finally, we note existing challenges in the prediction of chromatin interaction and organization and provide possible solutions for future improvements.
Computational methods for the prediction of chromatin interaction
The integrative analysis of DNA sequence and epigenomic features, such as transcription factor binding sites (TFBSs), chromatin accessibility and histone modification, enables the identification of active cis-regulatory elements (CREs) [20, 21]. Many computational methods have been developed to predict chromatin interaction between CREs via the consideration of features such as the characteristic arrangement of nucleosomes [22] and evolutionary conservation [23]. Here, we summarize 12 unsupervised and 21 supervised machine learning methods for the prediction of chromatin interaction based on the analysis of sequence and epigenomic profiles (Figure 2, Table 1; code availability and programming language of these methods are summarized in Table S1). Besides, we categorize the predicted chromatin interaction mainly into three categories: enhancer–promoter interaction (EPI), enhancer–target genes (ETGs) and 3D interaction. Comparatively, computational methods for EPI prediction focus more on extensive regulatory relationships, methods for ETGs prediction fit better to study the regulatory elements for specific genes and 3D chromatin interaction concerns more about chromatin interaction within specific chromatin folding structure, such as TADs.
Figure 2 .
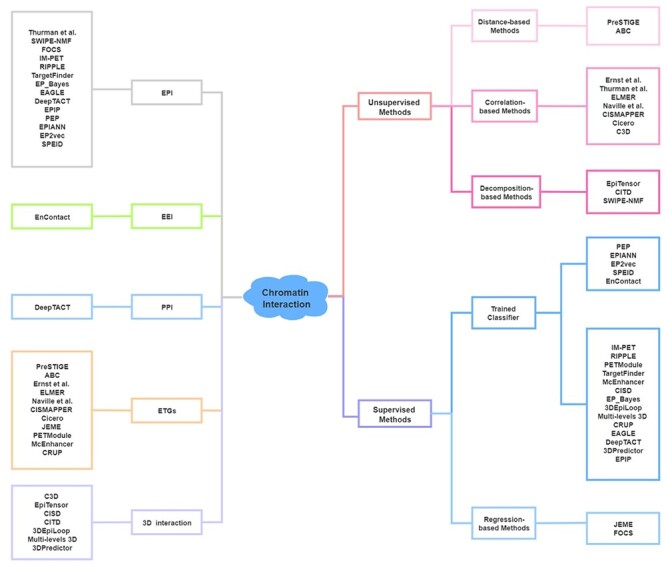
Computational methods for the prediction of chromatin interaction. Left: Category of methods based on output. Right: Category of methods based on algorithm. Sequence-based methods (PEP, EPIANN, EP2vec, SPEID and EnContact) of trained classifier methods are listed separately.
Table 1.
Computational methods for the prediction of chromatin interaction
| Tool Name | Year | Algorithms | Input features | Predictions | Reference | |
|---|---|---|---|---|---|---|
| Unsupervised Methods | ||||||
| PreSTIGE | 2014 | Distance-based Methods | Linear Domain Model | Distance, H3K4me1, RNA-seq | ETGs | [25] |
| ABC | 2019 | Activity-by-contact Model | DHS, distance, histone marks | ETGs | [28] | |
| Ernst et al. | 2011 | Correlation-based Methods | Pearson Correlation | CTCF, histone marks, TF binding | ETGs | [29] |
| Thurman et al. | 2012 | Pearson Correlation | DHS | EPI | [30] | |
| ELMER | 2015 | Pearson Correlation | DNA methylation, RNA-seq | ETGs | [33, 35] | |
| Naville et al. | 2015 | Linkage Scoring | DHS, histone marks, TFBSs | ETGs | [23] | |
| CISMAPPER | 2016 | Pearson Correlation | DHS, histone marks, RNA-seq | ETGs | [34] | |
| Cicero | 2018 | Graphical Lasso | scATAC-seq | ETGs | [31] | |
| C3D | 2019 | Pearson Correlation | DHS | 3D chromatin interaction | [32] | |
| EpiTensor | 2016 | Decomposition-based Methods | Tensor Decomposition | DHS, histone marks, RNA-seq | 3D chromatin interaction | [36] |
| CITD | 2016 | Wavelet Transformation | Histone marks | 3D chromatin interaction | [37] | |
| SWIPE-NMF | 2017 | Matrix Factorization | DHS, eQTL | EPI | [37] | |
| Supervised Methods | ||||||
| JEME | 2017 | Regression-based Methods | Linear Regression | DHS, distance, eRNA, histone marks | ETGs | [39] |
| FOCS | 2018 | Linear Regression | CAGE, DHS, GRO-seq | EPI | [40] | |
| IM-PET | 2014 | Trained Classifier Methods | Random Forest | DNA, histone marks, TFBSs, RNA-seq | EPI | [41] |
| RIPPLE | 2015 | Random Forest, Group Lasso | DHS, histone marks, TFBSs, RNA-seq | EPI | [43] | |
| PETModule | 2016 | Random Forest | CSS, DHS, distance, FSS | ETGs | [42] | |
| TargetFinder | 2016 | Gradient Tree Boosting | DHS, DNA methylation, TFBSs, histone marks, CAGE | EPI | [27] | |
| McEnhancer | 2017 | Markov Chain Model | DHS | ETGs | [49] | |
| CISD | 2017 | Logistic Regression | MNase-seq | 3D chromatin interaction | [22] | |
| EP_Bayes | 2017 | Naive Bayes Classifier | Histone marks, RNA Pol II, RNA-seq | EPI | [44] | |
| 3DEpiLoop | 2017 | Random Forest | Histone marks, TFBSs | 3D chromatin interaction | [51] | |
| Multi-levels 3D | 2017 | Random Forest | Histone marks | 3D chromatin interaction | [52] | |
| CRUP | 2019 | Random Forest | Histone marks, gene expression | ETGs | [45] | |
| EAGLE | 2019 | Ensemble Classifier | Histone marks, DHS, TF binding, RNA-seq | EPI | [46] | |
| DeepTACT | 2019 | Bootstrapping Deep Learning | DHS, DNA | EPI and PPI | [50] | |
| EPIP | 2019 | Adaboost | DHS, distance, histone marks | EPI | [47] | |
| 3DPredictor | 2020 | Gradient Boosting | CTCF, distance, RNA-seq | 3D chromatin interaction | [53] | |
| PEP | 2017 | Gradient Tree Boosting | DNA | EPI | [17] | |
| EPIANN | 2017 | CNN, Attention Model | DNA | EPI | [55] | |
| EP2vec | 2018 | Word2vec, Gradient Boosted Regression Trees | DNA | EPI | [55] | |
| SPEID | 2019 | CNN | DNA | EPI | [54] | |
| EnContact | 2019 | CNN, RNN | DNA, HiChIP data | EEI | [57] | |
Unsupervised methods
Unsupervised learning methods uncover naturally occurring patterns to predict chromatin interaction based on distance or correlations between regulatory elements [24], which are objective without the use of ‘handpicked’ features. Based on the strategies used to link distal CREs to target genes, we divide them into distance-based, correlation-based and decomposition-based methods.
Distance-based methods
EPI can be predicted effectively by assigning enhancers to their nearest genes via the computation of linear distances between regulatory elements. Distance-based methods serve as the baseline method in many studies, given their simple rationale. PreSTIGE is a multiple linear domain model that integrates H3K4me1 ChIP-seq and RNA-seq data to link cell type–specific enhancers to their target genes, considering CTCF binding sites as insulators [25]. It is available as an online application through Galaxy (http://prestige.case.edu/) [26]. As it is based on a simple correlation strategy, PreSTIGE may have less predictive accuracy than machine learning models that focus on regulatory links with multiple genomic features, such as TargetFinder [27]. By integrating the distance effect with the contact frequency of enhancer–promoter pairs and enhancer activity, measured by DNase I hypersensitive signals (DHSs) and H3K27ac ChIP-seq data, ABC provides a framework for the genome-wide mapping of enhancer–gene connections across many cell types based on epigenomic datasets [28].
Correlation-based methods
Improvements on the distance-based approach via the correlation of histone modification with enhancer and promoter DHSs or promoter transcript levels in given genomic domains have been introduced [29, 30]. Using correlations between TFs expression and motif enrichment, Ernst et al. [29] predicted cell type–specific activators and repressors that modulate putative target genes. Thurman et al. [30] identified EPI based on DHSs at regulatory regions. Cicero links enhancers to their target genes based on single-cell ATAC-seq data [31]. Similarly, C3D predicts chromatin interaction between CREs using correlations between open chromatin regions based on DHSs [32], and Naville et al. [23] estimated enhancer–gene associations based on correlations among chromatin accessibility, histone modification and TF binding. Methods based on correlations between the epigenomic modification of active regulatory elements and gene expression have also been developed to identify chromatin interaction, such as ELMER and CISMAPPER [33, 34]. ELMER identifies transcriptional targets by correlating methylation-affected enhancers with the expression of nearby genes [33]. Silva et al. [35] presented a revised version 2 of ELMER that provides an optional web-based interface and a new supervised analysis mode, which shows better performance. CISMAPPER predicts EPI using the correlation of histone marks at TFBSs with gene expression. It requires no training step and is more accurate than simple distance-based methods [34].
Decomposition-based methods
Different decomposing strategies have been used to extract underlying traits from high-dimensional signals for the identification of chromatin interaction based on relationships between these traits. EpiTensor uses 18 assays to model epigenomic data from five cell types as a 3rd-order tensor in which the dimensions represent genomic loci, assay type and cell type, respectively [36]. Based on the associations between subspaces decomposed from the tensor, EpiTensor can identify interaction hotspots in which promoters and enhancers are located in genomic regions with significantly more transcriptional activity and TFBSs enrichment across cell types [36]. CITD enables the de novo prediction and mapping of chromatin interaction via the integration of one-dimensional (1D) histone modification data by wavelet decomposition and histone reconstruction [37]. SWIPE-NMF builds a matrix factorization framework that integrates heterogeneous data and has been used to reconstruct enhancer–promoter networks in 127 human cell lines [38].
Supervised methods
Supervised learning methods include random forest, neural network, decision tree, logistic regression and linear regression analyses. They enable the training of a model using a cell line with specific sequence and epigenomic data, then applying the model to make predictions in another cell line. Based on the algorithms applied in the models, supervised methods can be classified as regression-based and trained classifier methods.
Regression-based methods
Regression-based methods identify interactional relationships between enhancers and promoters or target genes by associating enhancer features with promoter features or gene expression; JEME and FOCS are examples of these methods [39, 40]. JEME identifies ETGs in specific samples by considering the joint effect of multiple enhancers and integrated global and sample-specific information [39]. Its application to reconstruct the enhancer–promoter network in 935 samples of human primary cells, tissues and cell lines enables the systematic investigation of gene regulation in normal and disease states [39]. FOCS is a statistical framework used to infer enhancer–promoter links correlated with activity patterns across samples from ENCODE, the Roadmap Epigenomics project and FANTOM5 [40]. Like JEME, FOCS employs eRNA as a marker of enhancer activity. The application of FOCS to massive genomic datasets yields extensive enhancer–promoter maps for the derivation of biological hypotheses [40].
Trained classifier methods
By leveraging genomic or epigenomic features of true positive enhancer–promoter pairs, a trained classifier can be established to determine whether a pair of interest is involved in interaction or whether a putative pair engages in high-confidence contact. Many supervised classifiers have been designed to identify chromatin interaction. IM-PET integrates a set of discriminative features, including correlation among enhancer and promoter activity, TFs and target promoters; evolutionary constraint between interacting enhancer–promoter pairs and distance constraint between enhancer and target promoters to predict EPI; however, calculating these features is not easy due to the requirement of multiple input and complicated process [41]. PETModule is a motif module-based approach for ETGs prediction using similar features to IM-PET [42]. Based on multiple epigenomic signals and expression data, comprehensive classifiers have been established to predict EPI and ETGs, which include RIPPLE [43], TargetFinder [27], EP_Bayes [44], CRUP [45], EAGLE [46] and EPIP [47]. RIPPLE and TargetFinder can identify cell type–specific EPI based on numerous features, such as measures of open chromatin, gene expression, TFs, architectural proteins and modified histones [27, 43]. However, the requirement for various inputs makes it difficult to apply RIPPLE and TargetFinder to train specific classifiers for additional cell types [48]. EAGLE can be applied to data from various species and many cell types with a high degree of accuracy using a small number of genomic features [46]. EPIP predicts condition-specific EPI using a feature partitioning strategy, grouping features into 11 partitions or overlapping feature sets, which enables the use of sets with missing data [47]. EP_Bayes uses ChIP-seq RNA polymerase II data as the input and is not ideal for the identification of intronic enhancers [44]. Using DHSs as informative features, McEnhancer [49], CISD [22] and DeepTACT [50] can be used to predict multiple types of chromatin interaction. McEnhancer links enhancers to putative target genes with considerable accuracy. CISD identifies genome-wide chromatin interaction sites based on characteristic nucleosome arrangement patterns [22]. DeepTACT applies a bootstrapping deep-learning model to integrate genome sequence and chromatin accessibility data to predict EPI and promoter–promoter interaction (PPI) [50]. Several other trained classifiers have been developed to predict 3D chromatin interaction. 3DEpiLoop predicts high-resolution (1 kb) 3D physical chromatin interaction in TADs using 1D epigenomic and TF-binding profiles [51]. Bkhetan and Plewczynski [52] presented a random forest classifier that predicts multilevel 3D chromatin interaction using epigenomic profiles. The 3Dpredictor classifier can provide high-quality prediction of chromatin interaction based on only CTCF-binding signals and gene expression data [53].
Methods have also been developed to explore the possibility of predicting long-range interaction between regulatory elements based on sequence data alone (Figure 2, Table 1). For example, PEP integrates two strategies—PEP-Motif and PEP-Word—to predict EPI by extracting sequence features from given locations of putative enhancers and promoters in specific cell types [17]. SPEID applies deep neural networks (DNN) to predict EPI based solely on sequence features in enhancer and promoter regions [54]. Given the lack of expectation of a universal sequence-based EPI prediction mechanism, SPEID can only effectively predict EPI in training cell lines, which makes it inapplicable to other datasets [54]. EPIANN is an attention-based neural network model, which can focus more on features contributing to EPI and predict EPI more accurately [55]. EP2vec uses an unsupervised deep-learning method with natural language processing to transform enhancer and promoter sequences into sequence-embedding features and a supervised classifier to predict EPI [56]. EnContact, a context-specific deep-learning model, can predict enhancer–enhancer interaction (EEI) using only genomic sequence data [57].
Computational methods for the prediction of 3D chromatin organization
Recent studies suggest that DNA sequence and 1D epigenomic modification are also informative to predict 3D chromatin architecture. Based on these data, machine learning and polymer physics simulations methods have been applied to predict 3D chromatin organizations [58]. Jost et al. [59] introduced a block copolymer model to build chromatin folding from the epigenomic landscape, which can explain the formation and dynamics of TADs. By applying a reduced model based on histone modification in ENCODE data, Di Pierro et al. [60] illustrated that such modification alone carries sufficient information for the prediction of chromatin arrangement. Sefer and Kingsford [61] provided a comprehensive model of the joint effect of histone markers on TADs and demonstrated that the incorporation of sequence features significantly improved chromatin organization prediction. Zhang et al. [62] demonstrated that the consideration of sequence-based features alone can accurately predict whether convergent CTCF motif pairs will form loops. Also, Fudenberg et al. [63] and Schwessinger et al. [64] demonstrated that DNA sequence alone is sufficient to predict 3D genome folding structure with high accuracy. Here, we summarize 17 computational methods for the prediction of chromatin organization based on sequence and epigenomic profiles (Figure 3, Table 2) (code availability and programming language of these methods are summarized in Table S1).
Figure 3 .
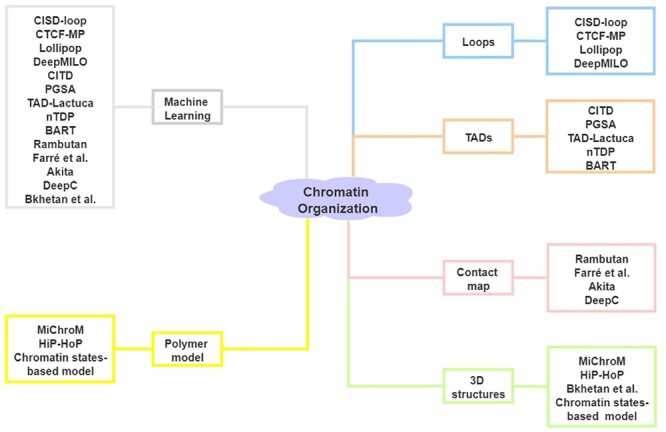
Computational methods for the prediction of chromatin organization. Left: Category of methods based on algorithm. Right: Category of methods based on output.
Table 2.
Computational methods for the prediction 3D chromatin organization
| Tool Name | Year | Method category | Algorithms | Input features | Predictions | Reference |
|---|---|---|---|---|---|---|
| Loops | ||||||
| CISD-loop | 2017 | Machine Learning | Logistic Regression | Mnase-seq | Loops | [22] |
| CTCF-MP | 2018 | Machine Learning | Word2Vec, Boosted Trees | CTCF, DHS, distance, DNA | CTCF-mediated loops | [62] |
| Lollipop | 2018 | Machine Learning | Random Forest | CTCF, histone marks, Rad21, RNA-Seq | CTCF-mediated loops | [65] |
| DeepMILO | 2020 | Machine Learning | CNN, RNN | CTCF, DNA | CTCF-mediated loops | [18] |
| TADs | ||||||
| CITD | 2016 | Machine Learning | Decomposition | Histone marks | TADs | [37] |
| PGSA | 2017 | Machine Learning | Position-specific Linear Model, Population Greedy Search Algorithm | DHS, histone marks, TFBSs | TADs | [66] |
| TAD-Lactuca | 2018 | Machine Learning | Random Forest, Artificial Neural Network | CTCF, DNA, histone marks | TADs | [67] |
| nTDP | 2019 | Machine Learning | Bernstein Polynomials | Histone marks | TADs | [61] |
| BART | 2015 | Machine Learning | Bayesian Additive Regression Trees | CTCF, histone marks | TADs and interaction hubs | [16] |
| Contact map | ||||||
| Rambutan | 2017 | Machine Learning | CNN | DHS, DNA | Contact map | [68] |
| Farré et al. | 2018 | Machine Learning | DNN | DHS, DNA | Contact map | [69] |
| Akita | 2020 | Machine Learning | CNN | DNA | Contact map | [63] |
| DeepC | 2020 | Machine Learning | CNN | DNA | Contact map | [64] |
| 3D structures | ||||||
| MiChroM | 2017 | Polymer model | Energy Landscape Model, Neural Network | Histone marks | 3D structures | [60] |
| HiP-HoP | 2018 | Polymer model | Heteromorphic Polymer model | ATAC-seq, CTCF, histone marks | 3D Structures | [73] |
| Chromatin states-based model | 2019 | Polymer model | Maximum Entropy | CTCF, DNA, histone marks | 3D Structures | [74] |
| Bkhetan et al. | 2019 | Machine Learning | Random Forest, Gradient Boosting Machine, Deep Learning Models, Polymer Simulation | DNA, histone marks, TFBSs | 3D structures | [75] |
Loop prediction
Based on the hypothesis that physical chromatin interaction can alter the characteristic flanking nucleosome arrangement patterns [22], CISD-loop identifies intra-TAD chromatin loops at kilobase resolution by integrating sequencing of micrococcal nuclease sensitive sites (MNase-seq) and low-resolution Hi-C data [22]. CISD-loop can be applied widely across human cell lines with high accuracy because these patterns are conservative among different cell types. CTCF-MP can be used to predict CTCF-mediated chromatin loops using functional genomic signals from CTCF ChIP-seq and DNase-seq data based on word2vec and boosted trees [62]. Word2vec is a two-layer neural network for processing natural language in which CTCF-MP apply it to extract DNA sequence features and boosted tree is an ensemble learning method which can promote the performance of CTCF-MP [62]. Lollipop is a random forest classifier that distinguishes CTCF-mediated loops from noninteracting ones based on a set of features generated from genomic and epigenomic data [65]. DeepMILO presents a deep learning network for the modeling of CTCF-mediated insulator loops and predicts the effects of noncoding variants on these loops using DNA sequences alone [18].
TAD prediction
CITD can estimate chromatin interaction frequency, TADs and their states (e.g. active or repressive), via the integration of 1D histone modification data [37]. It integrates correlations between histone modifications at interaction locus pairs and the power law that chromatin interaction frequency declines with distance for the inference of cell type–specific chromatin interaction matrices, which can be used to complement the topological domains derived from limited Hi-C data [37]. PGSA predicts TAD boundaries with a focus on associated predictive genomic elements, such as CTCF, ZNF143 and YY1 [66]. Similarly, TAD-Lactuca infers whether genome loci are at TAD boundaries based on contextual information from DNA sequences and eight histone marks [67]. nTDP is a semi-nonparametric method for TAD prediction based on a small set of histone marks, including H3K36me3, H3K4me1, H3K4me3 and H3K9me3, which are the most informative modification at TAD boundaries [61]. BART can be used to predict chromatin interaction hubs and TAD boundaries in which cell type–specific histone modification information is required [16].
Contact map prediction
Contact maps obtained from Hi-C have enable the discovery of multilevel hierarchical 3D chromatin organizations. Rambutan is a deep convolutional neural network used to predict Hi-C contacts at 1-kb resolution in cell types with no available Hi-C data, using only nucleotide sequences and DHSs as inputs [68]. Although the genomic distance effect creates major difficulties in the prediction of long-range Hi-C contacts, Rumbutan performs well for all distances [68]. Farré et al. [69] trained a dense neural network to predict chromatin conformation at the intrachromosomal scale using 1D sequences of DNA-bound chromatin factors and vice versa. They highlighted the importance of chromatin contexts and states in larger neighborhood conformations, along with critical alterations, for contact formation [69]. Akita is a convolutional neural network (CNN) for the prediction of 3D genome folding using only DNA sequence as input [63]. Similarly, DeepC can predict 3D genome folding using megabase-scale DNA sequence based on DNN [64]. DeepC tends to predict more pronounced interdomain interactions, stripes and dots, by learning the sequence determinants of genome folding [64].
3D chromatin structure simulation
MiChroM is an effective energy landscape model for de novo prediction of 3D chromatin structure using only ChIP-seq data on histone modification as input, based on certain sequence-to-structure relationships between patterns of histone modification and genome architecture [60]. HiP-HoP is a heteromorphic polymer model based on TF [70, 71], switching [72] and LE [7, 8] models used to analyze 3D chromatin structure via DNA accessibility, H3K27ac and CTCF/Rad21 signals [73]. Qi and Zhang [74] presented a predictive and transferable polymer model for the simulation of 3D chromatin structure at 5 kb resolution, which takes genomic location, epigenetic marks and CTCF orientation as inputs. Bkhetan et al. [75] proposed a pipeline that integrates an improved version of 3DEpiLoop and the spring model to construct visualized 3D chromatin structures based on molecular mechanics.
Evaluation of computational methods based on input features and algorithms
To validate the performance in the prediction of chromatin interaction and organization, numerous computational methods have been evaluated using different validation strategies and datasets. Performance metrics used commonly for this purpose include the F1 score, the area under the receiver operating characteristic curve (AUROC) and the area under the precision recall curve (AUPR). The F1 score could be interpreted as the harmonic mean of the precision and recall; precision is the number of correct positive results divided by the number of all positive results, and recall is the number of correct positive results divided by the number of positive results that should have been returned: F1 = 2 * (precision * recall)/(precision + recall); precision = true positives/(true positives + false positives); recall = true positives/(true positives + false negatives). The AUROC and AUPR do not depend on a particular classifier threshold and the AUPR is sensitive to unbalanced data in which instances are unequal for different classes [54]. Systematic evaluation of the performance of all available computational methods is difficult due to the diversity of rationales and gold standard definitions for prediction. Here, we compare the performance of computational methods based on different input features and algorithms.
Performance of computational methods based on different input features
Many computational methods for the prediction of chromatin interaction use diverse sequence and epigenomic data. For instance, TargetFinder and RIPPLE require multiple genomic features to determine whether putative enhancer–promoter pairs interact [27, 43], whereas EP2vec, PEP and SPEID take DNA sequences alone as the input [17, 54, 56]. Zeng et al. [56] evaluated the performance of EP2vec, TargetFinder and SPEID based on the labeled enhancer/promoter/window (region between enhancer and promoter) (E/P/W) training datasets used in TargetFinder. They found that EP2vec yields slightly better F1 scores, with lower AUROCs and AUPRs, than TargetFinder and that both methods outperform SPEID in 10-fold cross-validation experiments (Table 3). Using TargetFinder extended enhancer/promoter datasets, Yang et al. [17] showed that PEP and TargetFinder have comparable performance and that both methods outperform RIPPLE, with higher F1, AUROC and AUPR scores (Table 3). These evaluation results demonstrate that sequence data alone are sufficiently informative for the prediction of chromatin interaction. By extensively studying the performance of various deep learning models that use local sequence and epigenomic data around enhancer–promoter pairs, Xiao et al. [76] demonstrated that local epigenomic features are more informative than local sequence data for EPI prediction and that the integration of epigenomic features with sequence data can improve performance [77, 78].
Table 3.
Performance of sequence-based and epigenomic-based methods
| Datasets | Methods | F1 | AUROC | AUPR | |
|---|---|---|---|---|---|
| TargetFinder (E/P/W) datasets | Epigenomic-based methods | TargetFinder (E/P/W) | 0.884 (0.030) | 0.951 (0.019) | 0.961 (0.016) |
| Sequence-based methods | EP2vec | 0.892 (0.028) | 0.918 (0.023) | 0.918 (0.019) | |
| SPEID | 0.846 (0.040) | 0.901 (0.029) | 0.904 (0.024) | ||
| TargetFinder (EE/P) datasets | Epigenomic-based methods | TargetFinder (EE/P) | 0.809 (0.050) | 0.963 (0.013) | 0.853 (0.048) |
| RIPPLE | 0.772 (0.055) | 0.951 (0.012) | 0.807 (0.056) | ||
| Sequence-based methods | PEP | 0.815 (0.041) | 0.964 (0.010) | 0.864 (0.038) | |
The mean values and the SDs of F1, AUROC and AUPR scores of six cell lines (GM12878, K562, IMR90, HeLa-S3, HUVEC and NHEK) for TargetFinder (E/P/W), EP2vec, SPEID, TargetFinder (EE/P), RIPPLE and PEP. Performance of TargetFinder, EP2vec (k = 6, s = 1, m = 20, d = 100) and SPEID on six cell lines were evaluated on TargetFinder’s E/P/W datasets by Zeng et al. [56]. TargetFinder (EE/P), RIPPLE and PEP (K = 6) on six cell lines were evaluated on TargetFinder’s EE/P data by Yang et al. [17].
Unlike SPEID, which measures the cost of removing a feature, thereby identifying features that are necessary for prediction [54], TargetFinder measures the benefit of adding a feature, meaning that it identifies features that are sufficient for prediction [27]. Considering there are often limited datasets for a cell line, a diverse collection of required features will make it less flexible in practice. We assessed the computational performance of TargetFinder using its source code and labeled training datasets (E/P/W) on six cell lines [27]. We first output predictive importance of genomic features for each dataset within enhancer, promoter and window regions, then we performed the 10-fold cross-validation and output the F1 score by recursively eliminating genomic features according to their predictive importance (Figure 4). The results revealed redundancy of features used in TargetFinder, especially in GM12878 and K562. TargetFinder performs better when more genomic signals are available, whereas optimal or moderate performance can be achieved when only core features are available. Moore et al. [48] implemented TargetFinder in GM12878 using 303 features from 101 epigenomic datasets and four core epigenomic features: DNase-seq, H3K4me3, H3K27ac and CTCF data integrating distance features, respectively. They obtained an average AUPR reduction of 23% across 13 datasets for the four core models relative to the full models, which still represents better performance than the baseline distance-based method [48].
Figure 4 .
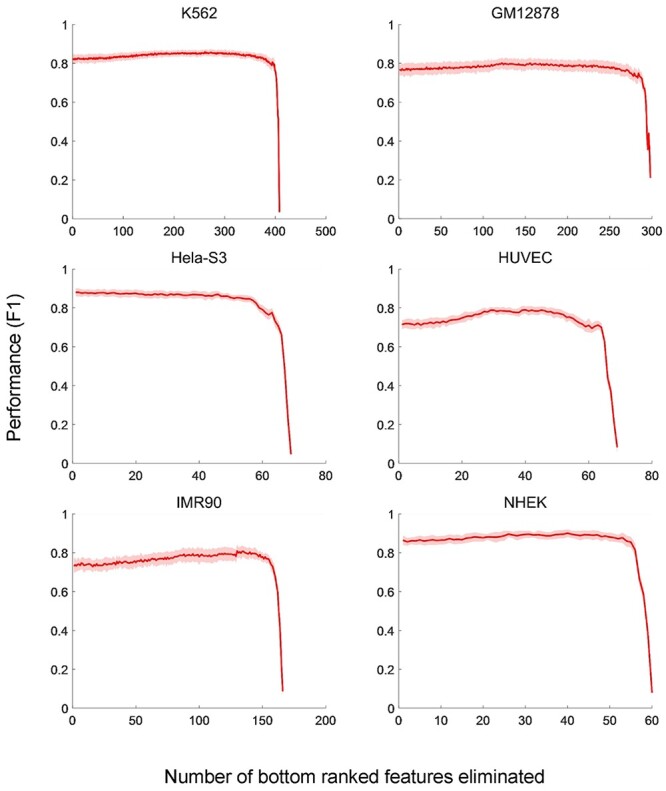
Performances of TargetFinder using different sets of genomic features by feature elimination. On six cell lines, the F1 scores of TargerFinder are evaluated by 10-fold cross-validation with the input genomic features eliminated one by one recursively according to their predictive importance. The mean values and the SDs of F1 scores in each 10-fold cross-validation are presented.
Performance of supervised and unsupervised methods
Moore et al. [48] developed the Benchmark of candidate Enhancer–Gene Interactions (BENGI) and used it to compare the performance of unsupervised and supervised methods (Figure 5A). TargetFinder performs best among distance-based method, three correlation-based [30, 79, 80] and two supervised methods [17, 27]), but it do not outperform the baseline distance-based method when tested across cell lines [30]. The DNase–DNase and DNase–expression correlation-based methods also do not outperform the baseline distance-based method, perhaps due to the unstable correlation across cell lines [30]. In addition, TargetFinder significantly outperforms PEP-motif when tested using the six BENGI datasets in GM12878 by cross-validation. Distance-based methods depend on precise CREs identification and neglect the possibility of enhancers to skip over nearby genes to link more distal targets [81]. Theoretically, correlation-based methods should outperform distance-based methods, whereas some correlations may be unstable across cell lines, which can lead to suboptimal performance.
Figure 5 .
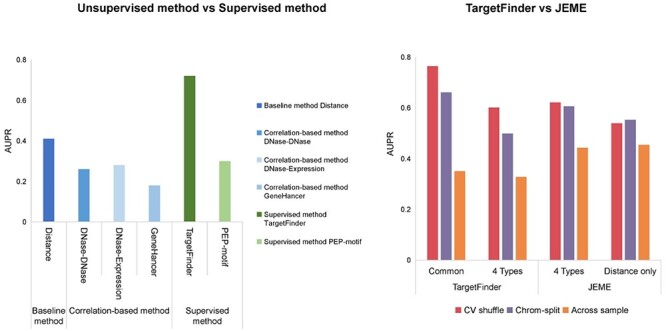
Performance of unsupervised and supervised methods. (A) Comparison of performance between distance-based, correlation-based and supervised methods using BENGI datasets. The AUPR scores of these six methods were evaluated by Moore et al [48]. (B) Comparison of regression-based model and trained classifier methods. Performances of TargetFinder and JEME on JEMEs ‘random target’ datasets using cross-validation with shuffling and chromosome-split strategies [82]. TargetFinder was validated with all features common in K562 and GM12878, and four epigenomic features (DNase-seq data and ChIP-seq data of H3K4me1, H3K27ac and H3K27me3) applied in JEME. JEME was validated with its four input features and distance feature, respectively. Across sample validation: TargetFinder and JEME were trained with K562 data and tested with GM12878 data. The AUPR scores of TargetFinder and JEME were evaluated by Cao and Fullwood [82].
Some supervised methods are overfitted due to the use of a problematic cross-validation group strategy in which samples are randomly split into training and test sets [48]. Using TargetFinder’s E/P/W datasets for six cell lines, Cao and Fullwood [82] found that a high degree of similarity between window features and the splitting of similar samples into training and test sets are likely to inflate cross-validation results, which may also explain the inability of TargetFinder to generalize across cell lines. To break the dependence between samples in training and test sets, they introduced a chromosome-split strategy by which all samples on the same chromosome are allocated to training or test set [82]. Comparison of TargetFinder and JEME on JEME’s random target datasets using cross-validation yields that the AUPRs of chromosome-split strategies are much lower than shuffling (Figure 5B) [82]. In addition, TargetFinder performs better with the use of all features common to K562 and GM12878 than with the use of the four types of features (DNase-seq and ChIP-seq of H3K4me1, H3K27ac and H3K27me3), whereas JEME using four features achieved similar performance as using distance alone (Figure 5B) [82].
DeepMILO outperforms CTCF-MP in the prediction of nonanchor loops, demonstrating that deep learning models can better learn complex sequence features to effectively predict the effects of mutations on insulator loops than common machine learning models [18]. Deep learning, one of the most active fields in machine learning research, has been applied recently to a variety of tasks in genomics, such as functional genome annotation and the prediction of gene expression [83]. Many elaborated deep-learning architectures, such as SPEID and DeepMILO, have been used to study regulatory interaction and 3D chromatin architecture [18, 54]. Deep learning can automatically extract sophisticated and meaningful features from massive high-dimensional datasets by training complex networks with multiple layers, which enables the integration of diverse types of input data [84, 85]. Deep learning not only improves predictive performance and accuracy over traditional models, but also provides insight into the mechanism of chromosome spatial organization by exploring internal representations in each layer of deep learning architecture [76, 86]. Hopefully, it will provide additional understanding of 3D genome organization and the mechanisms of gene regulation due to its powerful information-processing and abstraction capacities.
In general, supervised learning methods are limited by the uncertainty of labels used for training in which the definition of labels requires biology expertise. A positive labeled enhancer–promoter pair for training may be not physically interacted, which may influence the accuracy of prediction. Comparatively, unsupervised learning methods can be used to discover new patterns, such as PPI and EEI, without using prior knowledge [27, 36]. However, the verification of new patterns found by unsupervised learning poses a new challenge [21]. Semi-supervised methods have been developed to predict chromatin interaction and organization; they enable the extraction of features from unlabeled genomic and epigenomic profiles and training of models that integrate initial and extracted features, which results in much better algorithm performance than achieved with fully supervised methods [18, 56].
User guide
Generally, the prediction of chromatin interaction and organization using computational methods comprises three steps: (1) input data collection from publicly available databases or experiments, (2) input data processing using computational methods and (3) output of results representing chromatin interaction or spatial chromatin structures. In order to facilitate choose appropriate methods for different situations, we provide a user guide to facilitate the selection and application of these methods according to the prediction target and available data (Figure 6). To choose an appropriate method, the prediction target (chromatin interaction or organization) should first be determined; then the users can further select a method based on the available data or target output. Input data can be preferentially considered, if users have already obtained experimental data. Detailed input information of 48 methods are summarized in Table S2. If target output is preferentially considered, the user can select a method based on output and then prepare the required input data. Output categories are shown in Figures 2 and 3.
Figure 6 .
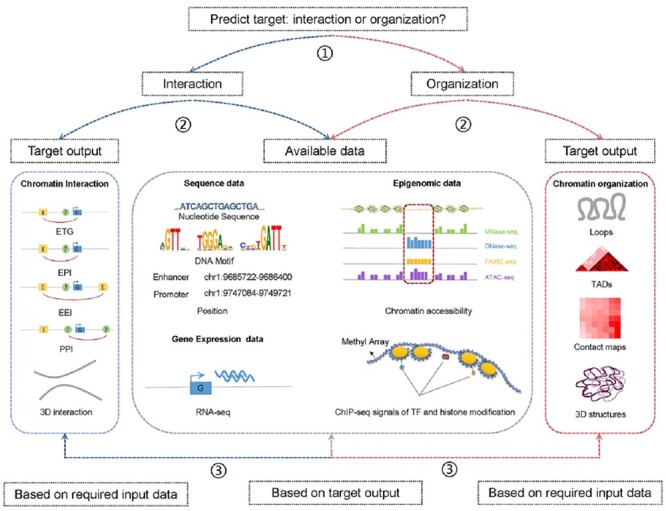
Pipeline of selecting computational methods for the prediction of chromatin and organization. ① Determine the predict target. ② Select method based on target output or available data. ③ Choose method based on required input data or target output within methods filtered from the second step.
Considering that such computational methods are numerous and that some tools are not publicly available, we list several methods that can be applied easily with small sets of input data (Table 4). PreSTIGE is an easy-to-use web-based tool for the prediction of ETGs based on H3K4me1 signals and RNA-seq data [25]. Cicero can be used to predict ETGs based on single-cell ATAC-seq data [31]. C3D and McEnhancer can be used to predict EPI and 3D chromatin interaction based on DNase-seq data [32, 49]. ELMER can be applied to predict ETGs when methylation and gene expression data are available [33]. EPIANN and EP2vec can be used to identify EPI based on DNA sequence data alone. For the prediction of chromatin conformation, DeepMILO can predict CTCF-mediated loops based on sequence data and CTCF signals. TAD-Lactuca can be applied to predict TAD boundaries using histone modification information. Akita can predict chromatin contact map based on DNA sequence data alone [63]. HiP-HoP can be used to predict 3D chromatin architecture with the integration of ATAC-seq or DNase-seq, H3K27ac, CTCF and Rad21 data to define loop anchors [73].
Table 4.
Recommended methods for the prediction of chromatin interaction and organization
| Method | Function | Input data | Available code | Implement | Reference |
|---|---|---|---|---|---|
| PreSTIGE | ETGs | H3K4me1, RNA-seq | http://prestige.case.edu | Web-based interface | [25] |
| ELMER | ETGs | DNA methylation, Gene expression | https://github.com/lijingya/ELMER; http://bioconductor.org/packages/ELMER/ | R/Web-based interface | [33, 35] |
| Cicero | ETGs | scATAC-seq | https://github.com/cole-trapnell-lab/cicero-release | R | [31] |
| McEnhancer | ETGs | DHS | https://ohlerlab.mdc-berlin.de/software/McEnhancer_134/ | Python | [49] |
| C3D | 3D chromatin interaction | DNase-seq | https://github.com/LupienLab/C3D | BASH, R | [32] |
| EPIANN | EPI | DNA sequence | https://github.com/wgmao/EPIANN | Python, R | [55] |
| EP2vec | EPI | DNA sequence | https://github.com/wanwenzeng/ep2vec | Python | [56] |
| DeepMILO | CTCF-mediated loops | CTCF, DNA sequence | https://github.com/khuranalab/DeepMILO | Python | [18] |
| TAD-Lactuca | TAD boundaries | CTCF, DNA sequence, Histone marks | https://github.com/LoopGan/TAD-Lactuca | Python | [67] |
| Akita | Contact map | DNA sequence | https://github.com/calico/basenji/tree/master/manuscripts/akita | Python | [63] |
| HiP-HoP | 3D chromatin structure | ATAC-seq, CTCF, Histone marks | https://www.sciencedirect.com/science/article/pii/S1097276518307871?via%3Dihub#sec4.1c | LAMMPS, Bowtie2, SAMtools, BEDtools, MACS2, etc. | [73] |
Applications in biomedicine
Applying computational methods to predict chromatin interaction and organization has been indispensable in the exploration of many biomedical problems. Some successful applications of these methods in biomedical fields are reviewed in this section, including the influence of genetic variants on gene expression, the mechanisms underlying gene regulation and disease development.
Influence of genetic variants on gene expression
Recent studies have shown that many noncoding single nucleotide polymorphisms (SNPs), identified in genome-wide association study (GWAS), are frequently located in cell line-specific enhancers and associated with risks of numerous common diseases [87, 88]. PreSTIGE provides evidence that multiple enhancer variants cooperatively contribute to the altered expression of their gene targets and demonstrates how GWAS-identified SNPs confer risks of given traits and noncoding variants confer susceptibility to common traits [25]. ABC interprets the functions of noncoding genetic variants that influence human traits [28]. Ernst et al. [29] proposed a method by which candidate regulatory functions are assigned to disease-associated variants. CTCF-MP can account for sequence changes by mutation and predict the impacts of these mutations on loops [62]. DeepMILO can be used to predict the impacts of variants identified by GWAS of samples on CTCF-mediated insulator loops from the cell type of interest [18]. Based on C3D, Johnston et al. [89] validated the effects of large structural variants on 3D genome architecture and transcriptional output. PRISMR can identify alterations in chromatin contacts induced by disease-associated structural variations, which may reconfigure TADs, thereby causing gene misexpression [90]. DeepC can predict the impact of both large-scale structural and single base-pair variations on genome folding [64].
Mechanisms underlying gene regulation
Computational methods also have been used to explore patterns underlying gene regulation and chromatin organization. For instance, Cicero can be used to investigate how changes in chromatin accessibility influence the expression of nearby genes and to dissect the mechanisms of cis-regulation at a genome-wide scale [31]. Cicero also can be used to predict chromatin hubs, which are involved in looping interaction [31]. DeepTACT can be used to identify hub promoters, which are active across cell lines and enriched in housekeeping genes [50]. Applying DeepTACT to identify disease-related genes showed that IFNA2 may be a significant autoimmune gene target [50]. EnContact can be used to identify hub enhancers. Moquin et al. [91] extended TargetFinder to investigate whether Epstein–Barr virus uses similar mechanisms of transcriptional regulation with the human genome by considering one region of interacting pair in the human genome, while the other is in the EBV genome. Using Rambutan to generate predictive chromatin contact maps for 53 human primary tissues, Schreiber et al. [68] showed that cell types of similar function had similar structures, whereas cancer cells did not.
Mechanisms underlying disease development
Disease-related mechanisms have also been investigated using computational methods. ELMER, a powerful algorithm for the examination of cis-regulatory interfaces between cancer-associated TFs and their functional target genes, can be used to investigate cancer-specific enhancers and paired gene promoters with TCGA datasets [33]. Ravi et al. [92] performed an mRNA expression and DNA methylation analysis of anaplastic thyroid cancer with ELMER v.2 [35], showing that aberrant DNA methylation affects gene expression and likely contributes to tumorigenesis in this disease. Besides, Naville et al. [23] provided new insight into the genetic basis of diseases caused by the misregulation of gene expression based on their correlation-based method. Using ChIP-seq time-course data from the estrogen-stimulated MCF7 breast cancer cell line, Dzida et al. [44] characterized the early response to estradiol in these cells. Sin-Chan et al. [93] used C3D with ATAC-seq data on embryonal tumors with multilayered rosettes to analyze long-range chromatin interaction and thus explored potential therapeutic vulnerability.
In addition, some methods provide available predictions that are often applied to related biomedical problems. For example, PreSTIGE supplies 2695 ENCODE annotated lncRNA transcripts expressed from active tissue-specific enhancers, as well as a database of predictions for 13 cell lines. The predictions have been used to infer putative eRNA–target pairs in squamous cell carcinoma of the head and neck [94], identify genetic interaction impacting body mass index [95], predict potential regulatory interaction related to the development and maturation of sensory epithelium [96] and analyze the associations of SNPs in regulatory regions with related diseases [97–99]. JEME reconstructs enhancer–promoter networks in 935 samples of human primary cells, tissues and cell lines, and the resulting predictions have been used to study biomedical problems related to asthma [100], bone density [101], attention deficit/hyperactivity disorder [102], serum levels of prostate-specific antigen [103], schizophrenia [104], osteoarthritis [105] and the immune response [106]. Other databases and platforms that incorporate predictive results obtained with the use of computational methods to enable better use of these data for the examination of biomedical problems include SEdb [107] and OncoBase [108]. We summarize such available predictions that could be applied directly in biomedical studies in Table S3.
Discussion
The precise prediction of 3D chromatin interaction and organization is crucial to deciphering gene regulation, cell differentiation and disease mechanisms. Computational methods based on sequence and epigenomic data have dramatically advanced our understanding of 3D chromatin architecture and its role in transcriptional regulation. In this paper, we described current computational methods based on DNA sequence and epigenomic data for the prediction of chromatin interaction and organization and provided a user guide aiding selection of the proper method based on the available data or target output. We reviewed the performance of such methods to highlight their merits and demerits. We also described their biomedical applications to facilitate biologists’ selection of methods suitable for their research.
Advice on improvement in user experience
Although numerous methods have been developed for the prediction of chromatin interaction and organization, their application in biomedical research is still not extensive. The biggest obstacle is that the complex algorithms applied in these methods are incomprehensible to biologists who lack computation-related knowledge, and their associations with biological functions might not be clear due to the black-box nature of some methods. These methods employ different software and programming languages, including python, java, C++ and R, and the instructions supplied may not be sufficiently explicit to allow users to reproduce procedures. Beyond the realization of predictive function and performance, the developers of computational methods could pay more attention to the improvement of user experience, which would help to promote the application of these methods. Convenient web-based methods that integrate raw data processing and analysis, which allow users to upload input data and obtain output file with simple operations, are preferable. To facilitate the execution of computational methods, developers should provide more detailed instructions for aspects such as raw data processing and the software required for specific versions, with the inclusion of file descriptions and simple examples of analyses. In addition, feedback on progress, summary reports and supporting information can be provided to make the computational process more transparent.
Existing challenges in the prediction of chromatin interaction and organization
Challenges remain in this filed [81]. First, high-resolution spatial proximity data remain limited, and the acquisition of such data remains difficult in terms of expenditure and sensitivity. Data resolution restricts the exploration of finer 3D chromatin organization [109]. Many computational algorithms, such as DeepHiC [110], HIFI [111], Boost-HiC [112], deDoc [113], hicGAN [114], HiCNN [115] and HiCPlus [11], have been developed to enhance the resolution of Hi-C data. Second, limitations remain regarding the strategies applied in state-of-art computational methods. Distance-based methods neglect the distal regulatory interaction and the case that multiple enhancers target the same promoter [40, 48]. The high sensitivity of Pearson correlation, used to identify enhancer–promoter pairs, also generates comparatively high false positive [40]. Besides, the overlap of TADs is not considered by some methods for TAD identification [116]. Third, methods for the exploration of fine-scale 3D chromatin organization in single cells remain lacking. Chromosome structures exhibit high variability in the interphase and high heterogeneity across multiple cell lines [58]. Most existing methods use population-averaged contact data and focus on fitting single consensus structures, while ignoring single cell variants and cell-to-cell variability in their models [117]. More effort should be put into developing computational methods for such purpose using single-cell genomic and epigenomic data Fourth, DNA–DNA ligation experiments are still suboptimal to identify physical chromatin interaction. Hi-C contacts obtained without loop-calling postprocessing do not provide strong evidence for physical interaction, and the confidence of predictions is highly influenced by experimental noise [51]. Conventional 3C-based processing cannot capture simultaneous or cooperative interaction [118, 119]. Unlike Hi-C measurement of the physical proximity of genomic loci, computational methods using sequence and epigenomic data can predict 3D chromatin organization and interaction based on various biological hypotheses, such as those regarding characteristic nucleosome arrangements, correlations of chromatin states, sequence motif features and evolutionarily conserved principles, which will provide complementary insight into chromatin organization. Fifth, the resolution of Hi-C experiment is limited by the restriction enzymes. A recently developed Hi-C variant technique, Micro-C, overcomes the limited resolution by fragmenting the chromatin by micrococcal nuclease [120, 121]. Micro-C exhibits improved signal-to-noise relative to Hi-C, and it can finer-scaled chromatin organizational features in mammalian cells, such as enhancer–promoter stripes, dots and domains [122, 123]. Lastly, current methods remain suboptimal. The most desirable application is the use of a model trained in a cell line with sequence and epigenomic data to make predictions for another cell line; current methods, however, perform undesirably across cell lines. Work to improve the accuracy and precision of predictions of regulatory interaction and spatial chromatin conformation via the integration of multiple data types and experimental methods is ongoing; examples are BART, which integrates Hi-C and epigenetic signatures [16], and GEM-FISH, which integrates fluorescence in situ hybridization and Hi-C data [124].
Future prospective
Recently, the mechanism by which DNA molecules pack into the nucleus in folded layers and ensure the precise regulation of genes has become a topic of interest in 3D genome research. With increasing availability of genomic, epigenomic and transcriptomic data, and advances in data-processing power, computational methods for the inference of 3D chromatin interaction and organization have dramatically facilitated the study of gene regulation mechanisms and 3D genome organization, which has provided new insight into cell differentiation and disease patterns. Studies of chromatin architecture alterations that could affect the expression of disease-related genes are ongoing [125]. For example, Kloetgen et al. [126] discovered the complexity and dynamic nature of the 3D chromatin architecture in human acute leukemia using various computational methods based on Hi-C, RNA-seq and CTCF ChIP-seq data and demonstrated that some changes in 3D interaction in leukemia could be inhibited by targeted small-molecular drugs. An understanding of disruptions to 3D chromatin organization will provide novel insight into the mechanisms responsible for disease-associated mutations and rearrangements and facilitate the treatment of genetic diseases. In this process, computational tools are indispensable for the prediction of regulatory element interaction and analysis of 3D chromatin organization. We believe that the influence of 3D chromatin architecture on gene regulation and disease development could be elaborated more exhaustively in the future with the extensive application of computational methods.
Key Points
We summarized 48 computational methods for the prediction of chromatin interaction and organization based on sequence and epigenomic profiles and compared the performance based on input data and algorithms.
To facilitate the application of these computational methods, we introduced their current biomedical applications and listed methods that could be applied easily with small sets of input data.
We summarized existing challenges affecting the inference of chromatin interaction and organization using computational methods and presented directions for its future improvement.
Supplementary Material
Acknowledgements
The authors wish to thank Prof. Cheng Li for the critical feedback and discussions.
Hao Li is a research assistant at the Beijing Institute of Radiation Medicine.
Huan Tao, Kang Xu and Junting Wang are master’s students at the Beijing Institute of Radiation Medicine.
Hao Hong, Shuai Jiang, Guifang Du, Yu Sun and Xin Huang are PhD students at the Beijing Institute of Radiation Medicine, Department of Biotechnology.
Yang Ding is a post-doc at the Beijing Institute of Radiation Medicine.
Fei Li is an associate professor at Chinese Academy of Sciences, Department of Computer Network Information Center.
Xiaofei Zheng is a professor at the Beijing Institute of Radiation Medicine.
Hebing Chen is an associate professor at the Beijing Institute of Radiation Medicine.
Xiaochen Bo is a professor at the Beijing Institute of Radiation Medicine.
Contributor Information
Huan Tao, Beijing Institute of Radiation Medicine, Beijing 100850, China.
Hao Li, Beijing Institute of Radiation Medicine, Beijing 100850, China.
Kang Xu, Beijing Institute of Radiation Medicine, Beijing 100850, China.
Hao Hong, Beijing Institute of Radiation Medicine, Beijing 100850, China.
Shuai Jiang, Beijing Institute of Radiation Medicine, Beijing 100850, China.
Guifang Du, Beijing Institute of Radiation Medicine, Beijing 100850, China.
Junting Wang, Beijing Institute of Radiation Medicine, Beijing 100850, China.
Yu Sun, Beijing Institute of Radiation Medicine, Beijing 100850, China.
Xin Huang, Beijing Institute of Radiation Medicine, Beijing 100850, China.
Yang Ding, Beijing Institute of Radiation Medicine, Beijing 100850, China.
Fei Li, Computer Network Information Center, Chinese Academy of Sciences, Beijing 100190, China.
Xiaofei Zheng, Beijing Institute of Radiation Medicine, Beijing 100850, China.
Hebing Chen, Beijing Institute of Radiation Medicine, Beijing 100850, China.
Xiaochen Bo, Beijing Institute of Radiation Medicine, Beijing 100850, China.
Funding
This work is supported by the National Natural Science Foundation of China [http://www.nsfc.gov.cn; nos. 31801112] and the Beijing Nova Program of Science and Technology [https://mis.kw.beijing.gov.cn; no. Z191100001119064] to Hebing Chen; the National Natural Science Foundation of China [http://www.nsfc.gov.cn; nos. 31900488] and the Beijing Natural Science Foundation [http://kw.beijing.gov.cn/; no. 5204040] to Hao Li; the National Natural Science Foundation of China [http://www.nsfc.gov.cn; nos. 61873276, 91540202, 81973244 to Xiaochen Bo, Xiaofei Zheng and Fei Li, respectively].
References
- 1.Dekker J, Rippe K, Dekker M, et al. . Capturing chromosome conformation. Science 2002;295:1306–11. [DOI] [PubMed] [Google Scholar]
- 2.Simonis M, Klous P, Splinter E, et al. . Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat Genet 2006;38:1348–54. [DOI] [PubMed] [Google Scholar]
- 3.Dostie J, Richmond TA, Arnaout RA, et al. . Chromosome conformation capture carbon copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res 2006;16:1299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lieberman-Aiden E, Berkum N, Williams L, et al. . Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009;326:289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Forcato M, Nicoletti C, Pal K, et al. . Comparison of computational methods for hi-C data analysis. Nat Methods 2017;14:679–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dixon J, Selvaraj S, Yue F, et al. . Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012;485:376–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fudenberg G, Imakaev M, Lu C, et al. . Formation of chromosomal domains by loop extrusion. Cell Rep 2016;15:2038–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanborn AL, Rao SS, Huang SC, et al. . Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci U S A 2015;112:E6456–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mora A, Sandve GK, Gabrielsen OS, et al. . In the loop: promoter-enhancer interactions and bioinformatics. Brief Bioinform 2016;17:980–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sati S, Cavalli G. Chromosome conformation capture technologies and their impact in understanding genome function. Chromosoma 2017;126:33–44. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, An L, Xu J, et al. . Enhancing Hi-C data resolution with deep convolutional neural network HiCPlus. Nat Commun 2018;9:750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rao SS, Huntley MH, Durand NC, et al. . A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014;159:1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 2004;306:636–40. [DOI] [PubMed] [Google Scholar]
- 14.Bernstein BE, Stamatoyannopoulos JA, Costello JF, et al. . The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol 2010;28:1045–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002;30:207–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang J, Marco E, Pinello L, et al. . Predicting chromatin organization using histone marks. Genome Biol 2015;16:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Y, Zhang R, Singh S, et al. . Exploiting sequence-based features for predicting enhancer-promoter interactions. Bioinformatics 2017;33:i252–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trieu T, Martinez-Fundichely A, Khurana E. DeepMILO: a deep learning approach to predict the impact of non-coding sequence variants on 3D chromatin structure. Genome Biol 2020;21:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lambert SA, Jolma A, Campitelli LF, et al. . The human transcription factors. Cell 2018;172:650–65. [DOI] [PubMed] [Google Scholar]
- 20.Ghandi M, Lee D, Mohammad-Noori M, et al. . Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput Biol 2014;10:e1003711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Shi W, Wasserman WW. Genome-wide prediction of cis-regulatory regions using supervised deep learning methods. BMC Bioinformatics 2018;19:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang H, Li F, Jia Y, et al. . Characteristic arrangement of nucleosomes is predictive of chromatin interactions at kilobase resolution. Nucleic Acids Res 2017;45:12739–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naville M, Ishibashi M, Ferg M, et al. . Long-range evolutionary constraints reveal cis-regulatory interactions on the human X chromosome. Nat Commun 2015;6:6904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu H, Zhang S, Yi X, et al. . Exploring 3D chromatin contacts in gene regulation: the evolution of approaches for the identification of functional enhancer-promoter interaction. Comput Struct Biotechnol J 2020;18:558–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corradin O, Saiakhova A, Akhtar-Zaidi B, et al. . Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Res 2014;24:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goecks J, Nekrutenko A, Taylor J, et al. . Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 2010;11:R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whalen S, Truty RM, Pollard KS. Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat Genet 2016;48:488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fulco CP, Nasser J, Jones TR, et al. . Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat Genet 2019;51:1664–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ernst J, Kheradpour P, Mikkelsen TS, et al. . Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011;473:43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thurman RE, Rynes E, Humbert R, et al. . The accessible chromatin landscape of the human genome. Nature 2012;489:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pliner HA, Packer JS, McFaline-Figueroa JL, et al. . Cicero predicts cis-regulatory DNA interactions from single-cell chromatin accessibility data. Mol Cell 2018;71:858–871.e858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mehdi T, Bailey SD, Guilhamon P, et al. . C3D: a tool to predict 3D genomic interactions between cis-regulatory elements. Bioinformatics 2019;35:877–9. [DOI] [PubMed] [Google Scholar]
- 33.Yao L, Shen H, Laird PW, et al. . Inferring regulatory element landscapes and transcription factor networks from cancer methylomes. Genome Biol 2015;16:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Connor T, Boden M, Bailey TL. CisMapper: predicting regulatory interactions from transcription factor ChIP-seq data. Nucleic Acids Res 2017;45:e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Silva TC, Coetzee SG, Gull N, et al. . ELMER v.2: an R/Bioconductor package to reconstruct gene regulatory networks from DNA methylation and transcriptome profiles. Bioinformatics 2019;35:1974–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu Y, Chen Z, Zhang K, et al. . Constructing 3D interaction maps from 1D epigenomes. Nat Commun 2016;7:10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Y, Wang Y, Xuan Z, et al. . De novo deciphering three-dimensional chromatin interaction and topological domains by wavelet transformation of epigenetic profiles. Nucleic Acids Res 2016;44:e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu D, Davila-Velderrain J, Zhang Z, et al. . Integrative construction of regulatory region networks in 127 human reference epigenomes by matrix factorization. Nucleic Acids Res 2019;47:7235–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao Q, Anyansi C, Hu X, et al. . Reconstruction of enhancer-target networks in 935 samples of human primary cells, tissues and cell lines. Nat Genet 2017;49:1428–36. [DOI] [PubMed] [Google Scholar]
- 40.Hait TA, Amar D, Shamir R, et al. . FOCS: a novel method for analyzing enhancer and gene activity patterns infers an extensive enhancer-promoter map. Genome Biol 2018;19:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He B, Chen C, Teng L, et al. . Global view of enhancer–promoter interactome in human cells. Proc Natl Acad Sci 2014;111:E2191–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao C, Li X, Hu HH. PETModule: a motif module based approach for enhancer target gene prediction. Sci Rep 2016;6:30043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roy S, Siahpirani AF, Chasman D, et al. . A predictive modeling approach for cell line-specific long-range regulatory interactions. Nucleic Acids Res 2015;43:8694–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dzida T, Iqbal M, Charapitsa I, et al. . Predicting stimulation-dependent enhancer-promoter interactions from ChIP-Seq time course data. Peerj 2017;5:e3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ramisch A, Heinrich V, Glaser LV, et al. . CRUP: a comprehensive framework to predict condition-specific regulatory units. Genome Biol 2019;20:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gao T, Qian JEAGLE. An algorithm that utilizes a small number of genomic features to predict tissue/cell type-specific enhancer-gene interactions. PLoS Comput Biol 2019;15:e1007436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Talukder A, Saadat S, Li X, et al. . EPIP: a novel approach for condition-specific enhancer–promoter interaction prediction. Bioinformatics 2019;35:3877–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moore JE, Pratt HE, Purcaro MJ, et al. . A curated benchmark of enhancer-gene interactions for evaluating enhancer-target gene prediction methods. Genome Biol 2020;21:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hafez D, Karabacak A, Krueger S, et al. . McEnhancer: predicting gene expression via semi-supervised assignment of enhancers to target genes. Genome Biol 2017;18:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li W, Wong WH, Jiang R. DeepTACT: predicting 3D chromatin contacts via bootstrapping deep learning. Nucleic Acids Res 2019;47:e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Al Bkhetan Z, Plewczynski D. Three-dimensional epigenome statistical model: genome-wide chromatin looping prediction. Sci Rep 2018;8:5217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Al Bkhetan Z, Plewczynski D. Multi-levels 3D Chromatin Interactions Prediction Using Epigenomic Profiles. Cham: Springer International Publishing, 2017, 19–28. [Google Scholar]
- 53.Belokopytova PS, Nuriddinov MA, Mozheiko EA, et al. . Quantitative prediction of enhancer-promoter interactions. Genome Res 2020;30:72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singh S, Yang Y, Póczos B, et al. . Predicting enhancer-promoter interaction from genomic sequence with deep neural networks. Quantitative Biology 2019;7:122–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mao W, Kostka D. Chikina M, Modeling enhancer-promoter interactions with attention-based neural networks. bioRxiv 2017;219667. DOI: 10.1101/219667. [DOI] [Google Scholar]
- 56.Zeng W, Wu M, Jiang R. Prediction of enhancer-promoter interactions via natural language processing. BMC Genomics 2018;19:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gan M, Li W, Jiang R. EnContact: predicting enhancer-enhancer contacts using sequence-based deep learning model. Peerj 2019;7:e7657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cheng RR, Contessoto VG, Lieberman Aiden E, et al. . Exploring chromosomal structural heterogeneity across multiple cell lines. Elife 2020;9:e60312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jost D, Carrivain P, Cavalli G, et al. . Modeling epigenome folding: formation and dynamics of topologically associated chromatin domains. Nucleic Acids Res 2014;42:9553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Di Pierro M, Cheng RR, Lieberman Aiden E, et al. . De novo prediction of human chromosome structures: epigenetic marking patterns encode genome architecture. Proc Natl Acad Sci U S A 2017;114:12126–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sefer E, Kingsford C. Semi-nonparametric modeling of topological domain formation from epigenetic data. Algorithms Mol Biol 2019;14:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang R, Wang Y, Yang Y, et al. . Predicting CTCF-mediated chromatin loops using CTCF-MP. Bioinformatics 2018;34:i133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fudenberg G, Kelley DR, Pollard KS. Predicting 3D genome folding from DNA sequence with Akita. Nat Methods 2020;17:1111–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schwessinger R, Gosden M, Downes D, et al. . DeepC: predicting 3D genome folding using megabase-scale transfer learning. Nat Methods 2020;17:1118–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kai Y, Andricovich J, Zeng Z, et al. . Predicting CTCF-mediated chromatin interactions by integrating genomic and epigenomic features. Nat Commun 2018;9:4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hong S, Kim D. Computational characterization of chromatin domain boundary-associated genomic elements. Nucleic Acids Res 2017;45:10403–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gan W, Luo J, Li YZ, et al. . A computational method to predict topologically associating domain boundaries combining histone marks and sequence information. BMC Genomics 2019;20:980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schreiber J, Libbrecht M, Bilmes J, et al. . Nucleotide sequence and DNaseI sensitivity are predictive of 3D chromatin architecture. bioRxiv 2018;103614. DOI: 10.1101/103614. [DOI] [Google Scholar]
- 69.Farré P, Heurteau A, Cuvier O, et al. . Dense neural networks for predicting chromatin conformation. BMC Bioinformatics 2018;19:372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brackley CA, Brown JM, Waithe D, et al. . Predicting the three-dimensional folding of cis-regulatory regions in mammalian genomes using bioinformatic data and polymer models. Genome Biol 2016;17:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brackley CA, Johnson J, Kelly S, et al. . Simulated binding of transcription factors to active and inactive regions folds human chromosomes into loops, rosettes and topological domains. Nucleic Acids Res 2016;44:3503–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brackley CA, Liebchen B, Michieletto D, et al. . Ephemeral protein binding to DNA shapes stable nuclear bodies and chromatin domains. Biophys J 2017;112:1085–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Buckle A, Brackley CA, Boyle S, et al. . Polymer simulations of heteromorphic chromatin predict the 3D folding of complex genomic loci. Mol Cell 2018;72:786–797 e711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Qi Y, Zhang B. Predicting three-dimensional genome organization with chromatin states. PLoS Comput Biol 2019;15:e1007024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Al Bkhetan Z, Kadlof M, Kraft A, et al. . Machine learning polymer models of three-dimensional chromatin organization in human lymphoblastoid cells. Methods 2019;166:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xiao M, Zhuang Z, Pan W. Local Epigenomic data are more informative than local genome sequence data in predicting enhancer-promoter interactions using neural networks. Genes (Basel) 2019;11:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jing F, Zhang S, Cao Z, et al. . An integrative framework for combining sequence and epigenomic data to predict transcription factor binding sites using deep learning. IEEE/ACM Trans Comput Biol Bioinform 2019. DOI: 10.1109/TCBB.2019.2901789. [DOI] [PubMed] [Google Scholar]
- 78.Nair S, Kim DS, Perricone J, et al. . Integrating regulatory DNA sequence and gene expression to predict genome-wide chromatin accessibility across cellular contexts. Bioinformatics 2019;35:i108–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shen Y, Yue F, McCleary DF, et al. . A map of the cis-regulatory sequences in the mouse genome. Nature 2012;488:116–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fishilevich S, Nudel R, Rappaport N, et al. . GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford) 2017;2017:bax028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hariprakash JM, Ferrari F. Computational biology solutions to identify enhancers-target gene pairs. Comput Struct Biotechnol J 2019;17:821–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cao F, Fullwood MJ. Inflated performance measures in enhancer-promoter interaction-prediction methods. Nat Genet 2019;51:1196–8. [DOI] [PubMed] [Google Scholar]
- 83.Cao C, Liu F, Tan H, et al. . Deep learning and its applications in biomedicine. Genomics Proteomics Bioinformatics 2018;16:17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Min S, Lee B, Yoon S. Deep learning in bioinformatics. Brief Bioinform 2017;18:851–69. [DOI] [PubMed] [Google Scholar]
- 85.Wainberg M, Merico D, Delong A, et al. . Deep learning in biomedicine. Nat Biotechnol 2018;36:829–38. [DOI] [PubMed] [Google Scholar]
- 86.Park Y, Kellis M. Deep learning for regulatory genomics. Nat Biotechnol 2015;33:825–6. [DOI] [PubMed] [Google Scholar]
- 87.Smemo S, Campos LC, Moskowitz IP, et al. . Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Hum Mol Genet 2012;21:3255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Andersson R, Gebhard C, Miguel-Escalada I, et al. . An atlas of active enhancers across human cell types and tissues. Nature 2014;507:455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Johnston MJ, Nikolic A, Ninkovic N, et al. . High-resolution structural genomics reveals new therapeutic vulnerabilities in glioblastoma. Genome Res 2019;29:1211–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bianco S, Lupianez DG, Chiariello AM, et al. . Polymer physics predicts the effects of structural variants on chromatin architecture. Nat Genet 2018;50:662–7. [DOI] [PubMed] [Google Scholar]
- 91.Moquin SA, Thomas S, Whalen S, et al. . The Epstein-Barr virus Episome Maneuvers between nuclear chromatin compartments during reactivation. J Virol 2018;92:e01413–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ravi N, Yang M, Mylona N, et al. . Global RNA expression and DNA methylation patterns in primary anaplastic thyroid cancer. Cancers (Basel) 2020;12:680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sin-Chan P, Mumal I, Suwal T, et al. . A C19MC-LIN28A-MYCN oncogenic circuit driven by hijacked super-enhancers is a distinct therapeutic vulnerability in ETMRs: a lethal brain tumor. Cancer Cell 2019;36:51–67.e57. [DOI] [PubMed] [Google Scholar]
- 94.Gu X, Wang L, Boldrup L, et al. . AP001056.1, a prognosis-related enhancer RNA in squamous cell carcinoma of the head and neck. Cancers (Basel) 2019;11:347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dong SS, Yao S, Chen YX, et al. . Detecting epistasis within chromatin regulatory circuitry reveals CAND2 as a novel susceptibility gene for obesity. Int J Obes (Lond) 2019;43:450–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yizhar-Barnea O, Valensisi C, Jayavelu ND, et al. . DNA methylation dynamics during embryonic development and postnatal maturation of the mouse auditory sensory epithelium. Sci Rep 2018;8:17348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Molineros JE, Singh B, Terao C, et al. . Mechanistic characterization of RASGRP1 variants identifies an hnRNP-K-regulated transcriptional enhancer contributing to SLE susceptibility. Front Immunol 2019;10:1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Michailidou K, Lindström S, Dennis J, et al. . Association analysis identifies 65 new breast cancer risk loci. Nature 2017;551:92–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ghoussaini M, French JD, Michailidou K, et al. . Evidence that the 5p12 variant rs10941679 confers susceptibility to Estrogen-receptor-positive breast cancer through FGF10 and MRPS30 regulation. Am J Hum Genet 2016;99:903–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Olafsdottir TA, Theodors F, Bjarnadottir K, et al. . Eighty-eight variants highlight the role of T cell regulation and airway remodeling in asthma pathogenesis. Nat Commun 2020;11:393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Styrkarsdottir U, Stefansson OA, Gunnarsdottir K, et al. . GWAS of bone size yields twelve loci that also affect height, BMD, osteoarthritis or fractures. Nat Commun 2019;10:2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cheng B, Du Y, Wen Y, et al. . Integrative analysis of genome-wide association study and chromosomal enhancer maps identified brain region related pathways associated with ADHD. Compr Psychiatry 2019;88:65–9. [DOI] [PubMed] [Google Scholar]
- 103.Gudmundsson J, Sigurdsson JK, Stefansdottir L, et al. . Genome-wide associations for benign prostatic hyperplasia reveal a genetic correlation with serum levels of PSA. Nat Commun 2018;9:4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wu C, Pan W. Integration of enhancer-promoter interactions with GWAS summary results identifies novel schizophrenia-associated genes and pathways. Genetics 2018;209:699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu Y, Chang JC, Hon CC, et al. . Chromatin accessibility landscape of articular knee cartilage reveals aberrant enhancer regulation in osteoarthritis. Sci Rep 2018;8:15499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lecellier CH, Wasserman WW, Mathelier A. Human enhancers Harboring specific sequence composition, activity, and genome organization are linked to the immune response. Genetics 2018;209:1055–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jiang Y, Qian F, Bai X, et al. . SEdb: a comprehensive human super-enhancer database. Nucleic Acids Res 2019;47:D235–d243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li X, Shi L, Wang Y, et al. . OncoBase: a platform for decoding regulatory somatic mutations in human cancers. Nucleic Acids Res 2019;47:D1044–d1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Park J, Lin S. Impact of data resolution on three-dimensional structure inference methods. BMC Bioinformatics 2016;17:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hong H, Jiang S, Li H, et al. . DeepHiC: a generative adversarial network for enhancing hi-C data resolution. PLoS Comput Biol 2020;16:e1007287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cameron CJ, Dostie J, Blanchette M. HIFI: estimating DNA-DNA interaction frequency from Hi-C data at restriction-fragment resolution. Genome Biol 2020;21:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Carron L, Morlot JB, Matthys V, et al. . Boost-HiC: computational enhancement of long-range contacts in chromosomal contact maps. Bioinformatics 2019;35:2724–9. [DOI] [PubMed] [Google Scholar]
- 113.Li A, Yin X, Xu B, et al. . Decoding topologically associating domains with ultra-low resolution Hi-C data by graph structural entropy. Nat Commun 2018;9:3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu Q, Lv H, Jiang R. hicGAN infers super resolution Hi-C data with generative adversarial networks. Bioinformatics 2019;35:i99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liu T, Wang Z. HiCNN: a very deep convolutional neural network to better enhance the resolution of Hi-C data. Bioinformatics 2019;35:4222–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Weinreb C, Raphael BJ. Identification of hierarchical chromatin domains. Bioinformatics 2016;32:1601–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Carstens S, Nilges M, Habeck M. Bayesian inference of chromatin structure ensembles from population-averaged contact data. Proc Natl Acad Sci U S A 2020;117:7824–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schoenfelder S, Furlan-Magaril M, Mifsud B, et al. . The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res 2015;25:582–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kolovos P, Brouwer RWW, Kockx CEM, et al. . Investigation of the spatial structure and interactions of the genome at sub-kilobase-pair resolution using T2C. Nat Protoc 2018;13:459–77. [DOI] [PubMed] [Google Scholar]
- 120.Hsieh TH, Weiner A, Lajoie B, et al. . Mapping nucleosome resolution chromosome folding in yeast by micro-C. Cell 2015;162:108–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hsieh TS, Fudenberg G, Goloborodko A, et al. . Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. Nat Methods 2016;13:1009–11. [DOI] [PubMed] [Google Scholar]
- 122.Hsieh TS, Cattoglio C, Slobodyanyuk E, et al. . Resolving the 3D landscape of transcription-linked mammalian chromatin folding. Mol Cell 2020;78:539–553.e538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Krietenstein N, Abraham S, Venev SV, et al. . Ultrastructural details of mammalian chromosome architecture. Mol Cell 2020;78:554–565.e557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Abbas A, He X, Niu J, et al. . Integrating Hi-C and FISH data for modeling of the 3D organization of chromosomes. Nat Commun 2019;10:2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Achinger-Kawecka J, Clark SJ. Disruption of the 3D cancer genome blueprint. Epigenomics 2017;9:47–55. [DOI] [PubMed] [Google Scholar]
- 126.Kloetgen A, Thandapani P, Ntziachristos P, et al. . Three-dimensional chromatin landscapes in T cell acute lymphoblastic leukemia. Nat Genet 2020;52:388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.