

Abstract
Regulatory T cells (Tregs) that promote tumor immune evasion are enriched in certain tumors and correlate with poor prognosis. However, mechanisms for Treg enrichment remain incompletely understood. We described a mechanism for Treg enrichment in mouse and human tumors mediated by the αvβ8 integrin. Tumor cell αvβ8 bound to latent transforming growth factor–β (L–TGF-β) presented on the surface of T cells, resulting in TGF-β activation and immunosuppressive Treg differentiation in vitro. In vivo, tumor cell αvβ8 expression correlated with Treg enrichment, immunosuppressive Treg gene expression, and increased tumor growth, which was reduced in mice by αvβ8 inhibition or Treg depletion. Structural modeling and cell-based studies suggested a highly geometrically constrained complex forming between αvβ8-expressing tumor cells and L–TGF-β–expressing T cells, facilitating TGF-β activation, independent of release and diffusion, and providing limited access to TGF-β inhibitors. These findings suggest a highly localized tumor-specific mechanism for Treg enrichment.
INTRODUCTION
Regulatory T cells (Tregs) are enriched in subsets of cancers and associated with poor clinical prognoses (1–3). Tregs suppress antitumor immune responses, with Treg depletion promoting effector CD8+ T cell immunity (4–6). Tumor-specific Treg enrichment likely contributes to resistance to current immunotherapies (7). How Treg enrichment occurs in tumors is not well understood, and a better understanding of this may improve antitumor therapies.
Tregs are induced by self-antigens in the thymus (tTregs) or foreign antigens in extrathymic peripheral tissues (pTregs) (8). tTregs are recruited to tumors through chemokines specific to individual tumor types, whereas pTregs are generated within tumors in response to signals generated within the tumor microenvironment (TME) (9, 10). The cytokine transforming growth factor–β (TGF-β) may contribute to pTreg enrichment in tumors (11). TGF-β is critical for pTreg generation because of its essential role in forkhead box P3 (FOXP3) expression during Treg differentiation; the role of TGF-β in tTreg generation is less clear (9, 12). Elucidating the role of TGF-β in pTreg enrichment in tumors could help identify therapies specifically targeting the immunosuppressive effects of TGF-β while minimizing toxicities associated with systemic inhibition of TGF-β itself, TGF-β receptors (TGF-βRs), or Treg depletion (6, 13–15).
TGF-β and its receptors are widely expressed in the TME (16, 17). TGF-β is always expressed in an inactive form within a latent complex, latent TGF-β (L–TGF-β), formed by noncovalent association of TGF-β with its prodomain, latency-associated protein (LAP) (18). On Tregs, TGF-β is presented at the cell surface through association with the scaffolding molecule glycoprotein A repetitions pre-dominant (GARP) (19). Within L–TGF-β, mature TGF-β cannot interact with its receptors unless undergoing “activation,” functionally defined as the acquired ability to initiate signaling through TGF-βRs.
TGF-β is cleaved intracellularly by furin from LAP, which has led to the widespread assumption that mature TGF-β must be physically released and diffused from the L–TGF-β complex for activation and induction of signaling (20). However, why L–TGF-β needs to be presented on the cell surface by GARP if TGF-β diffusion gradients account for Treg enrichment in tumors is unclear. L–TGF-β is present on the cell surface of CD4+ T cells isolated from murine tumors (21), suggesting an alternative model where local regulation of TGF-β activation only on specific L–TGF-β–presenting CD4+ T cells induces conversion to pTregs.
The integrin αvβ8 may be an important mediator of tumor-specific regulation of TGF-β function (21). αvβ8 is highly expressed in multiple cancer types that have high numbers of Tregs (6, 21, 22). The LAPs of L–TGF-β1 and L–TGF-β3 contain arginine-glycine-aspartate integrin-binding sites, which are recognized by several integrins, particularly αvβ8, which is critical for TGF-β activation in immune cell function (23–26). L–TGF-β is the only physiologically relevant ligand for αvβ8, and thus, targeting αvβ8 selectively inhibits TGF-β function (27, 28). Anti-αvβ8 inhibits tumor growth of β8-expressing tumors, correlating with increased immune cell numbers and reversal of effector T cell exclusion; combination with anti–programmed cell death protein 1 (PD-1) improves these antitumor effects (21).
A structure-based model predicts that αvβ8 most efficiently activates L–TGF-β when αvβ8 expressed by one cell binds to cell-surface L–TGF-β presented by another. Within the cell-cell αvβ8/L–TGF-β complex, active TGF-β exclusively signals to the L–TGF-β–presenting cell because active TGF-β is not released and does not diffuse from the complex (29). Here, we found that a αvβ8/L–TGF-β complex formed between αvβ8-expressing tumor cells and L–TGF-β–presenting T cells was associated with Treg enrichment in tumors. The αvβ8/L–TGF-β complex limited access to TGF-β inhibitors that would have otherwise been free to bind if active TGF-β was diffusible. These findings modify the conceptual framework for understanding how TGF-β functions in immune differentiation and affects therapeutic approaches to effectively and selectively inhibit it.
RESULTS
Tregs contributed to β8-dependent tumor growth
We investigated whether Tregs were required for protumorigenic effects of tumor cell αvβ8 using the syngeneic orthotopic β8-Lewis lung carcinoma (LLC) model (Fig. 1, A and B) (21). β8-LLC tumors had significantly more Tregs than non–β8-expressing mock-LLC tumors (Fig. 1, C and D). We depleted CD25+ T cells with intraperitoneal injection of anti-CD25 (clone PC-61.5.3) 1 day after tumor implantation (30). PC-61.5.3 and anti-αvβ8 (clone C6D4) both reduced FOXP3+ cells from β8-LLC tumors (Fig. 1, C and D). Neither antibody significantly affected FOXP3+ cell numbers from mock-LLC tumors implanted on the contralateral side within the time frame in which euthanasia was required because of the size of the primary β8-LLC tumor (Fig. 1, C and D).
Fig. 1. Treg depletion specifically inhibited β8-LLC but not mock-LLC tumor outgrowth.
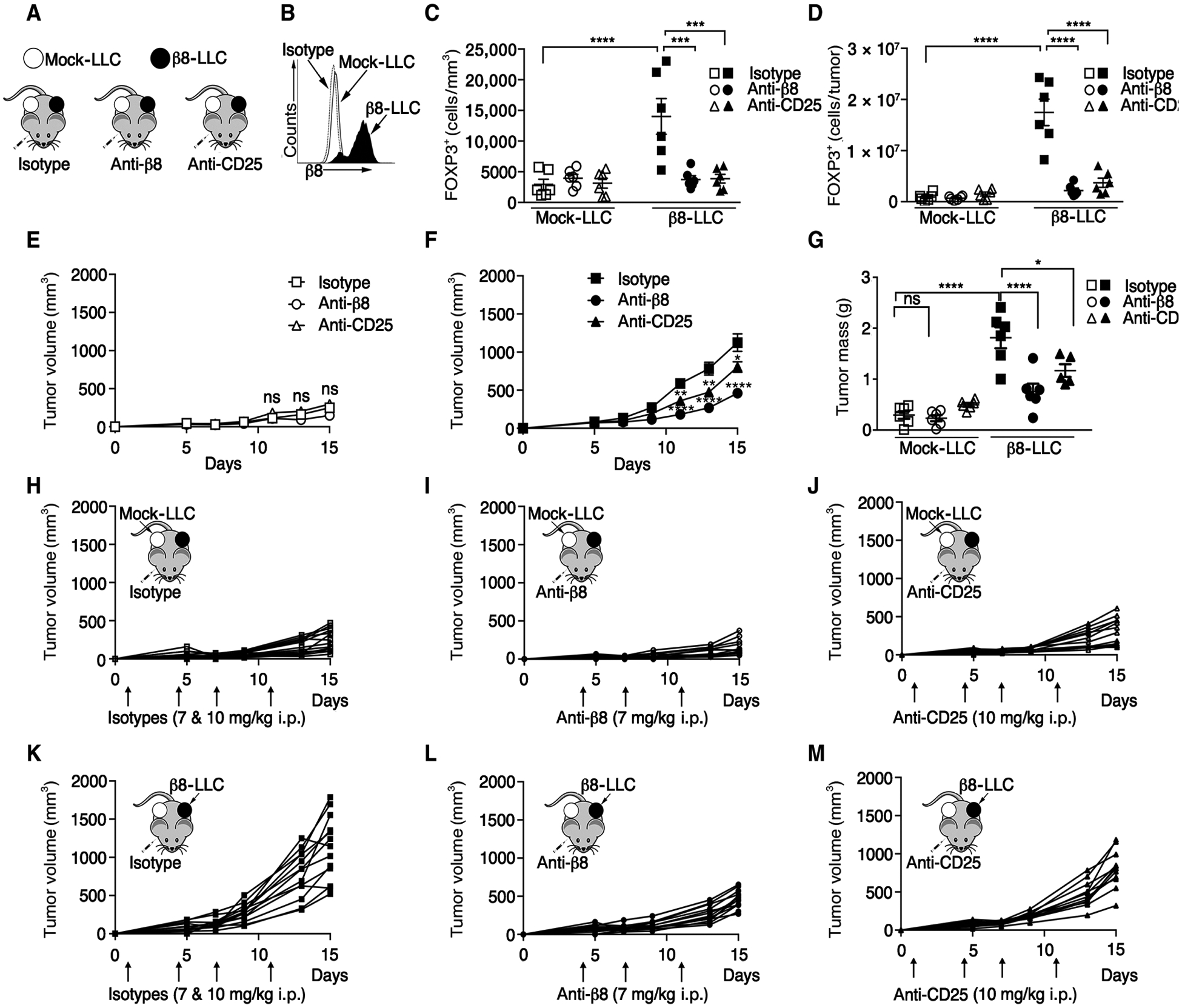
(A) Cartoon of tumor model. (B) Representative surface staining: anti-β8 (C6D4) or isotype control of mock-LLC or β8-LLC cells. (C to M) Mock-LLC and β8-LLC tumors were established on opposing flanks of C57BL/6 mice. (C to G, I, and L) Mice were treated [7 mg/kg, intraperitoneally (i.p.)] with anti-β8 (C6D4) after tumor establishment. (J and M) Tregs were depleted with anti-CD25 (clone PC-61.5.3) starting 1 day after tumor cell injection. (H and K) Isotype controls. (C and D) Intratumoral Treg numbers (outliers removed) confirmed by immunohistochemistry of FOXP3 of mock (open) or β8-LLC (filled) tumors shown by FOXP3+ cells/tumor surface area I or FOXP3+ cells/tumor (D). Average LLC tumor volumes for mock-LLC (E) and β8-LLC (F), with day 15 tumor weights (G). Corresponding spider plots for mock-LLC (H to J) treated with isotype (H), anti-β8, C6D4 (I), anti-CD25 (J), or β8-LLC treated with isotype (K), anti-β8, C6D4 (L), or anti-CD25 (M). Arrows indicate antibody injection days. (H to M) Cartoons show tumor type (arrow) in accompanying plots. One-way ANOVA was used for multiple comparisons followed by Tukey’s post-test. Student’s unpaired t test was used for comparing two datasets. Shown is SE, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. ns, not significant.
β8-LLC cells formed tumors significantly faster than mock-LLC tumors grown on opposite flanks, which was reduced by PC-61.5.3 or C6D4 (Fig. 1, E to M). PC-61.5.3 or C6D4 did not affect mock-LLC tumor volume or mass at day 14 after tumor cell injection (Fig. 1, E and G), consistent with the inability of PC-61.5.3 or C6D4 to reduce FOXP3+ cells from mock-LLC tumors (Fig. 1, C and D). We confirmed the protumorigenic effects of tumor cell αvβ8 in naturally β8-expressing TRAMP-C2 murine prostate carcinoma cells using a knockdown approach with ITGB8 short hairpin RNA (shRNA) (fig. S1, A to H).
The mechanism of action of C6D4 could be from functional blockade of αvβ8-mediated TGF-β activation or effector function (i.e., antibody-dependent cellular cytotoxicity). To test the role of antibody effector function, we performed β8-LLC tumorigenicity assays using a Fab of C6D4 lacking the Fc region required for effector function (31). C6D4-Fab blocked β8-LLC tumor growth, similar to intact C6D4, compared with isotype control (fig. S1, I to L). These results demonstrated that C6D4 functions in the TME by blocking αvβ8 function, not by antibody effector function.
Tumor cell αvβ8 expression caused Treg enrichment and an immunosuppressive Treg transcriptome
PC-61.5.3 and C6D4 specifically reduced Treg numbers and growth of β8-LLC but not mock-LLC tumors, suggesting that Tregs in β8-LLC tumors were distinct from Tregs in mock-LLC tumors. We hypothesized that αvβ8-expressing tumor cells participated in the local conversion of CD25+FOXP3− T cells to FOXP3+ pTreg. The absence of an abscopal effect of Tregs generated in β8-LLC on contralateral mock-LLC tumor growth suggests that Tregs generated within αvβ8-expressing tumors are either retained or are phenotypically unstable outside the αvβ8-expressing TME because pTreg may be less stable compared with tTreg (32).
To test whether αvβ8-expressing tumor cells locally mediated conversion of CD25+FOXP3− T cells to pTregs, we determined the transcriptome of Tregs generated in β8-LLC compared with mock-LLC tumors. β8-LLC tumors had more Tregs than mock-LLC tumors, and anti-β8 reduced Treg numbers in β8-LLC tumors, suggesting that tumor cell expression of αvβ8 correlated with CD4+CD25+FOXP3+ Treg enrichment (Fig. 2, B and C). We next performed RNA sequencing (RNAseq), evaluating the transcriptome of sorted Treg pools from mock-LLC or β8-LLC tumors from mice treated with isotype (SV5) or C6D4 (Fig. 2, A, D, and E, and fig. S2A). Similarly high read counts for FoxP3 and Il2ra (CD25) were seen in all Treg pools, consistent with high Treg purity (fig. S2B). Filtering of the dataset revealed 118 genes most highly and variably expressed across groups (Fig. 2, D and E).
Fig. 2. Tumor cell expression of αvβ8 drove a distinct immunosuppressive Tregtranscriptome.
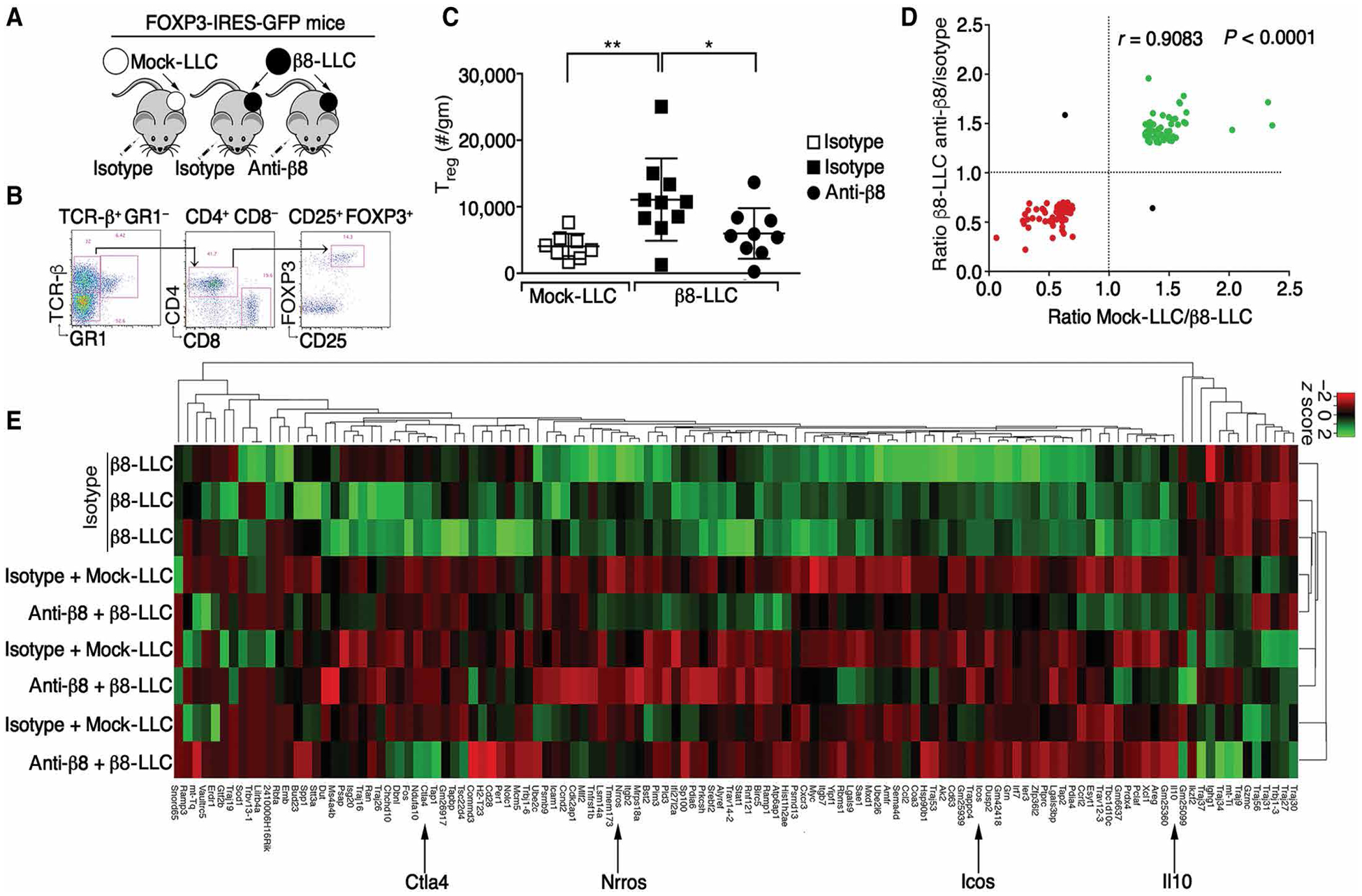
(A) Cartoon of model. (B) Gating strategy for FOXP3+CD25+ cells, enumerated in (C) as FOXP3+CD25+ cells/g (outliers removed) of mock (open boxes) or β8-LLC tumors treated with isotype (filled boxes) or anti-β8 (C6D4, filled circles). (D) Bulk RNAseq of sorted pools (9 to 10 mice per group in three pools) of CD4+GFP+ cells. Differential expression plot of 118 most highly expressed genes [>50 average fragments per kilobase million (FKPM)] increased (green) or decreased (red) in expression by at least 30% in Treg groups treated with anti-β8 compared with isotype control or mock-LLC compared with β8-LLC, with Pearson R and two tailed P value. (E) Hierarchal clustering and heatmap of 118 most highly and variably expressed genes shown in (D). Note that Tregs from β8-LLC isotype–treated tumors are distinct (top three rows) from mock or β8-LLC Treg treated with anti-β8 (C6D4). Arrows indicate key genes. For multiple comparisons, one-way ANOVA was used followed by Tukey’s post-test. *P < 0.05 and **P < 0.01.
Tregs from β8-LLC tumors had distinct transcriptional profiles that could be blocked by C6D4 to resemble Tregs from mock-LLC tumors. Comparison of Tregs from C6D4-treated β8-LLC tumors with Tregs from isotype-treated β8-LLC tumors revealed 116 of the 118 most variably expressed genes changed in the same direction (R = 0.908, P < 0.0001; Fig. 2D). Therefore, systemic blockade of αvβ8 with C6D4 had the same effect on the Treg transcriptome as absence of β8 expression by tumor cells, demonstrating the importance of tumor cell αvβ8 to locally control Treg gene expression. Hierarchal clustering of this gene set revealed tumor cell β8-dependent increases in expression of genes associated with Treg immunosuppressive function and differentiation (i.e., Il10, Ctla4, and Icos; Fig. 2E) (6, 33). Tregs from β8-LLC tumors treated with isotype clustered separately from Tregs from C6D4-treated β8-LLC tumors or mock-LLC tumors (Fig. 2E). In contrast, Tregs from mock or β8-LLC tumors treated with C6D4 clustered together and were indistinguishable (Fig. 2E). These data are consistent with the idea that tumor cell αvβ8 contributed to the enrichment of a Treg population distinct from those infiltrating mock-LLC tumors.
The mechanisms underlying Treg enrichment in tumors were likely to involve recruitment of Tregs and local Treg conversion (2, 34). Recruited Tregs have increased representation of tTregs, which potentially allows them to be distinguished from pTregs by high expression of two markers, Helios (Ikzf2) and neuropilin 1 (Nrp1) (9, 34–38). However, both β8-LLC and mock-LLC tumor Tregs had low or un-detectable levels of Ikzf2 and Nrp1 in contrast to splenic Tregs, which consist of about 70% tTregs and had high levels of these markers (fig. S2F) (36). Together, these data indicated that Tregs in β8-LLC tumors were skewed toward pTregs rather than tTregs.
L–TGF-β was expressed on the surface of Treg and non-Treg CD4+ T cells
We next determined whether non-Treg CD4+ T cells expressed L–TGF-β on their cell surface and were therefore “primed” for Treg conversion. There is cell surface expression of L–TGF-β on Tregs and activated non-Treg CD4+ T cells; however, no function has been attributed to this expression (19, 39, 40). Activated non-Treg CD4+ T cells [CD4+CD25+GFP− T cells from spleens of FoxP3-IRES-GFP mice (41)]] expressed increased cell surface L–TGF-β compared with nonactivated CD4+ T cells (Fig. 3, A, B, and D, and fig. S3, A to H). Consistent with other reports, there were more L–TGF-β+ cells in the CD25+FOXP3+GFP+ T cell population (~8 to 22%) (Fig. 3, C and D). Thus, significant fractions of both non-Treg CD4+ T cells and Tregs express cell surface L–TGF-β. The physiological relevance of these findings is highlighted by the detection of L–TGF-β on the surface of both non-Treg CD4+ T cells and Tregs isolated from β8-LLC tumors (21).
Fig. 3. Contact of L–TGF-β–presenting non-Treg CD4+ T cells with αvβ8 drove iTreg differentiation.
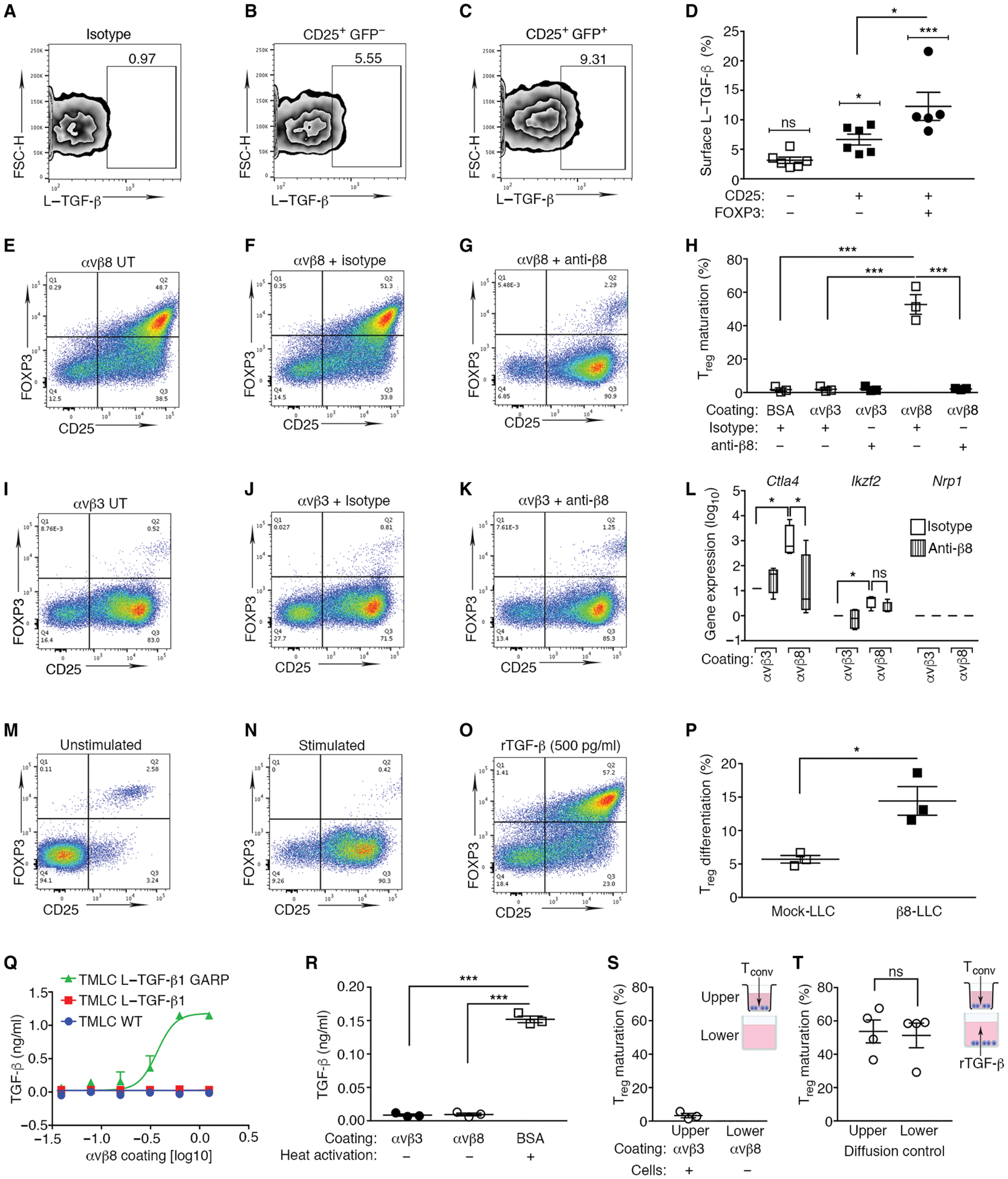
(A to C) CD4+ mouse splenocytes express L–TGF-β1 on the cell surface. (A) Isotype-matched negative control for (B) CD4+CD25+ FOXP3− T cells and (C) CD4+CD25+ FOXP3+ Treg stained with anti-LAP. (D) L–TGF-β1 surface staining (outlier removed) in CD4+CD25−FOXP3−, CD4+CD25+FOXP3−, and CD4+CD25+ FOXP3+ T cell subsets (brackets above each column indicate comparisons relative to isotype control). (E to G) CD4+ mouse splenocytes undergo iTreg differentiation when cultured on immobilized αvβ8, but not on (I to K) integrin αvβ3, or on (M and N) BSA. (E to K and N) CD4+ splenocytes from FOXP3-IRES-GFP mice were activated [anti-CD3 and interleukin (IL-2)] or (M) not stimulated. (O) As positive control, stimulated CD4+ T cells were treated with supraphysiologic levels of rTGF-β1 (500 pg/ml) (65). Representative experiment (n = 3) depicts CD4+ gated T cells stained with anti-CD25 (x axis) with FOXP3 expression determined by green fluorescent protein (GFP; y axis).Treg (CD4+CD25+ GFP+, upper right quadrant). Gating strategy is shown in fig. S3 (A to D). (E and I) Individual wells were untreated (UT), treated with isotype (F and J), or anti-β8 and C6D4 (1 μg/ml) (G and K). (H) Lower column (n = 3) coating substrate indicated as BSA, αvβ3, or αvβ8. (L) Ctla4, Ikzf2, or Nrp1 expression determined by qPCR demonstrates αvβ8-dependent Treg differentiation under identical culture conditions as in (F, G, J, and K). Results (log10) normalized to αvβ3 controls. Treatment with isotype (open) or C6D4 (vertically striped) are indicated. (P) Activated T cells cocultured with mock or β8-LLC cells significantly increased Treg differentiation compared with coculture with mock-LLC controls. (Q) TGF-β activation over range of αvβ8 coating concentrations reported by WT TMLC (blue), L–TGF-β1–transfected TMLC (red), or L–TGF-β1/GARP–transfected TMLC cells (green). (R) L–TGF-β within conditioned media of Tconv cultured under stimulatory conditions for 48 hours identical to conditions in (N). Reporter cells plated on control substrate αvβ3 or αvβ8 with secreted L–TGF-β. Heat (80°C) activation of conditioned media showed total amount of L–TGF-β present. (S) Transwell assay determined the importance of cell contact in αvβ8-mediated Treg differentiation. αvβ8 was coated on the lower chambers. CD4+ T cells were plated only into the upper chambers under stimulating conditions (IL-2 and anti-CD3). The upper chamber Transwell surface contains 0.4-μm pores, allowing diffusion of soluble mediators from the lower chamber, but not cells. (T) Active rTGF-β added to the medium in the lower chamber demonstrates that TGF-β freely diffuses from the lower to the upper chamber to induce CD25+FOXP3+ Treg differentiation. *P < 0.05 and ***P < 0.001 by one-way ANOVA for multiple comparisons followed by Sidak’s post-test or unpaired Student’s t test to compare two populations.
Tumoral αvβ8 directly drove Treg differentiation in vitro
We next tested whether binding of L–TGF-β on the surface of non-Treg CD4+ T cells to αvβ8 expressed on an opposing cell surface was sufficient to drive induced Treg (iTreg) conversion in vitro because iTregs approximate many qualities of pTregs generated in vivo (8). In this simplified system, iTreg differentiation is mediated by binding to immobilized integrin αvβ8 independent of paracrine factors secreted by tumor cells, which could be influenced by αvβ8 ligand binding. The αvβ8 ectodomain is also free of cytoskeletal interactions, which modulate integrin conformational changes involved in force transduction and are important for TGF-β activation by the closely related integrin αvβ6 but not αvβ8 (23, 29, 42, 43). Culturing activated non-Treg CD4+ T cells on immobilized αvβ8 (Fig. 3, E to G), but not αvβ3 [which does not mediate activation of TGF-β (Fig. 3, I to K) (29)], resulted in conversion to iTregs as determined by increased FOXP3 expression and acquisition of suppressor function (fig. S3, I to L). Treg conversion induced by αvβ8 was efficient (~60%) because it was similar to induction by a supraphysiologic concentration of recombinant TGF-β (rTGF-β; Fig. 3, F and O). Effects of αvβ8 on iTreg differentiation were not due to ligation of L–TGF-β/GARP on activated T cells, or cell attachment, because no increase in iTreg differentiation was seen when activated T cells expressing L–TGF-β were plated on wells coated with anti-LAP (fig. S3, M and N). Anti-β8 efficiently inhibited effects of immobilized αvβ8 on iTreg differentiation (Fig. 3, G and H). αvβ8-mediated conversion to iTreg depended on TGF-β, as demonstrated by blockade (~50%) with high concentrations of a pan–TGF-β isoform antibody, 1D11 (fig. S3, V and W). The source of the active TGF-β mediating non-Treg CD4+ T cell conversion to Treg on immobilized αvβ8 was not from secreted L–TGF-β in the media or from stimulated non-Treg CD4+ T cells (Fig. 3, Q and R). Last, αvβ8-mediated conversion to iTreg required contact of non-Treg CD4+ T cells with αvβ8 (Fig. 3, S and T).
The immunosuppressive Treg phenotype induced by immobilized αvβ8 was confirmed by significant β8-dependent induction of the immune checkpoint inhibitor Ctla4 (Fig. 3L). Ikzf2 and nrp1 were barely or not detected, indicating that in vitro generated iTregs were similar to the pTreg phenotype seen in Tregs isolated from murine LLC tumors (Figs. 2 and 3L). In addition, low levels of Ikzf2 and nrp1 in αvβ8-generated iTregs suggested that their origin was from non-Treg CD4+ T cells, not from the small (~1%) population of CD25+CD4+FOXP3+ T cells mostly consisting of tTreg (36). To directly address whether the iTreg in our system originated from the non-Treg CD4+ T cell population, we sorted CD4+CD25+GFP− T cells, which were cultured on immobilized αvβ8 or αvβ3 as a control. Robust αvβ8-mediated conversion of CD4+CD25+GFP− to CD4+CD25+GFP+ T cells was observed, demonstrating that the origin of iTreg in our system was CD4+CD25+ GFP− T cells and not expansion of the small population of CD4+CD25+GFP+ T cells (fig. S3, Q to W). In contrast, sorted CD4+CD25+GFP+ T cells from CD4+ splenocytes displayed minimal expansion when cultured on immobilized integrins or in response to rTGF-β (fig. S3, R to T). These results indicated that iTregs in our system originated from non-Treg CD4+ T cell populations rather than from expansion of existing Tregs.
To reproduce these results with αvβ8-expressing tumor cells, we cocultured activated non-Treg CD4+−T cells with β8-LLC cells and measured iTreg conversion. Coculture with β8-LLC significantly increased iTreg generation relative to mock-LLC cells (Fig. 3P and fig. S3, O and P). About 15 to 20% of non-Treg CD4+ T cells were converted to iTregs in the presence of β8-LLC cells (Fig. 3P), similar to the efficiency of conversion to iTreg on wells coated with αvβ8 at the same receptor density as β8-LLC cells (Fig. 5E). These results suggest that αvβ8 binding to L–TGF-β–presenting T cells, without any integrin cytoskeletal-mediated force transduction, was sufficient to mediate TGF-β activation and conversion of non-Treg CD4+ T cells to iTreg, consistent with the integrin force-independent mechanism of TGF-β activation (29).
Fig. 5. Formation of a localized tumor/T cell αvβ8/L–TGF-β signaling complex.
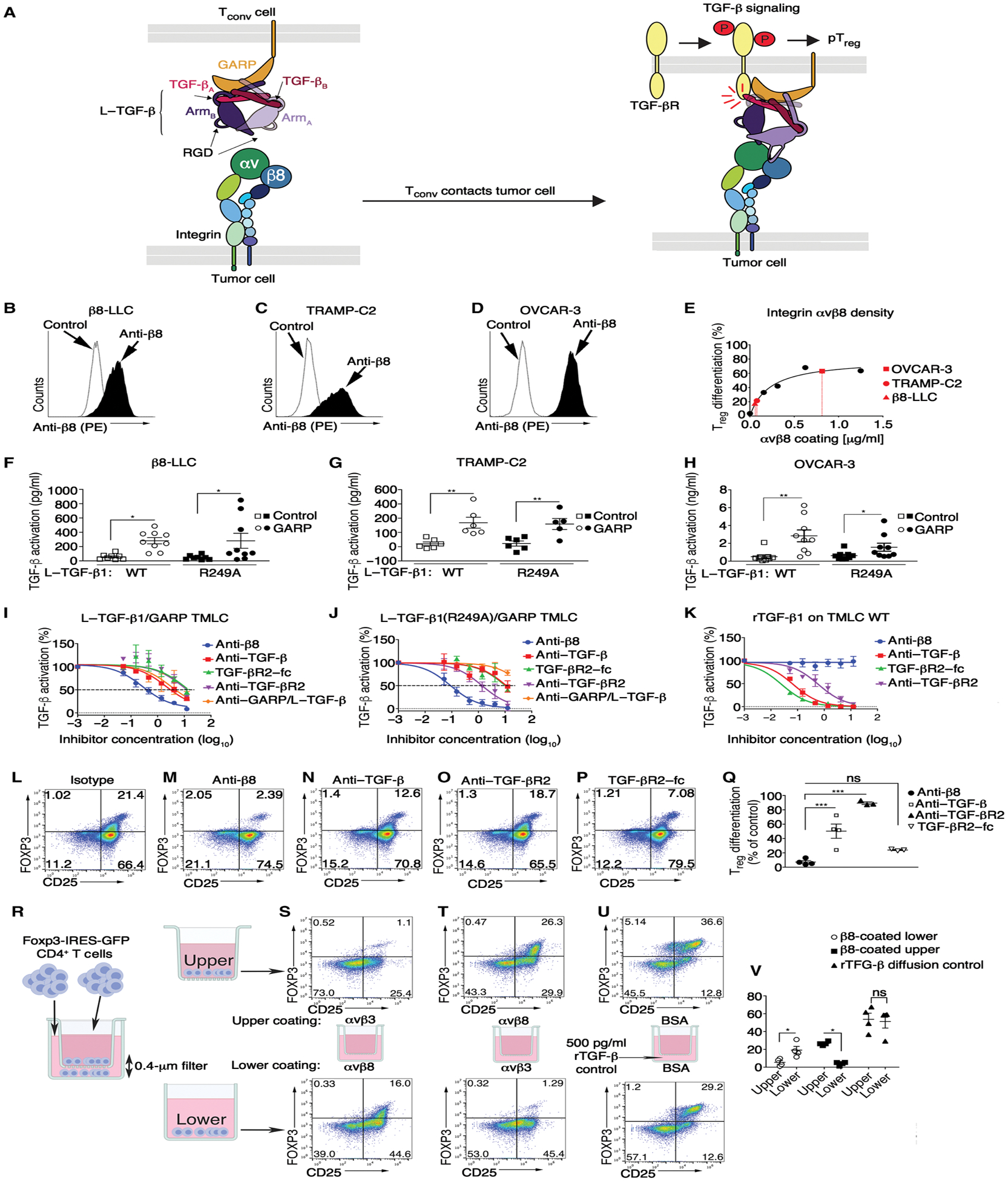
(A) Cartoon of structure-based model of αvβ8-mediated TGF-β activation and signaling based on structures of αvβ8/L–TGF-β (29, 43), L–TGF-β/GARP (51), and TGF-βR2/TGF-β1 (66). Integrin αv and β8 subunits, latency associated peptide (LAP) of dimeric L–TGF-β (subunit A + B), dimeric TGF-β (subunit A + B), TGF-βR2, and GARP color-coded matching annotations. Integrin and GARP/TGF-βR2 trans-membrane domains span tumor or Treg lipid bilayers, respectively. (B) αvβ8 surface expression in β8-LLC, (C) TRAMP-C2, and (D) OVCAR-3 stained with C6D4 (1 μg/ml) compared with iso-type. (E) Treg differentiation over a range of αvβ8 coating concentrations. Superimposed in red are β8-LLC (red triangle), TRAMP-C2 (red circle), and OVCAR-3 (red square) according to calculated αvβ8 cell surface receptor density. (F to H) WT human L–TGF-β1 or mutant incapable of producing diffusible TGF-β1 [L–TGF-β1(R249A)] expressed alone (square symbols) or coexpressed with human GARP (circles) in TGF-β reporter cells (TMLC) and cocultured with (F) β8-LLC, (G) TRAMP-C2, or (H) OVCAR-3. Outliers (Rout) were removed from (F and G). Shown is TGF-β activation (means ± SEM) determined using rTGF-β standard curve of each TMLC line (n ≥ 6). (I to K) Inhibition curves of anti-β8 (C6D4, blue line) compared with anti–pan–TGF-β (1D11, red line), TGF-βR2–Fc receptor trap (green line), anti-human GARP/L–TGF-β (MHG-8, purple line), or anti-human/mouse TGF-βR2 (clone 8322, orange line) generated using (I) WT human L–TGF-β1/human GARP TMLC, (J) human L–TGF-β1(R249A)/human GARP TMLC or control, and (K) WT TMLC cells with 500 pg of rTGF-β1. Shown is percent inhibition relative to no antibody control. Inhibitor concentrations are shown in μg/ml (log10). (L to P) iTreg differentiation of activated CD4+ T cells from foxp3-IRES-GFP splenocytes on immobilized αvβ8 in the presence of (L) isotype, (M) anti-β8 (C6D4), (N) anti–TGF-β1 (1D11), (O) anti–TGF-βR2 (clone 8322), (P) or TGF-βR2–Fc. (Q) Results enumerated in scatterplots (n ≥ 3). (R) Schematic of Transwell assay showing that diffusible TGF-β has no role in αvβ8-mediated iTreg differentiation. CD4+ T cells plated into upper and lower chambers under stimulating conditions. (S) αvβ8 coated on lower, αvβ3 control on upper, or (T) vice versa. (U) Active rTGF-β added to lower chamber media demonstrating diffusion of rTGF-β into the upper chamber inducing conversion of non-Treg CD4+ T cells (Tconv) to CD25+FOXP3+ Treg. (V) Scatterplots (n = 4) show gated CD4+ T cells stained with anti-CD25 (x axis) with FOXP3 expression determined by GFP (y axis). *P < 0.05, **P < 0.01, and ***P < 0.001 by one-way ANOVA for multiple comparisons followed by Sidak’s post-test.
αvβ8 expression by nontumor cells was not essential for conversion of T cells to Treg
We were unable to detect cell-surface αvβ8 on Tregs or conventional T cells (Tconvs) (fig. S4). These results mirror previous studies showing lack of αvβ8 expression on mouse CD4+ and CD8+ T cells, Tregs, macrophages, and dendritic cells (21, 29). However, it remains possible that levels of cell-surface αvβ8 on T cells or Tregs below the level of detection by cell surface staining are sufficient for Treg generation and function (44, 45). However, we could find no evidence of significant itgb8 gene or functional αvβ8 surface expression by T cells or Tregs in Treg generation, function, or immunosuppressive gene expression either in vitro or in vivo [Figs. 1 (C to E and G to I), 2 (C to E), and 3 (I to K and H) and fig. S2 (D and G)].
ITGB8 was most highly expressed by tumor cells
We next sought to translate our αvβ8 cell-type expression data to the human TME by measuring the relative expression of ITGB8 by various cell types in different human tumors. We performed bulk RNAseq of sorted immune (T cell, Treg, or myeloid) and nonimmune (stromal or tumor) cells from human lung, gynecologic, and head and neck carcinomas (Fig. 4 and fig. S5, A to E). ITGB8 was most highly and significantly expressed in tumor cells compared with immune cell populations in all three tumor types. ITGB8 was also significantly expressed by stromal cells, albeit generally at a lower level than tumor cells. Rare samples with signal above background were seen in immune cells, likely representing technical noise, because such data points were identified as statistical outliers (Fig. 4, B to D). Accordingly, no significant expression of ITGB8 was found in Tregs or other immune cell types known not to express ITGB8, such as CD4+ T cells and myeloid cells (Fig. 4, B to D) (21). Together, our mouse and human findings support our conclusion that tumor cells are a functionally important site for αvβ8 expression in the TME.
Fig. 4. ITGB8 was highly expressed in tumor cells, and TGFB1 was highly expressed in immune cells.
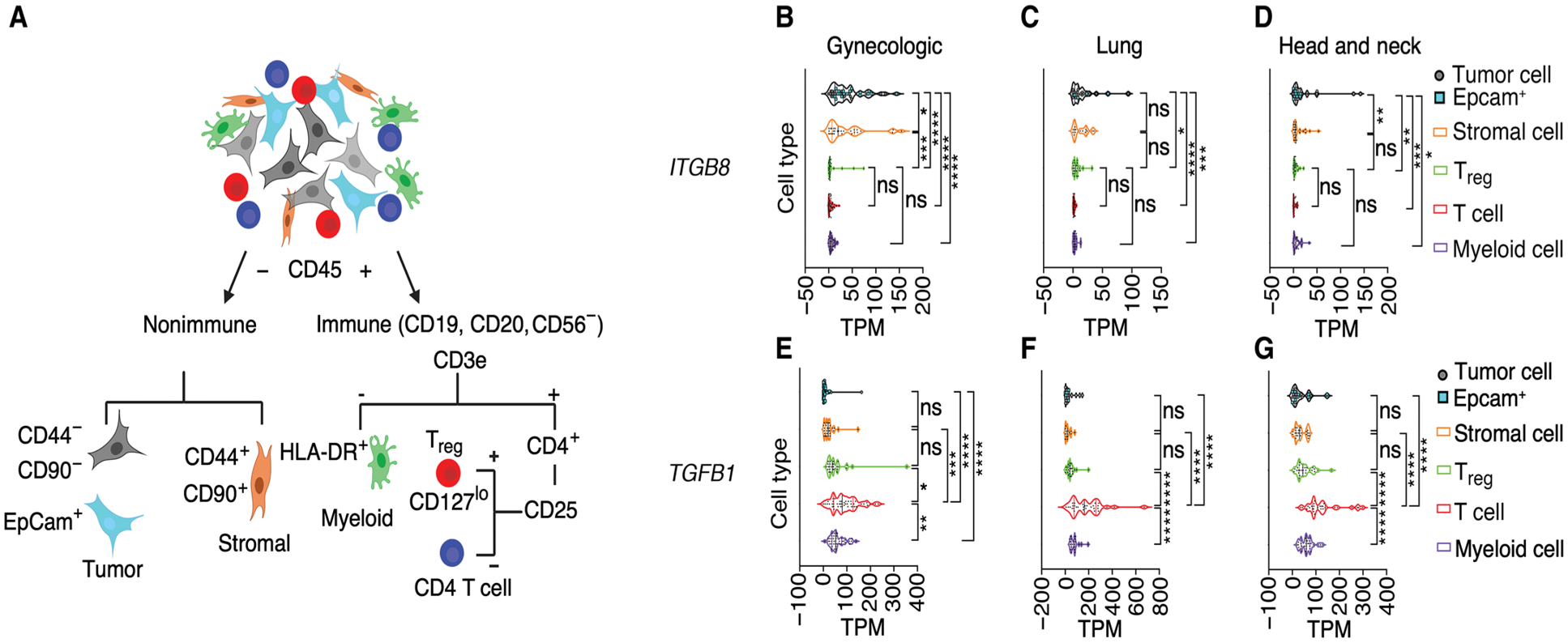
(A) Schematic of sorting strategy to purify tumor, stromal, myeloid, CD4+ T cell, and CD4+CD25+CD127lo Treg populations from disaggregated human tumors. (B to G). Bulk RNAseq performed on sorted cell populations from cohorts of human gynecologic (n = 53), non–small cell lung carcinoma (n = 41), or head and neck cancer specimens (n = 38) represented as transcript per million (TPM) (normalized read counts to gene length and scaling 1 × 106). Violin plots of normalized TPM of (B to D) ITGB8 and (E to G) TGFB1 of CD44−CD90− tumor cells (gray circles), some of which stain with anti-epithelial cell adhesion molecule (EpCam) (blue filled squares), CD44+CD90+ stromal cells (filled circles), CD4+CD25+ Treg (green) or CD4+ T cells (red), or major histocompatibility complex class II (MHCII+) (human leukocyte antigen DR isotype, HLA-DR+) myeloid cells (purple). All data points were included in the analysis without outlier exclusion and were analyzed for significance by one-way ANOVA for multiple comparisons followed by Dunnett’s post-test; means ± SD. ns, P > 0.05; *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
L–TGF-β/GARP was most highly expressed by immune cells
Abundant evidence suggests that TGF-β expression by T cells, not tumor cells, is important to maintain an immunosuppressive TME (46–48). T cell surface localization of TGF-β may also be important in the TME because conditional deletion of GARP in Treg inhibits growth of colitis-induced colon tumors (49). However, mechanisms of cell surface presentation of TGF-β in the TME have not been comprehensively studied. Thus, we next sought to determine which cells in the TME expressed TGF-β, GARP, or its functional homolog negative regulator of reactive oxygen species (NRROS or LRRC33) in the human TME.
TGFB1 was most highly and significantly expressed in CD4+ T cells, with levels decreasing in rank order of Tregs, myeloid cells, and stromal and tumor cells (Fig. 4, E to G). GARP and NRROS were both expressed above background in different cell types; Tregs and stromal cells significantly expressed GARP, whereas myeloid, Treg, and T cells significantly expressed NRROS (fig. S5). These data suggested multiple mechanisms for cell surface presentation of L–TGF-β; Tregs used both GARP and NRROS. Stromal cells used only GARP. Myeloid and T cells used NRROS, and tumor cells used neither (fig. S5). Our findings were consistent with previous identification of cell surface L–TGF-β on murine T cells, Treg, and myeloid cells but not tumor cells from orthotopic tumors (21). Together, our data supported that CD4+ T cells were a major source of cell surface L–TGF-β in the TME of human tumors.
Formation of a localized tumor/T cell αvβ8/L–TGF-β signaling complex
We next sought to test the physiological relevance of our structure-based model of TGF-β activation (Fig. 5A) (29) by asking whether αvβ8 expressed by tumor cell lines was sufficient to support TGF-β activation without release and diffusion of TGF-β from a cell-cell L–TGF-β/GARP complex. The respective αvβ8 cell surface receptor densities of β8-LLC (Fig. 5B), TRAMP-C2 (Fig. 5C), or human ovarian carcinoma (OVCAR-3; Fig. 5D) tumor cell lines were sufficient to efficiently support non-Treg CD4+ T cell–to–Treg conversion (Fig. 5E). Accordingly, these β8-expressing lines efficiently increased TGF-β reporter activity in L–TGF-β/GARP– or L–TGF-β (R249A)/GARP–expressing transformed mink lung cell (TMLC) reporter cell lines, with respective TGF-β activation efficiencies, correlating to αvβ8 surface receptor density (Fig. 5, E to H). These data demonstrated that αvβ8 expressed by tumor cells induced TGF-β signaling in L–TGF-β–presenting cells that they were in contact with.
Therapeutic implications of the tumor: T cell αvβ8/L–TGF-β complex
We developed a structural model of the αvβ8/L–TGF-β/GARP/TGF-βR2 signaling complex (29), which predicted numerous geometric constraints affecting binding of TGF-β protein inhibitors that have been developed to target freely diffusible mature TGF-β (Fig. 5A). Such inhibitors include antibodies to TGF-β, LAP, or GARP/L–TGF-β, as well as TGF-βR traps (29, 50–52). In contrast, the C6D4 antibody targets the αvβ8 ligand binding pocket and therefore would be predicted to efficiently prevent L–TGF-β binding to αvβ8 (29).
We used our TGF-β activation system to assess the relative ability of these inhibitors to block TGF-β activation. As predicted, anti-β8 (C6D4) efficiently inhibited TGF-β activation in wild-type (WT) L–TGF-β/GARP or L–TGF-β (R249A)/GARP reporter cells when plated on immobilized αvβ8, which, at low concentrations, was markedly more effective than antibody inhibitors to TGF-β, GARP, TGF-βR2, or TGF-βR2 receptor traps (Fig. 5, I and J). Decreased efficacy of antibody inhibitors to TGF-β, TGF-βR2, or TGF-βR2 receptor traps was due to the reduced ability to block mature TGF-β within the L–TGF-β complex because they were effective inhibitors of diffusible rTGF-β (Fig. 5K). These results were consistent with the inaccessibility of target epitopes for TGF-β, GARP, or TGF-βR2 within the αvβ8/L–TGF-β/GARP complex.
We next extended these findings to T cells themselves by using activated murine CD4+ T cells plated on immobilized αvβ8 versus control substrates. Treg generation on immobilized αvβ8 (Fig. 5L) was almost completely blocked by anti-β8 but was inhibited significantly less by other TGF-β protein inhibitors at the same concentration (Fig. 5, M to Q). The lack of efficacy of inhibitors preferentially targeting diffusible TGF-β supported our structure-based hypothesis that αvβ8-mediated T cell conversion to Treg was independent of diffusion of TGF-β. This hypothesis was further tested using a Transwell filter assay (Fig. 5R), which demonstrated that non-Treg CD4+ T cells binding to αvβ8 induced iTreg conversion only by non-Treg CD4+ T cells in direct contact and not those separated by the filter (Fig. 5, S to V). Together, our structural model of the αvβ8/L–TGF-β/GARP/TGF-βR2 signaling complex and comparative efficacy studies demonstrated a mechanism for Treg enrichment dependent on αvβ8-mediated TGF-β activation.
β8 expression in non–small cell lung cancer positively correlated with Treg density in the TME
Most of human cancers express αvβ8 in at least a fraction of tumor cells (21). We sought to test the hypothesis that a subpopulation of β8-expressing tumor cells was sufficient to drive local immunosuppressive Treg differentiation. We correlated CD4+FOXP3+ cell numbers with β8 tumor proportion scores (TPS), which estimates the percentage of tumor cells expressing β8, in a cohort of non–small cell lung cancers (NSCLCs) distinct from the cohort used for RNAseq (Figs. 4 and 6). We assessed cells with dual staining of CD4 and FOXP3 because most of these cells are Tregs (53). We found significant increases in CD4+FOXP3+ cell numbers with β8-TPS and CD4+FOXP3+ cell numbers and CD4+FOXP3+:CD4+FOXP3− cell ratios correlated significantly with β8-TPS (Fig. 6, A to F).
Fig. 6. Proportion of β8-expressing tumor cells correlated with CD4+ FOXP3+ T cell number in human and murine lung cancer.
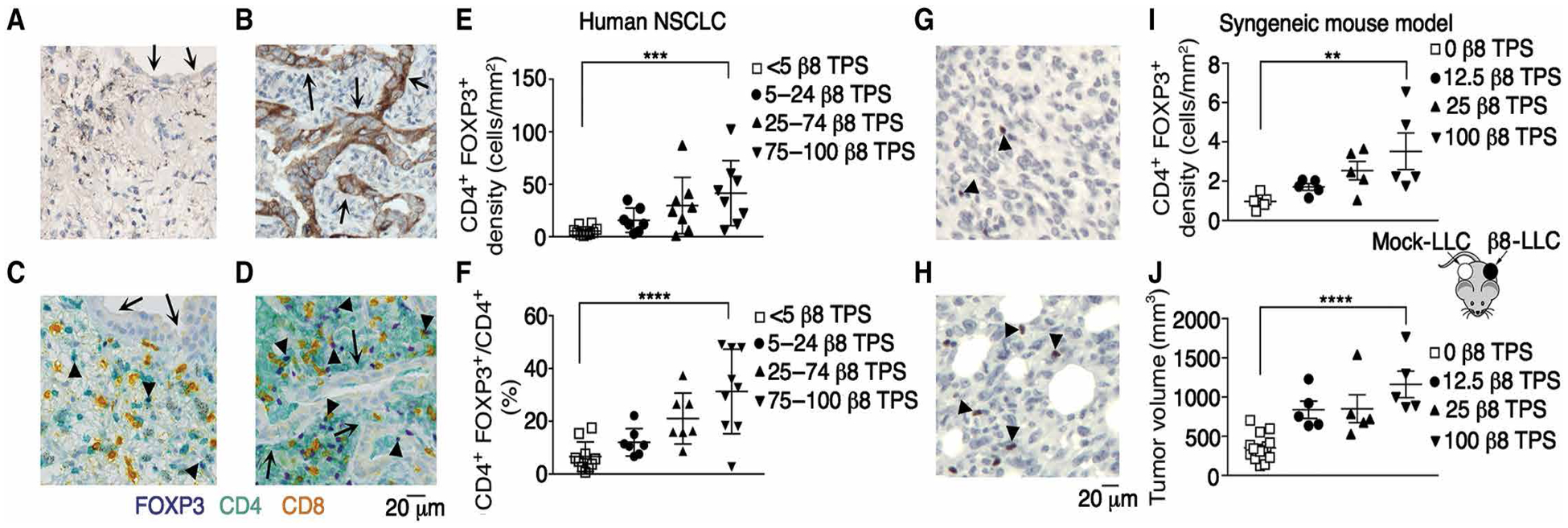
Representative images of immunohistochemical staining of human (n = 32) (A to D) or murine (n = 30) tumors (G and H). Immunohistochemical localization of integrin β8 in an independent cohort of human NSCLC with a β8 TPS of (A) <10% compared with (B) a high >50% TPS. Arrows in (B) indicate positively staining tumor cells. (C and D) Multiplex immunohistochemical staining of the same samples shown in (A and B) with anti-CD4 (teal), anti-CD8 (yellow), and anti-FOXP3 (purple). Arrows indicate tumor cells, and arrowheads indicate CD4+FOXP3+ cells. Scale bar, 20 μm. (E) CD4+FOXP3+ density according to TPS cutoffs <5%, 5 to 24%, 25 to 74%, and 75 to 100%. (F) Ratio of CD4+FOXP3+ to all CD4+ cells grouped according to the same cutoffs as in (E) (n = 36). (G and H) Immunohistochemical localization of FOXP3+ cells in (G) mock-LLC compared with (H) β8-LLC tumors. Arrowheads point to examples of stained nuclei. Scale bar, 20 μm. (I) Treg density depends on proportion of β8-expressing tumor cells. β8-LLC cells were mixed with mock-LLC cells in proportions of 1:0 (filled inverted triangles), 1:4 (filled upright triangles), and 1:8 (filled circles) and injected on the left flanks of mice. Mock-LLC (open squares) was injected on the right flank (see cartoon schematic). Shown are Treg (I) surface density (in mm2) and (J) tumor volume (in mm3) in mock-LLC tumors compared with tumors with various ratios of β8-LLC to mock-LLC. For multiple comparisons, one-way ANOVA and P test for trend were used. **P < 0.01, ***P < 0.001, and ****P < 0.0001.
To test the hypothesis that a limited proportion of β8-expressing tumor cells were sufficient to drive local Treg differentiation, β8-expressing LLC cells with mock-LLC cells were mixed in varying proportions. A significant trend for enrichment of FOXP3+ cells with increasing proportions of β8-expressing tumor cells was found (Fig. 6, G to J). Tumor growth correlated with FOXP3+ cell proportions, consistent with Tregs contributing to β8-mediated tumor immune evasion (Fig. 6J). These results suggested that expression of αvβ8 in a small fraction of tumor cells was sufficient to drive Treg enrichment in human tumors.
DISCUSSION
Here, we identified a mechanism of Treg enrichment in tumors, where αvβ8 expression on tumor cells caused Treg enrichment by increasing TGF-β activation in the TME. Our in vitro data suggested that the Treg enrichment in β8-expressing tumors occurred via contact of αvβ8-expressing tumor cells with L–TGF-β–presenting non-Treg CD4+ T cells, converting them to pTreg. We used cell-based assays to provide evidence that an intermolecular complex forms between αvβ8-expressing tumor cells and L–TGF-β–presenting non-Treg CD4+ T cell, where TGF-β was activated without release and diffusion of TGF-β and signaling was induced only on the L–TGF-β–presenting non-Treg CD4+ T cell.
Existing evidence supports two mechanisms of tumor Treg enrichment, either intratumoral non-Treg CD4+ T cell conversion to pTreg or recruitment of preexisting tTreg to tumors (Fig. 7, A and B) (22, 24). While exogenous TGF-β can lead to conversion of T cells to iTregs, no previous studies have addressed how TGF-β is activated in the TME or whether this activation leads to conversion of T cells to pTregs, as seen in the murine colon (54). However, in human tumors, the relative importance of conversion of non-Treg CD4+ T cells to pTreg is controversial because only a fraction of tumor Treg share common T cell receptor (TCR) clonotypes with non-Treg CD4+ T cells (55, 56). Our analysis of human tumors does not allow discrimination of tTreg from pTreg; thus, it is unclear whether tumors with high αvβ8 expression have increased overlap between the TCR repertoires of non-Treg CD4+ T cells and Tregs compared with those from tumors with low αvβ8 expression.
Fig. 7. Proposed mechanisms of Treg enrichment and differentiation in αvβ8-expressing tumors.
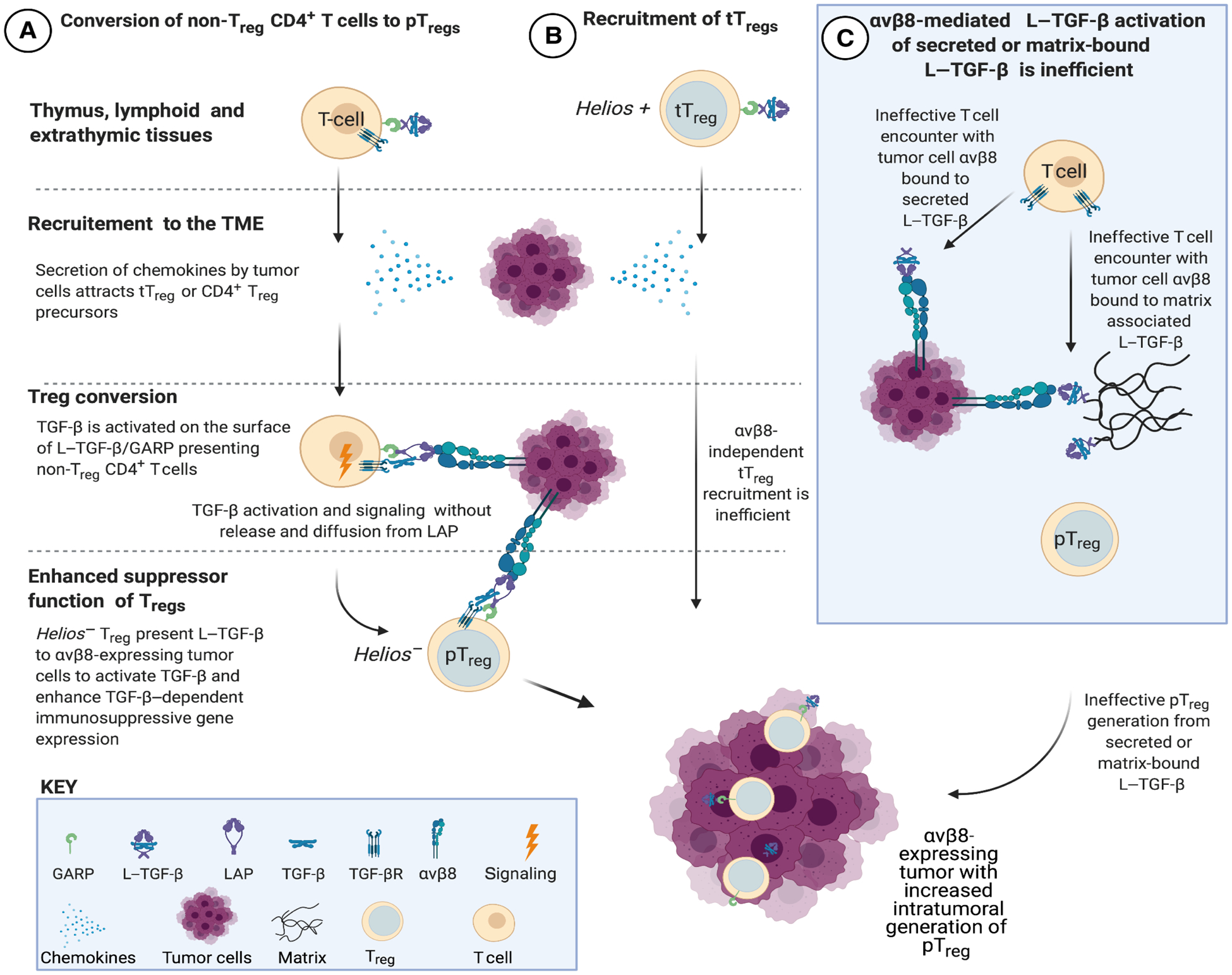
(A) Non-Treg CD4+ T cells expressing L–TGF-β/GARP infiltrate tumors in response to chemokines in the TME (21). TGF-β cannot interact with TGF-βR unless it undergoes activation. L–TGF-β/GARP–expressing non-Treg CD4+ T cells undergo Treg conversion to Helios− pTreg after binding to integrin αvβ8 expressed by tumor cells. The mechanism of TGF-β activation does not require the release and diffusion of TGF-β, ensuring that only T cells in contact with αvβ8-expressing tumor cells are converted to pTreg (29). (B) Thymically derived Helios+ Treg (tTreg) can potentially be recruited to the TME, but this is not evident in αvβ8-expressing tumors. (C) When L–TGF-β is soluble or matrix bound, TGF-βRs are not positioned on the same surface as L–TGF-β. Thus, if active TGF-β is not released from L–TGF-β when exposed to αvβ8 bearing tumor cells, then TGF-βR–expressing T cells need to find, orient, and overcome steric hindrance to bind to TGF-β exposed within the L–TGF-β complex. This activation process is less efficient than when L–TGF-β and TGF-βRs are on the same surface (29). Therefore, soluble or matrix-bound L–TGF-β is less likely to significantly contribute to αvβ8-mediated pTreg conversion in the TME. Created in BioRender.
Our study suggests that pTreg enriched in murine αvβ8-expressing tumors contributed to tumor immune evasion because both PC-61.5.3 and C6D4 reduced Tregs in β8-LLC. PC-61.5.3 depletes intratumoral CD4+CD25hi T cells when given after tumor cell injection but not CD4+CD25loFoxP3+ T cells (5, 30). Depleting Treg precursors (L–TGF-β+CD4+CD25hiFoxP3−) in β8-LLC tumors would be expected to have the same effect as blocking αvβ8 function on intratumoral conversion to pTregs. The mock-LLC Treg population likely consists of CD4+CD25loFoxP3+ T cells because PC-61.5.3 failed to deplete it. Reducing Treg by PC-61.5.3 or C6D4 decreased β8-LLC tumor growth, suggesting the importance of Treg-induced immunosuppression in αvβ8-mediated tumor growth promotion. Tregs inhibit expansion of effector CD8+ T cells (57). Our current and previous studies showed that C6D4 decreased the numbers of Tregs and increased the numbers of effector CD8+ T cells in β8-LLC tumors (21).
RNAseq data supported that tumor cells were the major αvβ8-expressing cell type, whereas T cells were the major TGF-β1–expressing cell in the human TME. Cell surface expression of L–TGF-β in non-Treg CD4+ T cells from murine tumors suggests that they are poised to differentiate to pTregs through activation of cell surface L–TGF-β (21). Our data supported a mechanism of Treg enrichment where αvβ8 expression by tumor cells caused L–TGF-β–presenting non-Treg CD4+ T cells to undergo local conversion to pTreg, rather than promoting recruitment of tTreg (Fig. 7, A and B). This conclusion was supported by in vitro studies demonstrating that immobilized or tumor cell αvβ8 drove the conversion of activated T cells to immunosuppressive Treg with low levels of tTreg markers, Helios and Nrp1, similar to in vivo studies, demonstrating that tumor cell αvβ8 led to the enrichment of pTregs.
Our inability to detect a functional role for αvβ8 expressed by Tregs contrasts with other reports proposing a cell-autonomous role for αvβ8 in Treg function using Tregs from nontumor sources (44, 45, 58). In these reports, surface expression of αvβ8 is not assessed; rather, the β8 subunit (itgb8) was detected by quantitative polymerase chain reaction (qPCR) in murine tTreg and effector Treg populations, which is at very low levels relative to housekeeping genes (44, 45). Such low mRNA expression is consistent with our inability to detect cell-surface αvβ8 because surface expression of αvβ8 is regulated at the level of itgb8 transcription (59). Thus, although it is possible that, in other tumor systems, αvβ8 can be expressed at functionally significant levels in Tregs, our current and past studies, which include surface expression surveys of both tumor and splenic Treg, transcriptomic analysis, and in vitro and in vivo functional assays failed to produce evidence that expression of αvβ8 by T themselves played a role in non-Treg CD4+ Treg cell–to–Treg conversion.
We propose a model (Fig. 7C) to explain why T cells require presentation of L–TGF-β on their cell surface for Treg conversion. In this model, the αvβ8 receptor and its ligand, L–TGF-β/GARP, are concentrated on opposing cell surfaces, not on the same cell surface, and are specifically directed only to the L–TGF-β/GARP–presenting T cell after binding TGF-β signaling (29). This process is more efficient and context-specific than a TGF-βR–bearing T cell encountering active TGF-β diffusing through the extracellular space. Furthermore, this model predicts that if tumor cell αvβ8 binds to either secreted or matrix-bound L–TGF-β, then a TGF-βR–expressing T cell would have to find and orient its receptors to mature TGF-β exposed within the latent complex (Fig. 7C) (29).
Current dogma suggests that actin cytoskeletal force generation through the β-integrin cytoplasmic domain is required to induce conformational changes through the integrin for force generation to L–TGF-β to release TGF-β (42). However, our structural studies show that αvβ8 does not undergo major conformational rearrangements, always remaining in a single extended-closed conformation poised for ligand binding but not force transduction (43, 60). Hence, αvβ8 on cell surfaces is always available for binding to an L–TGF-β–presenting cell, and this binding creates an anchor point to focus the inherent flexibility of L–TGF-β to allow active TGF-β to be sufficiently exposed to bind to its receptors but only on the cell-presenting surface L–TGF-β/GARP (29). Here, we tested this model in vitro using isolated T cells and showed that T cell TGF-β activation did not require cytoskeletal force transduction from the αvβ8 integrin because the αvβ8 ectodomain immobilized on a solid substrate was freely capable of inducing conversion of contacting non-Treg CD4+ T cells to Tregs. This conversion occurred without release and diffusion of TGF-β. The identification of a TGF-β activation mechanism that is diffusion-independent has biologic implications because any L–TGF-β–presenting cell type could potentially increase its TGF-β signaling when contacting an αvβ8-expressing cell (21, 60). Our model also has therapeutic implications because protein-based therapies directed at TGF-β currently in clinical trials are conceptually designed to inhibit diffusible TGF-β (61). Our data indicated that such therapies would poorly target αvβ8-mediated TGF-β activation, while still exposing patients to considerable risk.
Overall, this study highlights a mechanism of Treg enrichment regulated by TGF-β activation in specific tumors that express sufficient levels of αvβ8. We propose that targeting αvβ8 to prevent TGF-β activation will provide a highly effective and more selective approach to overcome Treg-mediated tumor immune evasion in patients with αvβ8+ tumors.
MATERIALS AND METHODS
Study design
This study tested the hypothesis that Tregs were required for integrin αvβ8–mediated tumor growth promotion using cell-based assays, in vivo tumor models, and correlative human studies. In vivo models used randomization and blinding maintained until end points were reached and data analysis was completed. Sample sizes for in vitro and in vivo experiments were estimated using power calculations with predetermined effect sizes and variances based on experience.
Mice and orthotopic tumor models
Syngeneic bilateral tumor models were performed in C57BL/6 mice expressing foxp3-IRES-GFP [B6.Cg-FOXP3tm2(EGFP)Tch/J, Jax] (41) and WT C57BL/6 mice (all female except for TRAMP-C2 experiments using male), 8 to 10 weeks of age, were purchased (the Jackson laboratory), as described (21). See Supplementary Materials and Methods for more information.
Human subjects
Patients were consented for tissue collection under University of California San Francisco Institutional Review Board (UCSF IRB)–approved protocols (UCSF CHR 10–04727, 14–15342, 11–06107, and 10–03413). The study enrollment period started from January 2015 to present, and the sample size was determined by the availability of specimens throughout this period. Samples were selected without regard to prior treatment. Two separate cohorts were developed: the first for cell sorting and RNAseq including samples from gynecologic (n = 53), lung (n = 41), and head and neck squamous cell carcinoma (n = 38) and the second for immunohistochemistry from patients undergoing resection for NSCLC (n = 32).
Study approval
Human tissues were obtained with full approval of the UCSF IRB in full accordance with Declaration of Helsinki principles. Written informed consent was received from participants before inclusion in the study. All animal studies have been approved by the UCSF Institutional Animal Care and Use Committee.
Cell lines
LL/2 (LLC1) [American Type Culture Collection (ATCC), CRL-1642] and TRAMP-C2 (gift from L. Fong, UCSF, San Francisco, CA, USA) were used in TGF-β reporter assays and syngeneic tumor models. OVCAR-3 (UCSF Cell and Genome Engineering Core) and transformed mink lung TGF-β reporter cells (TMLC) (62) were a gift from J. Munger (New York University Medical Center, New York, NY, USA) and were stably transfected with L–TGF-β WT or L–TGF-β (R249A), with or without GARP, as previously described (29). LLC cells were stably transfected to overexpress vector only (mock-LLC) or β8 (β8-LLC) as previously described (21). CHOlec3.2.8.1 (gift from P. Stanley, Albert Einstein College of Medicine, New York, NY, USA), CHO-K1 (ATCC, CCL-61), and human embryonic kidney (HEK) 293 cells (ATCC, CRL-1573) were used for recombinant integrin expression and immunoglobulin G2a (IgG2a) production. All cell lines were maintained in the appropriate media with selection agents and antibiotics, as previously described (21, 29, 43).
Reagents
Anti-CD25 (clone PC-61.5.3) and rat isotype control (HRPN, BP0088) were obtained from Bio X Cell. Anti-β8 (C6D4) is a highly specific engineered recombinant antibody to the specificity determining loop 2 (SDL2) domain of αvβ8 consisting of humanized V genes and CH1 domains, with murine linker and CH2/3 domains in an IgG2a format and is produced in CHO-K1 cells (21, 29). The αvβ8 and αvβ3 ecto-domains were expressed and purified as previously described (43, 60). For all other reagents, see Supplementary Materials and Methods.
DNA constructs
The following PCR products were produced using the following primers and templates: 5′-GATTGTGGGCCCTCTGGGCTCGTCC-GGATTGCTGGTGTTATATTCTTCTGAG-3′ and 5′-CT-GTGGACGCGTATCGCC-3′, human TGFBR2 expression vector (Sino Biological, HG10358-ACG) and CTCAGAAGAATATAACACCAGCAATCCGGACGAGCCCAGAGGGCCCACAATC and AACGGATCCTCATTTACCCGGAG, mouse IgG2a expression vector. The products were joined by splice overlap extension PCR and cloned into AbVec 2.0 (Addgene, plasmid no. 80795), as described (60). The resulting plasmid was transiently transfected into HEK-293 cells and protein purified as described (21).
Lentiviral transduction
For itgb8 knockdown, TRAMP-C2 cells were stably transfected with itgb8-specific shRNA or nonmammalian control shRNA via lentiviral transduction [Sigma-Aldrich MISSION lentiviral transduction particles TRCN0000067303 (itgb8) or SHC002V (nonmammalian control)].
Isolation, staining, and RNAseq of mouse tumor and immune cells
Mouse tumor immune cell isolation, RNA isolation, and sequencing were performed as described (21). Briefly, mouse tumors were digested; live tumor cells were negatively selected by magnetic beads, or infiltrating lymphoid cells were enriched by density gradient centrifugation. FOXP3+ cells were then sorted from the enriched lymphoid cells by staining with fluorochrome-labeled antibodies as described (21). Total RNA was isolated using kits as described (21), after which cDNA synthesis and amplification were performed. For specific details including library construction and analysis, see Supplementary Materials and Methods.
Isolation, staining, and RNAseq of human tumor and immune cells
Fresh patient tumor samples were dissociated, and the resulting cell suspension was enumerated and stained with the LIVE/DEAD stain and an extracellular antibody cocktail before cell sorting. RNA isolation, cDNA synthesis, library construction, sequencing, and analysis are described in Supplementary Materials and Methods.
Treg maturation assays
Briefly, CD4+ T cells from foxp3-IRES-GFP mice were purified and plated onto tissue culture plates coated with αvβ8tr or control substrate [αvβ3tr or bovine serum albumin (BSA)] under T cell stimulation conditions. CD4+ T cells were incubated for 72 hours before phenotyping via flow cytometric analysis. For Transwell diffusion assays, cells were plated into each chamber of the Transwell culture wells containing a 0.4-μm filter using the same conditions as above with further details provided in Supplementary Materials and Methods.
Lymphocyte suppression assays
Cells were cultured as above to create Treg pools and labeled with carboxyfluorescein diacetate succinimidyl ester fluorescent tracking dye exactly as described (63). Labeled cells were stimulated using anti–CD3/CD28 Dynabeads and plated in round-bottom 96-well culture plates. Labeled CD4+ cells were then cocultured with αvβ8-generated Tregs or control cells at ratios ranging between 1:1 and 8:1 (labeled CD4+ T cells:Tregs). Tconv proliferation was measured using flow cytometry after 4 days. For specific details, see Supplementary Materials and Methods.
TGF-β bioassays
TGF-β bioassays were performed as previously described (29) using the cell-intrinsic TGF-β activation reporter system described in (64). Reporter cells were seeded onto culture wells coated with αvβ8tr or control substrate (αvβ3tr or BSA). Cells were incubated for 18 hours before cell lysis and assessment of luciferase activity. For some experiments mock-LLC, β8-LLC, TRAMP-C2, or OVCAR-3 (3 × 104) cells were used in place of immobilized substrates.
Determination of receptor density
CHO WT or CHO transfected with human αvβ8 were plated onto culture plates over a concentration range of 5 × 103 to 30 × 103 cells per well, and recombinant αvβ8 ectodomain [phosphate-buffered saline (10 to 5000 ng/ml)] were coated onto separate wells on the same plate. After cell attachment, cells and coated receptor wells were fixed with 4% paraformaldehyde. Cell-associated or recombinant αvβ8 was detected with clone F9, followed by anti-mouse horseradish peroxidase and colorimetric detection, and cell surface receptor density was determined on the basis of standard curve of estimated recombinant αvβ8 receptor coating efficiency, as described in Supplementary Materials and Methods.
Immunohistochemical analysis of murine and human tumors
Immunohistochemistry was performed as previously described (21). Briefly, prepared formalin-fixed paraffin-embedded (FFPE) sections were stained with anti-mouse β8 (clone F9), B5 (anti-human β8, which does not work in FFPE immunostaining and thus used as isotype control for F9), anti-CD4, anti-CD8, or anti-FOXP3, followed by appropriate detection reagents. For multiplex immunostaining, the Ventana Discovery platform was used as described in Supplementary Materials and Methods.
Quantification and statistical analysis
All data are reported as means ± SEM unless otherwise specified. Comparisons between two different groups were determined using two-tailed Student’s t test. One-way analysis of variance (ANOVA) was used for multiple comparisons, and Tukey’s, Dunnett’s, or Sidak’s post hoc tests were used to test for statistical significance. Outliers were included unless stated otherwise. Significance was defined as P < 0.05. All statistical analyses, including outlier identification (Rout), were performed using the software package Prism 7.0b (GraphPad Software).
Supplementary Material
Acknowledgments:
We thank L. Fong (UCSF, San Francisco, CA, USA) and A. Craig (Queens University, Kingston, ON, Canada) for TRAMP-C2 cells and anti-human and anti-mouse TGF-βR2, respectively.
Funding:
Support was through the NIH (U54HL119893 and R01HL113032 to S.L.N., R01HL134183 to S.L.N. and Y.C., S10OD020054 to Y.C., and P41CA196276 to J.M.) and UC-CAI UCSF Catlyst2 (AB2664) to S.L.N., R01DK093646 (J.L.B.), UCSF Liver Center-P30DK026743 (J.L.B., J.P., S.L.N., and J.M.J.), the Ibrahim El-Hefni Technical Training Foundation (J.L.B.), NIH 1K01DK099405 (J.P.), NIH T32 AI 007334-27 and NIH F31 DK112607 (J.M.J.), and the Howard Hughes Medical Institute (Y.C.).
Competing interests:
S.L.N. and J.M. serve on the Scientific Advisory Board of Venn Therapeutics. S.L.N. is a consultant for Fleet Therapeutics. J.L. is a recipient of sponsored research funds from Venn Therapeutics. The antibodies C6D4 and F9 are included in U.S. Patent application no. 16/331,902 (S.L.N., Y.C., J.M., J.L., J.L.B., and N.T.).
Footnotes
SUPPLEMENTARY MATERIALS
immunology.sciencemag.org/cgi/content/full/6/57/eabf0558/DC1
Materials and Methods
Fig. S1. β8 expression by tumor cells drives tumor growth, which is blocked by C6D4 Fab in vivo.
Fig. S2. Tumor Treg isolation and expression of select Treg genes by RNAseq in this report compared with single-cell RNAseq from splenic Treg.
Fig. S3. Non-Treg CD4+ T cells express cell surface L–TGF-β, are converted to Treg by contact with β8-LLC cells, and are the source of suppressive iTreg generated on αvβ8.
Fig. S4. Murine nTregs do not express detectable levels of cell-surface αvβ8.
Fig. S5. LRRC32 (GARP) is most highly expressed by Treg and stromal cells and NRROS by myeloid cells.
Table S1. Raw data table (Excel spreadsheet).
Data and materials availability:
Antibodies and cell lines used in this manuscript will be available upon execution of a material transfer agreement. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. RNAseq datasets are deposited in the Gene Expression Omnibus (GSE158031).
REFERENCES AND NOTES
- 1.Fridman WH, Pagès F, Sautès-Fridman C, Galon J, The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 12, 298–306 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W, Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med 10, 942–949 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Curiel TJ, Tregs and rethinking cancer immunotherapy. J. Clin. Invest 117, 1167–1174 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shimizu J, Yamazaki S, Sakaguchi S, Induction of tumor immunity by removing CD25+CD4+ T cells: A common basis between tumor immunity and autoimmunity. J. Immunol 163, 5211–5218 (1999). [PubMed] [Google Scholar]
- 5.Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E, Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor α) monoclonal antibody. Cancer Res. 59, 3128–3133 (1999). [PubMed] [Google Scholar]
- 6.Tanaka A, Sakaguchi S, Targeting Treg cells in cancer immunotherapy. Eur. J. Immunol 49, 1140–1146 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Chen DS, Mellman I, Oncology meets immunology: The cancer-immunity cycle. Immunity 39, 1–10 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Yadav M, Stephan S, Bluestone JA, Peripherally induced Tregs—Role in immune homeostasis and autoimmunity. Front. Immunol 4, 232 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Valzasina B, Piconese S, Guiducci C, Colombo MP, Tumor-induced expansion of regulatory T cells by conversion of CD4+CD25- lymphocytes is thymus and proliferation independent. Cancer Res. 66, 4488–4495 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Paluskievicz CM, Cao X, Abdi R, Zheng P, Liu Y, Bromberg JS, T regulatory cells and priming the suppressive tumor microenvironment. Front. Immunol 10, 2453 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghiringhelli F, Puig PE, Roux S, Parcellier A, Schmitt E, Solary E, Kroemer G, Martin F, Chauffert B, Zitvogel L, Tumor cells convert immature myeloid dendritic cells into TGF-β–secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J. Exp. Med 202, 919–929 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klein L, Robey EA, Hsieh CS, Central CD4+ T cell tolerance: Deletion versus regulatory T cell differentiation. Nat. Rev. Immunol 19, 7–18 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Anderton MJ, Mellor HR, Bell A, Sadler C, Pass M, Powell S, Steele SJ, Roberts RRA, Heier A, Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol. Pathol 39, 916–924 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Vitsky A, Waire J, Pawliuk R, Bond A, Matthews D, LaCasse E, Hawes ML, Nelson C, Richards S, Piepenhagen PA, Garman RD, Andrews L, Thurberg BL, Lonning S, Ledbetter S, Ruzek MC, Homeostatic role of transforming growth factor-beta in the oral cavity and esophagus of mice and its expression by mast cells in these tissues. Am. J. Pathol 174, 2137–2149 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tolcher AW, Berlin JD, Cosaert J, Kauh J, Chan E, Piha-Paul SA, Amaya A, Tang S, Driscoll K, Kimbung R, Kambhampati SRP, Gueorguieva I, Hong DS, A phase 1 study of anti-TGFβ receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother. Pharmacol 79, 673–680 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elliott RL, Blobe GC, Role of transforming growth factor Beta in human cancer. J. Clin. Oncol 23, 2078–2093 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Massagué J, TGFβ in cancer. Cell 134, 215–230 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishimura SL, Integrin-mediated transforming growth factor-β activation, a potential therapeutic target in fibrogenic disorders. Am. J. Pathol 175, 1362–1370 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edwards JP, Fujii H, Zhou AX, Creemers J, Unutmaz D, Shevach EM, Regulation of the expression of GARP/latent TGF-β1 complexes on mouse T cells and their role in regulatory T cell and Th17 differentiation. J. Immunol 190, 5506–5515 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Batlle E, Massagué J, Transforming growth factor-β signaling in immunity and cancer. Immunity 50, 924–940 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takasaka N, Seed RI, Cormier A, Bondesson AJ, Lou J, Elattma A, Ito S, Yanagisawa H, Hashimoto M, Ma R, Levine MD, Publicover J, Potts R, Jespersen JM, Campbell MG, Conrad F, Marks JD, Cheng Y, Baron JL, Nishimura SL, Integrin αvβ8–expressing tumor cells evade host immunity by regulating TGF-β activation in immune cells. JCI Insight 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stockis J, Roychoudhuri R, Halim TYF, Regulation of regulatory T cells in cancer. Immunology 157, 219–231 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL, The integrin αvβ8 mediates epithelial homeostasis through MT1-MMP–dependent activation of TGF-β1. J. Cell Biol 157, 493–507 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tanaka A, Sakaguchi S, Regulatory T cells in cancer immunotherapy. Cell Res. 27, 109–118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aluwihare P, Mu Z, Zhao Z, Yu D, Weinreb PH, Horan GS, Violette SM, Munger JS, Mice that lack activity of αvβ6- and αvβ8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J. Cell Sci 122, 227–232 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Z, Mu Z, Dabovic B, Jurukovski V, Yu D, Sung J, Xiong X, Munger JS, Absence of integrin-mediated TGFβ1 activation in vivo recapitulates the phenotype of TGFβ1-null mice. J. Cell Biol 176, 787–793 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ozawa A, Sato Y, Imabayashi T, Uemura T, Takagi J, Sekiguchi K, Molecular basis of the ligand binding specificity of αvβ8 integrin. J. Biol. Chem 291, 11551–11565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kitamura H, Cambier S, Somanath S, Barker T, Minagawa S, Markovics J, Goodsell A, Publicover J, Reichardt L, Jablons D, Wolters P, Hill A, Marks JD, Lou J, Pittet JF, Gauldie J, Baron JL, Nishimura SL, Mouse and human lung fibroblasts regulate dendritic cell trafficking, airway inflammation, and fibrosis through integrin αvβ8–mediated activation of TGF-β. J. Clin. Invest 121, 2863–2875 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Campbell MG, Cormier A, Ito S, Seed RI, Bondesson AJ, Lou J, Marks JD, Baron JL, Cheng Y, Nishimura SL, Cryo-EM reveals integrin-mediated TGF-β activation without release from latent TGF-β. Cell 180, 490–501.e16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arce Vargas F, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, Miranda Rota E, Dahan R, Georgiou A, Sledzinska A, Ben Aissa A, Franz D, Werner Sunderland M, Wong YNS, Henry JY, O’Brien T, Nicol D, Challacombe B, Beers SA, Turajlic S, Gore M, Larkin J, Swanton C, Chester KA, Pule M, Ravetch JV, Marafioti T, Peggs KS, Quezada SA, Spain L, Wotherspoon A, Francis N, Smith M, Strauss D, Hayes A, Soultati A, Stares M, Spain L, Lynch J, Fotiadis N, Fernando A, Hazell S, Chandra A, Pickering L, Rudman S, Chowdhury S, Swanton C, Jamal-Hanjani M, Veeriah S, Shafi S, Czyzewska-Khan J, Johnson D, Laycock J, Bosshard-Carter L, Goh G, Rosenthal R, Gorman P, Murugaesu N, Hynds RE, Wilson G, Birkbak NJ, Watkins TBK, McGranahan N, Horswell S, Mitter R, Escudero M, Stewart A, van Loo P, Rowan A, Xu H, Turajlic S, Hiley C, Abbosh C, Goldman J, Stone RK, Denner T, Matthews N, Elgar G, Ward S, Biggs J, Costa M, Begum S, Phillimore B, Chambers T, Nye E, Graca S, al Bakir M, Hartley JA, Lowe HL, Herrero J, Lawrence D, Hayward M, Panagiotopoulos N, Kolvekar S, Falzon M, Borg E, Simeon C, Hector G, Smith A, Aranda M, Novelli M, Oukrif D, Janes SM, Thakrar R, Forster M, Ahmad T, Lee SM, Papadatos-Pastos D, Carnell D, Mendes R, George J, Navani N, Ahmed A, Taylor M, Choudhary J, Summers Y, Califano R, Taylor P, Shah R, Krysiak P, Rammohan K, Fontaine E, Booton R, Evison M, Crosbie P, Moss S, Idries F, Joseph L, Bishop P, Chaturved A, Quinn AM, Doran H, Leek A, Harrison P, Moore K, Waddington R, Novasio J, Blackhall F, Rogan J, Smith E, Dive C, Tugwood J, Brady G, Rothwell DG, Chemi F, Pierce J, Gulati S, Naidu B, Langman G, Trotter S, Bellamy M, Bancroft H, Kerr A, Kadiri S, Webb J, Middleton G, Djearaman M, Fennell D, Shaw JA, le Quesne J, Moore D, Nakas A, Rathinam S, Monteiro W, Marshall H, Nelson L, Bennett J, Riley J, Primrose L, Martinson L, Anand G, Khan S, Amadi A, Nicolson M, Kerr K, Palmer S, Remmen H, Miller J, Buchan K, Chetty M, Gomersall L, Lester J, Edwards A, Morgan F, Adams H, Davies H, Kornaszewska M, Attanoos R, Lock S, Verjee A, MacKenzie M, Wilcox M, Bell H, Iles N, Hackshaw A, Ngai Y, Smith S, Gower N, Ottensmeier C, Chee S, Johnson B, Alzetani A, Shaw E, Lim E, de Sousa P, Barbosa MT, Bowman A, Jorda S, Rice A, Raubenheimer H, Proli C, Cufari ME, Ronquillo JC, Kwayie A, Bhayani H, Hamilton M, Bakar Y, Mensah N, Ambrose L, Devaraj A, Buderi S, Finch J, Azcarate L, Chavan H, Green S, Mashinga H, Nicholson AG, Lau K, Sheaff M, Schmid P, Conibear J, Ezhil V, Ismail B, Irvin-sellers M, Prakash V, Russell P, Light T, Horey T, Danson S, Bury J, Edwards J, Hill J, Matthews S, Kitsanta Y, Suvarna K, Fisher P, Keerio AD, Shackcloth M, Gosney J, Postmus P, Feeney S, Asante-Siaw J, Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory T cells and synergizes with PD-1 blockade to eradicate established tumors. Immunity 46, 577–586 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pincetic A, Bournazos S, DiLillo DJ, Maamary J, Wang TT, Dahan R, Fiebiger BM, Ravetch JV, Type I and type II Fc receptors regulate innate and adaptive immunity. Nat. Immunol 15, 707–716 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thornton AM, Lu J, Korty PE, Kim YC, Martens C, Sun PD, Shevach EM, Helios+ and Helios− Treg subpopulations are phenotypically and functionally distinct and express dissimilar TCR repertoires. Eur. J. Immunol 49, 398–412 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Landuyt AE, Klocke BJ, Colvin TB, Schoeb TR, Maynard CL, Cutting edge: ICOS-deficient regulatory T cells display normal induction of Il10 but readily downregulate expression of Foxp3. J. Immunol 202, 1039–1044 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ondondo B, Jones E, Godkin A, Gallimore A, Home sweet home: The tumor microenvironment as a haven for regulatory T cells. Front. Immunol 4, 197 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bruder D, Probst-Kepper M, Westendorf AM, Geffers R, Beissert S, Loser K, von Boehmer H, Buer J, Hansen W, Neuropilin-1: A surface marker of regulatory T cells. Eur. J. Immunol 34, 623–630 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, Shevach EM, Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J. Immunol 184, 3433–3441 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, Xiong H, Dolpady J, Frey AB, Ruocco MG, Yang Y, Floess S, Huehn J, Oh S, Li MO, Niec RE, Rudensky AY, Dustin ML, Littman DR, Lafaille JJ, Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J. Exp. Med 209, 1723–1742 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, Anthony BA, Sverdrup FM, Head R, Kuster DJ, Ruminski P, Weiss D, Schack DV, Bluestone JA, Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J. Exp. Med 209, 1713–1722 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oida T, Weiner HL, TGF-β induces surface LAP expression on murine CD4 T cells independent of Foxp3 induction. PLOS ONE 5, e15523 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakamura K, Kitani A, Strober W, Cell contact–dependent immunosuppression by CD4+CD25+ regulatory T cells is mediated by cell surface–bound transforming growth factor β. J. Exp. Med 194, 629–644 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haribhai D, Lin W, Relland LM, Truong N, Williams CB, Chatila TA, Regulatory T cells dynamically control the primary immune response to foreign antigen. J. Immunol 178, 2961–2972 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Dong X, Zhao B, Iacob RE, Zhu J, Koksal AC, Lu C, Engen JR, Springer TA, Force interacts with macromolecular structure in activation of TGF-β. Nature 542, 55–59 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cormier A, Campbell MG, Ito S, Wu S, Lou J, Marks J, Baron JL, Nishimura SL, Cheng Y, Cryo-EM structure of the αvβ8 integrin reveals a mechanism for stabilizing integrin extension. Nat. Struct. Mol. Biol 25, 698–704 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edwards JP, Thornton AM, Shevach EM, Release of active TGF-β1 from the latent TGF-β1/GARP complex on T regulatory cells is mediated by integrin β8. J. Immunol 193, 2843–2849 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Worthington JJ, Kelly A, Smedley C, Bauché D, Campbell S, Marie JC, Travis MA, Integrin αvβ8-mediated TGF-β activation by effector regulatory T cells is essential for suppression of T-cell-mediated inflammation. Immunity 42, 903–915 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Azhar M, Yin M, Bommireddy R, Duffy JJ, Yang J, Pawlowski SA, Boivin GP, Engle SJ, Sanford LP, Grisham C, Singh RR, Babcock GF, Doetschman T, Generation of mice with a conditional allele for transforming growth factor beta 1 gene. Genesis 47, 423–431 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas DA, Massagué J, TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 8, 369–380 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Donkor MK, Sarkar A, Li MO, Tgf-β1 produced by activated CD4+T cells antagonizes T cell surveillance of tumor development. Onco. Targets. Ther 1, 162–171 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salem M, Wallace C, Velegraki M, Li A, Ansa-Addo E, Metelli A, Kwon H, Riesenberg B, Wu B, Zhang Y, Guglietta S, Sun S, Liu B, Li Z, GARP dampens cancer immunity by sustaining function and accumulation of regulatory T cells in the colon. Cancer Res. 79, 1178–1190 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moulin A, Mathieu M, Lawrence C, Bigelow R, Levine M, Hamel C, Marquette JP, le Parc J, Loux C, Ferrari P, Capdevila C, Dumas J, Dumas B, Rak A, Bird J, Qiu H, Pan CQ, Edmunds T, Wei RR, Structures of a pan-specific antagonist antibody complexed to different isoforms of TGFβ reveal structural plasticity of antibody-antigen interactions. Protein Sci. 23, 1698–1707 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lienart S, Merceron R, Vanderaa C, Lambert F, Colau D, Stockis J, van der Woning B, De Haard H, Saunders M, Coulie PG, Savvides SN, Lucas S, Structural basis of latent TGF-β1 presentation and activation by GARP on human regulatory T cells. Science 362, 952–956 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Gabriely G, da Cunha AP, Rezende RM, Kenyon B, Madi A, Vandeventer T, Skillin N, Rubino S, Garo L, Mazzola MA, Kolypetri P, Lanser AJ, Moreira T, Faria AMC, Lassmann H, Kuchroo V, Murugaiyan G, Weiner HL, Targeting latency-associated peptide promotes antitumor immunity. Sci. Immunol 2, eaaj1738 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gavin MA, Torgerson TR, Houston E, deRoos P, Ho WY, Stray-Pedersen A, Ocheltree EL, Greenberg PD, Ochs HD, Rudensky AY, Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proc. Natl. Acad. Sci. U.S.A 103, 6659–6664 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pratama A, Schnell A, Mathis D, Benoist C, Developmental and cellular age direct conversion of CD4+ T cells into RORγ+ or Helios+ colon Treg cells. J. Exp. Med 217, e20190428 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ahmadzadeh M, Pasetto A, Jia L, Deniger DC, Stevanović S, Robbins PF, Rosenberg SA, Tumor-infiltrating human CD4+ regulatory T cells display a distinct TCR repertoire and exhibit tumor and neoantigen reactivity. Sci. Immunol 4, eaao4310 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, Chudakov DM, Rudensky AY, Regulatory T cells exhibit distinct features in human breast cancer. Immunity 45, 1122–1134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kastenmuller W, Gasteiger G, Subramanian N, Sparwasser T, Busch DH, Belkaid Y, Drexler I, Germain RN, Regulatory T cells selectively control CD8+ T cell effector pool size via IL-2 restriction. J. Immunol 187, 3186–3197 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stockis J, Liénart S, Colau D, Collignon A, Nishimura SL, Sheppard D, Coulie PG, Lucas S, Blocking immunosuppression by human Tregs in vivo with antibodies targeting integrin αVβ8. Proc. Natl. Acad. Sci. U.S.A 114, E10161–E10168 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Markovics JA, Araya J, Cambier S, Jablons D, Hill A, Wolters PJ, Nishimura SL, Transcription of the transforming growth factor beta activating integrin beta8 subunit is regulated by SP3, AP-1, and the p38 pathway. J. Biol. Chem 285, 24695–24706 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Minagawa S, Lou J, Seed RI, Cormier A, Wu S, Cheng Y, Murray L, Tsui P, Connor J, Herbst R, Govaerts C, Barker T, Cambier S, Yanagisawa H, Goodsell A, Hashimoto M, Brand OJ, Cheng R, Ma R, Knelly KJM, Wen W, Hill A, Jablons D, Wolters P, Kitamura H, Araya J, Barczak AJ, Erle DJ, Reichardt LF, Marks JD, Baron JL, Nishimura SL, Selective targeting of TGF-β activation to treat fibroinflammatory airway disease. Sci. Transl. Med 6, 241ra79 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Derynck R, Turley SJ, Akhurst RJ, TGFβ biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol 18, 9–34 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, Rifkin DB, An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal. Biochem 216, 276–284 (1994). [DOI] [PubMed] [Google Scholar]
- 63.Lim JF, Berger H, Su IH, Isolation and activation of murine lymphocytes. J. Vis. Exp, 54596 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, Springer TA, Latent TGF-β structure and activation. Nature 474, 343–349 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Khan SA, Joyce J, Tsuda T, Quantification of active and total transforming growth factor-β levels in serum and solid organ tissues by bioassay. BMC. Res. Notes 5, 636 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Radaev S, Zou Z, Huang T, Lafer EM, Hinck AP, Sun PD, Ternary complex of transforming growth factor-beta1 reveals isoform-specific ligand recognition and receptor recruitment in the superfamily. J. Biol. Chem 285, 14806–14814 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Newsted D, Banerjee S, Watt K, Nersesian S, Truesdell P, Blazer LL, Cardarelli L, Adams JJ, Sidhu SS, Craig AW, Blockade of TGF-β signaling with novel synthetic antibodies limits immune exclusion and improves chemotherapy response in metastatic ovarian cancer models. Onco. Targets. Ther 8, e1539613 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hartigan-O’Connor DJ, Poon C, Sinclair E, McCune JM, Human CD4+ regulatory T cells express lower levels of the IL-7 receptor alpha chain (CD127), allowing consistent identification and sorting of live cells. J. Immunol. Methods 319, 41–52 (2007). [DOI] [PubMed] [Google Scholar]
- 69.Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, Szcześniak MW, Gaffney DJ, Elo LL, Zhang X, Mortazavi A, A survey of best practices for RNA-seq data analysis. Genome Biol. 17, 13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li B, Dewey CN, RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sorensen K, Brodbeck U, Assessment of coating-efficiency in ELISA plates by direct protein determination. J. Immunol. Methods 95, 291–293 (1986). [DOI] [PubMed] [Google Scholar]
- 73.Zemmour D, Zilionis R, Kiner E, Klein AM, Mathis D, Benoist C, Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat. Immunol 19, 291–301 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Antibodies and cell lines used in this manuscript will be available upon execution of a material transfer agreement. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. RNAseq datasets are deposited in the Gene Expression Omnibus (GSE158031).