

Abstract
CLN5 neuronal ceroid lipofuscinosis is a hereditary neurodegenerative disease characterized by progressive neurological decline, vision loss and seizures. Visual impairment in children with CLN5 disease is attributed to a progressive decline in retinal function accompanied by retinal degeneration as well as impaired central nervous system function associated with global brain atrophy. We studied visual system pathology in five Golden Retriever littermates homozygous for the CLN5 disease allele previously identified in the breed. The dogs exhibited signs of pronounced visual impairment by 21 to 22 months of age. Electroretinogram recordings showed a progressive decline in retinal function primarily affecting cone neural pathways. Altered visual evoked potential recordings indicated that disease progression affected visual signal processing in the brain. Aside from several small retinal detachment lesions, no gross retinal abnormalities were observed with in vivo ocular imaging and histologically the retinas did not exhibit apparent abnormalities by 23 months of age. However, there was extensive accumulation of autofluorescent membrane-bound lysosomal storage bodies in almost all retinal layers, as well as in the occipital cortex, by 20 months of age. In the retina, storage was particularly pronounced in retinal ganglion cells, the retinal pigment epithelium and in photoreceptor cells just interior to the outer limiting membrane. The visual system pathology of CLN5-affected Golden Retrievers is similar to that seen early in the human disease. It was not possible to follow the dogs to an advanced stage of disease progression due to the severity of behavioral and motor disease signs by 23 months of age. The findings reported here indicate that canine CLN5 disease will be a useful model of visual system disease in CLN5 neuronal ceroid lipofuscinosis. The baseline data obtained in this investigation will be useful in future therapeutic intervention studies. The findings indicate that there is a fairly broad time frame after disease onset within which treatments could be effective in preserving vision.
Keywords: Retinal degeneration, lysosomal storage, photoreceptor cells, autophagy, retinal pigment epithelium, phagocytosis, dog
1. Introduction
The neuronal ceroid lipofuscinoses (NCLs) are a group of inherited progressive neurodegenerative disorders characterized by accumulation of autofluorescent lysosomal storage bodies in neurons and other cell types. Variants in 14 genes have been associated with different forms of NCL (Beck-Wödl et al., 2018; Butz et al., 2020). The CLN5 form of the disease is associated with sequence variants in CLN5 which encodes a lysosomal protein whose function remains unclear (de Silva et al., 2015; Jules et al., 2017; Larkin et al., 2013; Lebrun et al., 2009; Leinonen et al., 2017; Mamo et al., 2012; Moharir et al., 2013; Schmiedt et al., 2010). 37 pathogenic variants have been reported in all 4 exons and in noncoding regions of CLN5 (“NCL Mutation and Patient Database,” n.d.; Savukoski et al., 1998). In most cases, the onset of clinical signs in CLN5 disease occurs in children between 2 to 8 years of age. Cognitive, motor and language function usually decline first followed by seizures and impaired vision progressing to blindness. However, there is some variability in visual signs between patients with different CLN5 disease alleles. In most patients, visual failure occurs between 5 to 10 years of age (Bessa et al., 2006; Pineda-Trujillo et al., 2005; Santavuori et al., 1982; Simonati et al., 2017; Xin et al., 2010), although a subset of affected individuals with certain variants exhibits visual signs that are minimal or begin later in the disease course (Ge et al., 2018; Xin et al., 2010). Vision loss in CLN5 disease has been attributed to a progressive decline in retinal function and associated retinal degeneration (Åberg et al., 2011). However, because CLN5 disease is characterized by widespread brain atrophy, central nervous system degeneration is also likely to play a significant role in vision loss.
Progress in understanding the pathogenesis of and developing treatments to prevent visual decline in CLN5 disease is likely to come from studying disease progression in large animal models. Naturally occurring CLN5 disease has been identified in Golden Retrievers, Border Collies and Australian Cattle Dogs as well as in Borderdale sheep and Devon cattle (Frugier et al., 2008; Gilliam et al., 2015; Houweling et al., 2006; Kolicheski et al., 2016; Melville et al., 2005). CLN5-affected Golden Retrievers with a predicted truncating mutation in CLN5 develop progressive neurological signs as early as 13 months of age with seizures and visual impairment starting at 18 months of age. In the Golden Retriever disorder, previous documentation of functional visual impairment consisted primarily of observations by the dogs’ owners; no objective assessments of retinal or visual function were performed. The affected companion dogs were euthanized due to disease progression at 30 to 34 months of age, 1.5 to 2 years after onset of clinical signs. Postmortem examination of the retinas revealed accumulations of autofluorescent storage bodies in several layers of the neural retina (Gilliam et al., 2015). In order to better characterize visual system pathology in this disorder, five Golden Retriever puppies homozygous for the previously identified CLN5 disease allele were evaluated for progression of retinal and brain pathology and changes with age in electroretinogram (ERG) and visual evoked potential (VEP) responses. A goal was to obtain baseline data that could be used in the design of potential therapeutic intervention studies.
2. Materials and methods
2.1. Animals
Five CLN5-affected Golden Retriever puppy littermates were evaluated in this study. The puppies were homozygous for the CLN5:c.934_935delAG variant previously identified in the breed that predicts a truncated protein lacking the 39 C-terminal amino acids (Gilliam et al., 2015). The dogs were maintained on a 12:12 daily light cycle in AALAC-accredited facilities at the University of Missouri – Columbia. They received routine husbandry and veterinary care and were socialized outside of their pens daily. The study was performed in accordance with the U.S. National Research Council Guide for the Care and Use of Laboratory Animals and was approved by the University of Missouri Animal Care and Use Committee.
2.2. Electrophysiology and retinal imaging
Complete ophthalmic exams were performed at 6.5, 15.5 and 17.5 months of age. Exams included tonometry and assessment of menace response, palpebral response and pupillary light reflex (PLR). Pupils were then dilated with 1% tropicamide ophthalmic solution (Alcon, Fort Worth, TX) and slit-lamp biomicroscopy (SL14; Kowa Co. Ltd., Tokyo, Japan) and indirect ophthalmoscopy were performed. Indirect ophthalmoscopy was done with an indirect wireless headset (12,500; Welch Allyn Inc., Skaneateles Falls, NY) and a 30-diopter clear handheld lens (Volk Optical Inc., Mentor, OH).
ERG recordings were performed at approximately 7, 13, 16, 20 and 23 months of age. Photopic and scotopic ERG responses were elicited bilaterally and recorded simultaneously as described previously (Ekesten et al., 2013; Katz et al., 2008; Whiting et al., 2013). Dogs were prepared for the procedure in ambient room light. Pupils were dilated with 1% tropicamide and dogs were sedated with up to 30 μg/kg intramuscular dexmedetomidine and 0.25 mg/kg intravenous midazolam. The eyes were held in place facing forward with conjunctival stay sutures. Recordings were performed with a bilateral full-field flash ERG unit (HMsERG model 1000, OcuScience, Henderson, NV) and the Dog Diagnostic Protocol recommended by the European College of Veterinary Medicine was used (Ekesten et al., 2013). After the ERG electrodes were placed, the room lights were switched off and subsequent procedures were performed in the dark. Five sets of ten 10.2 log photons/cm2/s (10 mcd/m2) flashes were repeated over 20 minutes (scotopic rod responses) followed by four 12.65 photons/cm2/s (3 cd/m2) flashes (scotopic low-intensity mixed rod and cone responses) and four 13.2 log photons/cm2/s (10 cd/m2) flashes (scotopic high-intensity mixed rod and cone responses). A 10- minute period of light adaption (13.65 log photons/cm2/s, 30 cd/m2) was followed by thirty-two 12.65 log photons/cm2/s (3 cd/m2) flashes (photopic cone responses) and a 3 cd/m2, 30 Hz photopic cone flicker. ERG waveforms were analyzed with ERGView software (OcuScience, Henderson, NV) using the average response of each set of flashes. The a- and b-wave amplitudes were measured as described previously (Marmor et al., 2004).
VEP recordings were performed immediately after the ERG. Room lights were turned on and needle electrodes were placed under the skin, with the active electrode placed over the rostral aspect of the occipital protuberance, the ground halfway between the lateral canthus and the base of the ear, and the reference on the dorsal aspect of the snout. The VEP protocol consisted of thirty 12.65 photons/cm2/s (3 cd/m2) bilateral flashes 1 second apart, and this was repeated three times. Data were analyzed in ERGView. A 100 Hz lowpass filter was applied and the latency and amplitude of peaks P1, N1, P2, N2 and P3 were measured as shown in Figure 1 (Kimotsuki et al., 2005; Strain et al., 1990). Latencies were measured as the time elapsed between the stimulus and each peak and amplitudes were measured from peak to peak (0 ms-P1, P1-N1, N1-P2, P2-N2 and N2-P3).
Figure 1.
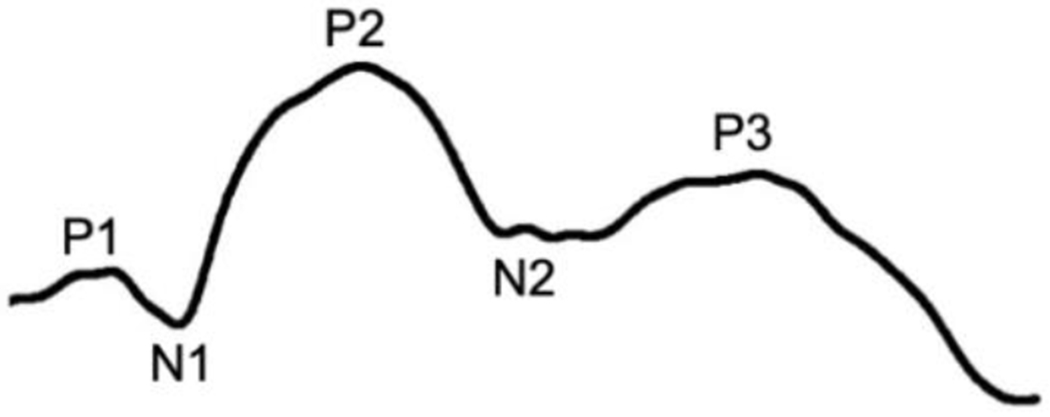
Elements of the VEP indicating the three positive peaks (P1, P2, and P3) and the two negative peaks (N1 and N2) that are typically present in normal healthy dogs when the recordings are performed as described for the present study (Strain et al., 1990).
Retinal imaging was performed immediately following each ERG and VEP recording session. Dogs were maintained under general anesthesia with isoflurane and in vivo scanning laser ophthalmoscopy (SLO) and optical coherence tomography (OCT) were performed using a Heidelberg Spectralis HRA/OCT (Heidelberg, Germany) equipped with a wide-angle 55° lens. SLO images of the fundus and serial OCT scans of the superior and inferior retina and optic nerve head were acquired as previously described (Whiting et al., 2015).
2.3. Retinal morphology
The five dogs evaluated in this study were euthanized at the ages indicated in Table 1. Each euthanasia procedure was performed between 5 and 6 hours after the onset of the light phase of the daily light cycle. Shortly after the last retinal imaging procedure was completed, the dogs were euthanized via intravenous infusion of pentobarbital (15 ml of 390 mg/ml pentobarbital Fatal-Plus solution, Dearborn, MI). Immediately after euthanasia, the left eyes were enucleated and immersed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer at pH 7.4 and the right eyes were enucleated and immersed in 3.5% depolymerized paraformaldehyde, 0.05% glutaraldehyde, 120 mM sodium cacodylate, 1 mM CaCl2, pH 7.4 (Immuno Fix). Both eyes were enucleated with approximately 1 cm of proximal optic nerve attached. Within minutes of being immersed in the fixatives, the corneas, irises and lenses were removed from the eyes, and fixative solution was injected into the vitreous. The eyecups with optic nerves were then incubated in the fixatives with gentle mixing at room temperature for a minimum of 24 hours before further dissections were undertaken.
Table 1.
Ages at which dogs were euthanized
| Dog | Sex | Age at Euthanasia (months) |
|---|---|---|
| Dog V | F | 20.1 |
| Dog W | F | 21.9 |
| Dog X | F | 23.1 |
| Dog Y | F | 23.4 |
| Dog Z | M | 23.4 |
After the initial fixation, the optic nerves were dissected from each eye and transferred to small vials of the same fixative used for the initial fixation. The eyecups were then dissected into 8 regions as shown in Figure 2 (Whiting et al., 2020a). All microscopic analyses were performed on sections obtained from the regions of the posterior retina indicated in Figure 1. A segment of each Immuno-fixed optic nerve was embedded in paraffin and sectioned at a thickness of 5 μm. Sections were stained with Masson’s Trichrome. A sample of posterior retina from each Immuno-fixed eye was washed in 170 mM sodium cacodylate, pH 7.4 and embedded in Tissue Tek cryo-embedding medium (Sakura FineTek, Torrance, CA). Sections of the cryo-embedded samples were cut with a Microm HM525 cryostat (Thermo Scientific) at a thickness of 8 μm. The sections were mounted on Superfrost Plus slides (Fisher Scientific) and were examined for NCL storage body-specific autofluorescence as described previously (Katz and Redmond, 2001).
Figure 2.

Diagram illustrating the eye dissections. N:nasal; T:temporal; S:superior; I:Inferior. Black circle: optic nerve head. The eyecups were dissected into the regions indicated by A – G. For this study, microscopic analyses were performed on the region of the retina indicated by the shaded rectangle.
A second sample of posterior retina from each Immuno-fixed eye was washed in 170 mM sodium cacodylate, pH 7.4 and embedded in paraffin. Sections of the paraffin-embedded retina samples along with sections of the optic nerves were cut a thickness of 5 μm and were immunostained for localization of lysosome-associated membrane protein 2 (LAMP2). The primary antibody used for immunostaining was anti-LAMP2A (Abcam ab18528; 1:300). Immunostaining was performed as described previously (Morgan et al., 2014).
The corresponding regions of the retinas from the eyes fixed in 2.5% glutaraldehyde were postfixed in osmium tetroxide and embedded in epoxy resin (Katz et al., 2005). Sections of these samples were cut at a thickness of 0.6 μm for light microscopy and 70 to 90 nm for electron microscopy. Sections for light microscopic examination were stained with Toluidine blue and sections for electron microscopic examination were mounted on thin-barred copper grids and stained with uranyl acetate and lead citrate. Light microscopy was performed using a Leica DMI 6000B microscope, and electron microscopy was performed using a JEOL JEM-1400 transmission electron microscope equipped with a Gatan digital camera.
Immediately after the eyes were enucleated the brain was collected from each dog and immersed in ice-chilled PlasmaLyte (Baxter Healthcare, Deerfield, IL) for 10 minutes. The brain was then cut into 4 mm thick coronal slices using an Adult Rhesus monkey brain matrix (Electron Microscopy Sciences, Hatfield, PA). Half of each brain slice was fixed in 2.0% glutaraldehyde, 1.12% paraformaldehyde, 130 mM sodium cacodylate, 1 mM CaCl2, pH 7.4 and the other half in Immuno Fix. Slices of occipital cortex were dissected from these samples and processed for and examined with fluorescence microscopy, immunohistochemistry (IHC), and electron microscopy as described above for the retina samples.
2.4. Visually-mediated behavior
Beginning at 21 months of age, visual tracking was assessed in dogs W, X, Y, and Z as part of routine neurologic examinations. For this assessment, a cotton ball was dropped from approximately eye-level height in front of the dog. This was repeated approximately 5-10 times per evaluation. The dog’s ability to track the cotton ball on its path from point of drop to the floor was assessed subjectively by watching eye fixation and head movements in relation to the falling cotton ball. Visual tracking ability at each assessment was assigned one of the following designations: good (consistently tracks cotton ball well), decreased (inconsistently tracks cotton ball from point of drop to floor), poor (occasionally reacts when cotton ball contacts floor but does not track path from drop to floor), or absent (does not track cotton ball at all). Cotton ball vision tests were repeated every 1-3 weeks at time of neurologic examinations. Vision was also subjectively evaluated at time of neurologic examinations through simple observation of behavior and ability to navigate around obstacles in a closed room.
2.5. Statistical Analyses
All statistical tests were performed using SigmaPlot (Systat Software Inc., San Jose, CA). Normality was confirmed with the Shapiro-Wilk test. A two-tailed Student’s t-test was used to compare ERG b-wave amplitudes, a-wave amplitudes and b:a wave ratios at 7 months and 22-23 months of age. A two-tailed Student’s t-test was also used to compare VEP P2 amplitudes and the ratio of P2 amplitudes to cone b-wave amplitudes between 7 months and 22-23 months of age and between 7, 12, 16 and 20 months of age combined and 22-23 months of age. Statistical power analyses indicated that sample sizes were too small to enable reliable pairwise comparisons of data between each of the time points at which ERG and VEP data were obtained.
3. Results
3.1. Ocular imaging
Ophthalmic exams were performed at 6.5, 15.5 and 17.5 months of age. Pupillary light reflex and menace response were intact through 17.5 months of age but were not able to be assessed at later ages due to behavioral changes and lack of compliance with examination. Dog W and Dog Z developed 1 to 3 small bullous retinal detachments per eye, first noted at 15.5 months of age on ophthalmic exam and 20 months of age by SLO imaging, respectively. In both cases, lesions were confirmed with OCT imaging (Figure 3F and 3G). No other abnormalities were observed.
Figure 3.

SLO and OCT images of Dog X (A), Dog Y (B) and Dog Z (C) at 7 months of age and Dog X (D), Dog Y (E), Dog W (F) and Dog Z (G) at 23 months of age. 1-3 small bullous retinal detachment lesions were present in both eyes of Dog W (F) and Dog Z (G) by 23 months of age. Green lines indicate the locations from which the OCT images were obtained. Arrows indicate the locations of the retinal lesions.
3.2. Electroretinographic and visual evoked potential findings
The scotopic b-wave response amplitudes elicited by a low-intensity flash after 20 minutes of dark adaptation did not change significantly between 7 and 23 months of age (Figure 4A, p=0.263). The b-wave amplitudes elicited by scotopic high intensity flash stimuli at 23 months were an average of 53% lower than the responses to the same stimuli elicited at 7 months of age (Figure 4B, p=0.004) while the a-wave amplitudes were unchanged (Figure 4C, 0.231), resulting in a decrease in the b- to a-wave ratio (Figure 4D, p=0.005). The b-wave amplitudes elicited by both photopic single flash (Figure 4E) and flicker (Figure 4H) stimuli were decreased at 23 months relative to the responses elicited by these stimuli at 7 months of age (p=0.006 and 0.029 for the flash and flicker responses respectively). Photopic a-wave amplitudes elicited by the single flash were also reduced by 23 months (Figure 4F, p=0.039) as was the b-wave to a-wave ratio (Figure 4G, p=0.042), indicating that reductions in the b-wave were greater than the decreases in a-wave amplitudes. Representative ERG responses of Dog X at 7, 16 and 23 months of age are shown in Figure 5.
Figure 4.
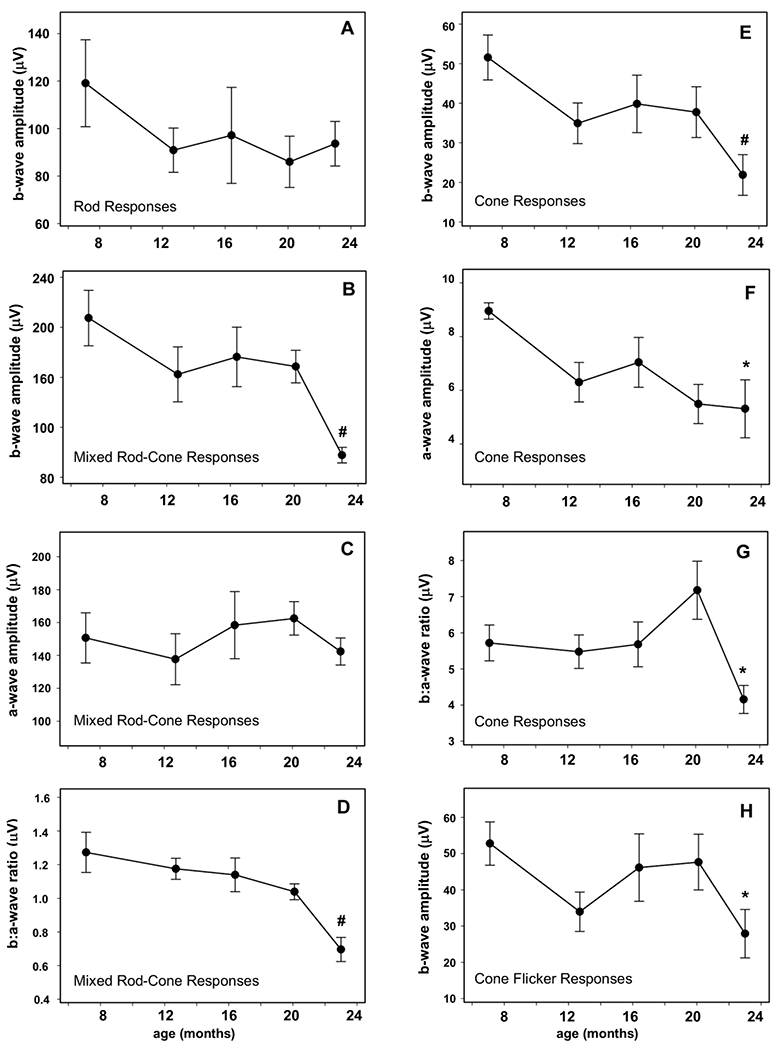
Mean ERG response amplitudes ±SEM for all 5 dogs. Cone function was primarily affected. Mean cone (#p<0.01, D), cone flicker (*p<0.05, H) and mixed rod and cone (#p<0.01, A) b-wave amplitudes were significantly reduced from baseline at 23 months of age. The mean cone a-wave amplitude (E) was significantly decreased at 23 months of age (*p<0.05) while the mixed rod and cone a-wave (B) was unchanged. Mean cone (*p<0.05, F) and mixed rod and cone (#p<0.01, C) b:a wave ratios were significantly decreased from baseline at 23 months of age. Mean rod response (G) was not significantly changed from baseline at 23 months of age.
Figure 5.

ERG waveforms from Dog X at 7, 16 and 23 months of age.
For the VEPs, the latencies and amplitudes of peaks P1, N1, N2 and P3 and the latencies of P2 did not change significantly with age (data not shown). The amplitude of P2 was reduced at 23 months of age relative to 7 months of age (p=0.042) and to amplitudes at 7, 12, 16 and 20 months combined (p=0.016) (Figure 6A). Because the VEP was elicited with a bright flash stimulus under photopic conditions and therefore mediated primarily by cones, we also evaluated the ratio of the P2 amplitude to the cone b-wave amplitude (Figure 6B). This ratio was increased at 23 months compared to both 7 months (p=0.038) and all other time points combined (p=0.027). Some of the VEP responses included one or more positive peaks following P2 (asterisks in Figure 7) that do not appear in the VEP responses of normal healthy dogs (Strain et al., 1990). These peaks were more pronounced at the later ages. VEP waveforms from Dog Y and Dog Z at 7, 16 and 23 months of age are shown in Figure 6.
Figure 6.

Mean VEP P2 amplitudes ±SEM for all 5 dogs (A) and the mean ratio of the P2 amplitude to the cone b-wave amplitude ±SEM for all 5 dogs (B). The mean amplitude of P2 was decreased by 23 months of age (*p<0.05). The mean ratio of the P2 amplitude to the cone b-wave amplitude was increased by 23 months (*p<0.05).
Figure 7.

VEP waveforms from Dog Y (A) and Dog Z (B) at 7, 16 and 23 months of age. Arrows indicate P2. P2 was identified as the first positive peak following N1, following the standard convention. In some of the recordings, there was an additional positive peak following P1 (asterisks) that has not been reported in normal dogs.
3.3. Visually-mediated behavior
Visually mediated behavior was assessed in part by determining whether the dogs visually tracked falling cotton balls that were dropped in front of them from above the dog’s head level. The assessments were performed at periodic intervals on four of the dogs starting at 21 months of age (the fifth dog was euthanized prior to the start of these evaluations). Tracking behavior was rated as good, decreased, or poor relative to the tracking by healthy dogs under the same conditions. For consistency, all ratings were performed by the same observer. The findings are summarized in Table 2. All of the dogs exhibited decreased visual tracking with disease progression. At the first cotton ball test performed at 21 months of age, two dogs were assessed to have good visual tracking ability and two dogs had decreased visual tracking ability. Subsequent evaluations showed progressive decline of ability to track the cotton ball for all dogs. Refer to Videos 1A and 1B in Supplemental Materials for a comparison of Dog Z’s performance on this test at 21 and 23.2 months of age.
Table 2 –
Cotton ball visual tracking assessments
|
|
|||||
|---|---|---|---|---|---|
| Age (months)* | |||||
|
|
|||||
| Dog | 21.0 | 21.9 | 22.4 | 23.0 | 23.2 |
|
| |||||
| Dog W | Decreased | Poor | |||
|
| |||||
| Dog X | Good | Good | Decreased | Decreased | |
|
| |||||
| Dog Y | Decreased | Poor | Poor | Poor | Poor |
|
| |||||
| Dog Z | Good | Decreased | Poor | Poor | Poor |
If no data, dogs were euthanized prior to this age.
Visual deficits in navigation ability in a closed room became apparent starting at 21.0 to 22.4 months of age in Dog W, Dog X, Dog Y, and Dog Z. Behavioral visual deficits subjectively worsened over variable periods of time, with Dog X Y and Z having markedly progressed visual impairment by 22.4 to 23.1 months of age (Video 2 in Supplemental Materials). Dog W’s visual deficits were subjectively less progressed by the time of euthanasia at 21.9 months of age (Video 3 in Supplemental Materials).
3.4. Retinal and occipital cortex morphology
Despite the decreased retinal and visual function, the overall morphology of the retinas remained relatively normal, even at 23.4 months of age (Figure 8). Inclusions with autofluorescence properties typical of ceroid and lipofuscin were observed in all retinal layers except the photoreceptor outer segments (Figure 9). The accumulation of these inclusions was most pronounced in the retinal ganglion cell bodies, along the outer limiting membrane, and in the retinal pigment epithelium (RPE). The accumulation of this autofluorescent material was already pronounced in the dog euthanized at 20 months of age and was not appreciably different in the dogs euthanized at later time points. In the occipital cortex, by 20 months of age almost every neuron contained significant amounts of autofluorescent storage material (Figure 10). The autofluorescent inclusions co-localized with immunostaining for the lysosomal marker LAMP2 in both the retina and the occipital cortex (Figure 11).
Figure 8.

Light micrograph of a section of the superior-central retina from Dog Y who was euthanized at 23.4 months of age. All layers of the retina were well preserved with no apparent morphological abnormalities.
Figure 9.

Fluorescence micrographs of the inner (A) and outer (B) layers of unstained cryostat sections of the retina from Dog Z, who was euthanized at 23.4 months of age. Bar in (B) indicates the magnification of both micrographs.
Figure 10.

Fluorescence micrographs of unstained cryostat sections of the occipital cortex from Dog V (A) and Dog Y (B), who were euthanized at 20.1 and 23.4 months of age respectively. Bar in (B) indicates the magnification of both micrographs.
Figure 11.

Light micrographs of sections of the retina (A) and occipital cortex (B) from Dog X. Sections were immunostained for LAMP2 localization. No counterstain was used. In the retina, LAMP2 immunostaining was most pronounced in the ganglion cells, along the outer limiting membrane, and in the RPE. In the occipital cortex most cells contained aggregates of LAMP2-positive inclusions. Bar in (B) indicates the magnification of both micrographs.
Upon electron microscopic examination, the storage bodies in the neural retina and neurons of the occipital cortex were seen to consist of membrane-bounded organelles with contents whose ultrastructure varied with cell type (Figures 12–14). Already by 20 months of age, almost every neuron in the in the occipital cortex contained the storage bodies, the contents of which consisted primarily of membrane-like structures that were vesicular or linear (Figure 12). In the retinal ganglion cells, in certain planes of section the storage body contents had a paracrystalline appearance (Figure 13). With electron microscopy, the storage bodies along the outer limiting membrane were found to be localized almost exclusively to the regions of the photoreceptor cells just interior to the junctional complexes that form the outer limiting membrane (Figure 14). The contents of these storage bodies consisted primarily of vesicular structures. These storage bodies appeared to be present in every photoreceptor cell. The storage bodies in the RPE were more heterogenous in appearance (Figures 15 and 16). Some of the storage bodies contained what appeared to be remnants of partially degraded photoreceptor outer segments or melanosomes (Figure 15). The contents of other storage bodies consisted of heterogenous mixtures of components of variable fine structure. Some of the inclusions were surrounded by double membranes characteristic of autophagosomes (Figure 15C).
Figure 12:

Electron micrograph of a neuron in the occipital cortex of an affected Golden Retriever (Dog V) euthanized at approximately 20 months of age.
Figure 14:

Electron micrograph showing an accumulation of disease-specific storage bodies (s) in a photoreceptor cell of an affected Golden Retriever (Dog V) euthanized at approximately 20 months of age. The storage bodies were present almost exclusively just interior to the retinal outer limiting membrane (arrows) and exterior to the photoreceptor nuclei (n).
Figure 13:

Electron micrographs of disease-specific storage bodies in retinal ganglion cells of an affected Golden Retriever (Dog V) euthanized at approximately 20 months of age. In certain planes of section some of the contents of the storage bodies had a paracrystalline appearance.
Figure 15:

Electron micrographs of disease-specific storage bodies in the retinal pigment epithelium of an affected Golden Retriever (Dog W) euthanized at approximately 22 months of age. The ultrastructural appearances of the storage bodies (arrows in A and inclusions in B and C) were quite heterogenous. Some inclusions contained melanosomes (m) and a small minority appeared to be phagocytosed photoreceptor outer segments (p). The contents of some inclusions had a membrane-like appearance (B). Some inclusions were surrounded by a double membrane characteristic of autophagosomes (arrows in C).
Figure 16.

Electron micrographs of additional disease-specific storage bodies in the retinal pigment epithelium of an affected Golden Retriever (Dog W) euthanized at approximately 22 months of age. The ultrastructural appearances of the storage bodies were quite heterogeneous, even within the same eye.
Optic nerve sections were stained with Masson’s trichrome that stains collagenous connective tissue components blue and most cellular components red or pink. In the normal dog retina, the axons are divided into fascicles surrounded by connective tissue trabeculae (Balaratnasingam et al., 2009; Donovan et al., 1974). In the CLN5-affected dogs, these septa were greatly diminished resulting reduced boundaries between fascicles (Figure 17).
Figure 17.

Light micrograph of a cross-section of the optic nerve from Dog Y stained with Masson’s trichrome. The blue-staining connective tissue septa (arrows) only partially surround axon fascicles.
4. Discussion
Most children afflicted with CLN5 disease exhibit progressive vision loss starting early in the disease process (Åberg et al., 2011; Pineda-Trujillo et al., 2005; Santavuori et al., 1991, 1982). Evidence from human studies indicates that visual decline in CLN5 disease results from a combination of progressive impairment of retinal and central nervous system function (Santavuori et al., 1982, 1991; Holmberg et al., 2000; Simonati et al., 2017). It is also well documented that children with CLN5 disease exhibit pathology of the retina and brain regions involved in visual signal processing (Santavuori et al., 1991, 1982; Autti et al., 1992; Tyynelä et al., 1997). In these respects the Golden Retriever disease appears to be a good model for the human disorder (Gilliam et al., 2015; Kolicheski et al., 2016; Melville et al., 2005; Villani et al., 2019).
To further assess whether the visual system pathology seen in children also occurs in affected dogs, we evaluated visual and retinal function in five CLN5-affected Golden Retrievers. Clinical assessments identified clear deficits in visually mediated behavior at 21 to 22 months of age, similar to what has been seen in Border Collies and Australian cattle Dogs with CLN5 disease (Gilliam et al., 2015; Kolicheski et al., 2016; Sisk et al., 1990; Studdert and Mitten, 1991). Visual deficits worsened over time and the dogs exhibited pronounced visual deficits by 23 months of age. It is challenging to assess visual capabilities in animals. We used a cotton ball visual tracking test that has been described previously as part of neuro-ophthalmic evaluations (Gelatt et al., 2013; Skerritt, 2018). While this is a good method of assessing vision in canine subjects, CLN5-affected Golden Retrievers are similar to children with CLN5 disease in that they exhibit both visual and cognitive deficits. In addition to functional impairment of the retina, progressive cognitive decline would be expected to alter performance on the visual tracking test. The progressive changes in tracking ability were striking (Video 1 in Supplemental Materials) and were consistent with observations that the dogs had difficulty with visually mediated navigation at the later ages, often bumping into objects when moving about in familiar surroundings under both bright and dim light conditions. Because visually mediated behavior involves both the visual pathway and neural pathways involved in cognition, it is not possible to assess the relative contributions of these two pathways using behavioral testing.
To objectively determine whether altered retinal function may have contributed to the visual behavior deficits, ERG recordings were performed over the course of the disease progression. ERG recordings showed a progressive decline in b-wave amplitudes in the dark-adapted state. After an initial decline in photopic ERG amplitudes between 8 and 12 months of age, the response amplitudes stabilized until approximately 20 months of age, after which they declined further by 23 months of age. Early in the disease progression, the photopic b- to a-wave amplitude ratios remained constant, but between 20 and 23 months of age there was a significant decline in this ratio. This could indicate that late in the disease progression inner retinal function was affected to a greater degree than photoreceptor cell function, or alternatively, that photoreceptor pathology inhibited signal transmission from these cells to the inner retina. It was not possible to follow the dogs past 23 months of age because at this age, in addition to having visual deficits, their coordination was severely impaired, they were hyperactive, and they had begun to suffer from mild seizures. This put the dogs at high risk for injury, making it necessary to euthanize them. Therefore, it was not possible to follow the dogs to the same degree of disease progression that affected children reach prior to succumbing to the disease.
To assess whether central nervous system components of the visual pathway are affected in the canine disorder, VEPs were recorded over the course of disease progression. The P2 component of the VEP, which reflects postsynaptic cortical responses, decreased in amplitude between 20 and 23 months of age. Because the VEPs are recorded with bright light stimuli under photopic conditions, they reflect responses from retinal cone inputs. To determine whether the decreased P2 amplitudes were due to decreased signal input to the visual cortex or to more central components of the visual pathway, the ratio of the P2 amplitudes to the cone b-wave amplitudes were determined for each dog at each age. This ratio actually increased over the course of the disease progression. Thus, for the same input from the retina, the cortical response reflected by the VEP P2 wave was increased. The VEP data therefore indicate that there are alterations in visual signal processing in the brain. The neural generators of the components of the VEP are not well defined, but a topographic mapping study in dogs reported that P2 reflects the conduction of signal from the retina to the lateral geniculate nucleus (Kimotsuki et al., 2005). In addition to the effects of the disease on VEP P2 amplitudes, starting at 16 months of age some of the affected dogs exhibited additional peaks in the VEP responses that are not normally present in the canine VEP. These peaks may be indicative of aberrant visual signal processing in the brain. This is not unexpected since, as in the human disorder, canine CLN5 disease is characterized by progressive global brain atrophy (Autti et al., 1992; Santavuori et al., 2001; Simonati et al., 2017; Tyynelä et al., 1997).
Ophthalmoscopically, the only retinal abnormalities that developed in the affected dogs were a small number of focal retinal detachments. Similar detachments, but larger in size and greater in number, develop in dogs with the CLN2 form of NCL (Whiting et al., 2015). Similar lesions have not been reported in children with either CLN5 or CLN2 disease, so these lesions may be specific to dogs. However, it is possible that these lesions occur in human subjects but have been overlooked. The development of these lesions is likely to be indicative of alterations in the important interactions between the photoreceptor cells and the apposed RPE. The optic atrophy reported in some children with late-stage CLN5 disease was not observed in the affected dogs. This may be due to the fact that the dogs were euthanized at an earlier stage in the disease progression than the stage at which optic atrophy was noted in affected children.
Consistent with the ophthalmoscopic findings, no gross abnormalities were observed histologically in the retinas of the affected dogs. However, there was extensive accumulation of autofluorescent membrane-bound inclusion bodies in almost all retinal layers, as well as in the occipital cortex, by 20 months of age. The lysosomal marker protein LAMP2 was localized to the inclusions, indicating that they were lysosomal storage bodies. Although the precise function of the CLN5 protein has not been determined, the protein localized to the lysosomal lumen and appears to be necessary for normal lysosome function (Basak et al., 2021; Isosomppi et al., 2002; Lebrun et al., 2009; Schmiedt et al., 2010). The ultrastructure of the storage bodies varied between cell types, and sometimes within the same cell type. This suggests that CLN5 deficiency results not in a failure of lysosomal degradation of a specific biochemical substrate, but in a more general impairment of lysosomal function. Thus, the material that accumulates in the lysosomes of a specific cell type appears to be dependent on what that cell type normally degrades via the lysosomal pathway. In the retinal ganglion cells, the contents of the storage bodies had a paracrystalline appearance, suggesting the failure to degrade primarily a single substrate or a small number of substrates. In the RPE, on the other hand, the ultrastructure of the storage body contents was much more heterogenous, consistent with impaired degradation of both phagocytosed photoreceptor outer segments and RPE cellular components incorporated into lysosomes via autophagy. The contents of some of the RPE inclusions appeared to consist of partially degraded photoreceptor outer segment fragments. Because the dogs were euthanized at a time in the daily light cycle at which outer segment disc shedding is expected to be minimal, the presence of these inclusions may be indicative of impairment in degradation of this phagocytosed material (Katz and Shanker, 1989). Storage material accumulation was especially pronounced in the RPE and in the retina along the outer limiting membrane. Although RPE accumulation of storage bodies has been reported in mouse models of the CLN3 and CLN6 forms of NCL, it has not been observed in animals or humans with other NCL types, nor in a human subject with CLN3 disease (Bensaoula et al., 2000; von Eisenhart-Rothe et al., 2018; Wavre-Shapton et al., 2015). The autofluorescence properties of the RPE storage material are similar to those of RPE age pigment (lipofuscin). However, the ultrastructural appearance of the storage material is distinctly different from that of lipofuscin (Boulton et al., 1990; Gouras et al., 2018; Katz et al., 1986; Katz and Robison, 1984).
The lysosomal storage material along the OLM was localized to photoreceptor cells just interior to the junctional complexes between the photoreceptor and Mueller cells. Accumulation of lysosomal storage material in photoreceptor cells has not been reported for any other form of NCL. These data indicate that the lysosomal turnover within photoreceptor cells is much more pronounced that has been previously recognized. The fact that accumulation of these storage bodies occurs in the CLN5 disease and not in other disorders affecting lysosome function indicates that the CLN5 protein plays a role that is central to normal lysosomal function in this cell type. The localization of the storage material to only the region of the photoreceptors between the outer nuclear layer and the outer limiting membrane indicates that there is specific functional compartmentalization of lysosomes to this part of the cells.
Pronounced lysosomal storage body accumulation was present in almost every neuron in the brain occipital cortex. It is not known whether the presence of this storage material is detrimental to cell function. However, the presence of this storage material strongly suggests that the CLN5 protein is normally expressed in this region of the brain involved in visual signal processing.
Although age- and breed-matched unaffected Golden Retrievers were not available for comparison, data from previous studies clearly indicate that the changes observed in the affected dogs over the time course of observation were disease-related. While absolute amplitude values should not be directly compared amongst different breeds, studies have shown that in healthy dogs of various breeds ERG amplitudes do not significantly decline over the age span included in this study. For example, no changes in ERG response amplitudes or funduscopic appearances of the retinas were observed in healthy Dachshunds and Briard dogs monitored over similar age ranges (Whiting et al., 2014; Whiting et al., 2016; Narfström et al., 2003; Narfström et al., 2005). Likewise, in healthy dogs VEPs do not change significantly over the age span over which the affected Golden Retrievers were evaluated (Kimotsuki, et a., 2006). In addition, the retinal pathology observed in the affected Golden Retrievers does not occur in healthy dogs of similar ages (Whiting et al., 2020a; Whiting et al., 2020b; Narfström et al., 2003; Narfström et al., 2005; Defour et al., 2020).
In addition to dogs, naturally occurring CLN5 diseases have been identified in sheep and cattle (Frugier et al., 2008; Houweling et al., 2006; Kolicheski et al., 2016; Melville et al., 2005; Villani et al., 2019). The disorder has been extensively characterized in Borderdale sheep (Amorim et al., 2015; Frugier et al., 2008; Perentos et al., 2016, 2015; Russell et al., 2018). Affected sheep have been bred and utilized in therapeutic intervention studies (Best et al., 2017; Hughes et al., 2014; Mitchell et al., 2018). Blindness is a prominent sign in CLN5-affected Borderdale sheep which exhibit impaired vision starting at 10 to 12 months of age and progressing to functional blindness (bilateral absence of menace response and loss of visual tracking) by 13 months of age. Slow PLR and severely diminished mixed receptor ERG responses are apparent at 15 to 17 months of age. Fundoscopy in affected sheep reveals extremely hyperreflective foci and thinned retinal vasculature (Mitchell et al., 2018). The sheep model has been a very useful large animal model for this disorder. A canine model would complement the sheep model and would be more accessible for research at the larger number of institutions that have resources for maintaining research dogs. Due to the risk of injury that accompanied behavioral changes and motor deficits, the dogs evaluated in this study were euthanized somewhat early in the disease progression. While the dogs that were evaluated in this report were euthanized before 24 months of age, previously studied affected companion Golden Retrievers that were maintained in home environments were kept alive as late as 30 months of age. For the canine model to be most useful for testing the long-term efficacy of therapeutic interventions, it would be advisable to breed the disease variant into a smaller breed such as the Dachshund that can be more easily maintained in a research environment even when significantly neurologically impaired (Katz et al., 2015, 2014).
Canine models have been proven invaluable in preclinical testing and can provide strong evidence to support clinical trials. We have used a Dachshund model of CLN2 disease extensively to test therapies targeting the brain and retina (Katz et al., 2015, 2014; Tracy et al., 2016; Whiting et al., 2020a, 2020b). An intrathecal enzyme replacement therapy approach tested in this model led to the first disease-modifying therapy for an NCL and data showing efficacy of intravitreal enzyme replacement therapy supported an investigational new drug application (Schulz et al., 2018). Intrathecal gene therapy data from the sheep model of CLN5 disease is promising (Mitchell et al., 2018). However, to preserve visual function it will likely be necessary to target both the brain and retina since pathology in both probably contributes to vision loss in this disease. Based on previous work and the findings reported here, it appears that canine CLN5 disease will be a useful model for testing therapies to preserve functional vision. It was recently reported that intravitreal injection of an AAV9. CLN5 vector prior to the onset of visual signs in a sheep model of CLN5 disease resulted in preservation of retinal structure and function for over 15 months post-injection (Murray et al., 2021). Therapeutic intervention studies using a canine model of the CLN2 form of NCL indicate that protein replacement and ex-vivo gene therapy are also promising approaches for treating the visual system pathology in CLN5 disease (Tracy et al., 2016; Whiting et al., 2020b, 2020a).
Supplementary Material
Highlights.
Progressive visual impairment occurs in canine CLN5 neuronal ceroid lipofuscinosis.
Disease pathology is present in the retinal and central nervous system visual pathways.
The canine disorder can serve as a model for testing therapeutic interventions for CLN5 diease.
Acknowledgments
Funding for this research was provided in part by U.S. National Institutes of Health grant EY031674 and a University of Missouri Life Sciences graduate fellowship (GRK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Åberg L, Autti T, Cooper JD, Elleder M, Haltia M, Jalanko A, Kitzmüller C, Kopra O, Mole SE, Nuutila A, Peltonen L, Punkari M-L, Rapola J, Tyynelä J, 2011. CLN5, in: Mole S, Williams R, Goebel H (Eds.), The Neuronal Ceroid Lipofuscinoses (Batten Disease). Oxford University Press, Oxford, UK. 10.1093/med/9780199590018.003.0009 [DOI] [Google Scholar]
- Amorim IS, Mitchell NL, Palmer DN, Sawiak SJ, Mason R, Wishart TM, Gillingwater TH, 2015. Molecular neuropathology of the synapse in sheep with CLN5 Batten disease. Brain and Behavior 5, 1–12. 10.1002/brb3.401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autti T, Raininko R, Launes J, Nuutila A, Santavuori P, 1992. Jansky-Bielschowsky variant disease: CT, MRI, and SPECT findings. Pediatric Neurology 8, 121–126. 10.1016/0887-8994(92)90032-T [DOI] [PubMed] [Google Scholar]
- Balaratnasingam C, Morgan WH, Johnstone V, Pandav SS, Cringle SJ, Yu D-Y, 2009. Histomorphometric measurements in human and dog optic nerve and an estimation of optic nerve pressure gradients in human. Experimental Eye Research 89, 618–628. 10.1016/j.exer.2009.06.002 [DOI] [PubMed] [Google Scholar]
- Basak I, Wicky HE, McDonald KO, Xu JB, Palmer JE, Best HL, Lefrancois S, Lee SY, Schoderboeck L, Hughes SM, 2021. A lysosomal enigma CLN5 and its significance in understanding neuronal ceroid lipofuscinosis. Cellular and Molecular Life Sciences. 10.1007/s00018-021-03813-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck-Wödl S, Harzer K, Sturm M, Buchert R, Rieß O, Mennel H-D, Latta E, Pagenstecher A, Keber U, 2018. Homozygous TBC1 domain-containing kinase (TBCK) mutation causes a novel lysosomal storage disease – a new type of neuronal ceroid lipofuscinosis (CLN15)? Acta Neuropathologica Communications 6, 145. 10.1186/s40478-018-0646-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaoula T, Shibuya H, Katz ML, Smith JE, Johnson GS, John SK, Milam AH, 2000. Histopathologic and immunocytochemical analysis of the retina and ocular tissues in batten disease1. Ophthalmology 107, 1746–1753. 10.1016/S0161-6420(00)00264-5 [DOI] [PubMed] [Google Scholar]
- Bessa C, Teixeira CAF, Mangas M, Dias A, Sá Miranda MC, Guimarães A, Ferreira JC, Canas N, Cabral P, Ribeiro MG, 2006. Two novel CLN5 mutations in a Portuguese patient with vLINCL: Insights into molecular mechanisms of CLN5 deficiency. Molecular Genetics and Metabolism 89, 245–253. 10.1016/j.ymgme.2006.04.010 [DOI] [PubMed] [Google Scholar]
- Best HL, Neverman NJ, Wicky HE, Mitchell NL, Leitch B, Hughes SM, 2017. Characterisation of early changes in ovine CLN5 and CLN6 Batten disease neural cultures for the rapid screening of therapeutics. Neurobiology of Disease 100, 62–74. 10.1016/j.nbd.2017.01.001 [DOI] [PubMed] [Google Scholar]
- Boulton M, Docchio F, Dayhaw-Barker P, Ramponi R, Cubeddu R, 1990. Age-related changes in the morphology, absorption and fluorescence of melanosomes and lipofuscin granules of the retinal pigment epithelium. Vision Research 30, 1291–1303. 10.1016/0042-6989(90)90003-4 [DOI] [PubMed] [Google Scholar]
- Butz ES, Chandrachud U, Mole SE, Cotman SL, 2020. Moving towards a new era of genomics in the neuronal ceroid lipofuscinoses. Biochimica et Biophysica Acta - Molecular Basis of Disease 1866, 165571. 10.1016/j.bbadis.2019.165571 [DOI] [PubMed] [Google Scholar]
- Dufour VL, Yu Y, Pan W, Ying GS, Aguirre GD, Beltran WA, 2020. In-vivo longitudinal changes in thickness of the postnatal canine retina. Experimental Eye Research. 192:107926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Silva B, Adams J, Lee SY, 2015. Proteolytic processing of the neuronal ceroid lipofuscinosis related lysosomal protein CLN5. Experimental Cell Research 338, 45–53. 10.1016/j.yexcr.2015.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan RH, Carpenter RL, Schepens CL, Tolentino FI, 1974. Histology of the normal collie eye. III. Lens, retina and optic nerve. Annals of Ophthalmology 6, 1299–1307. [PubMed] [Google Scholar]
- Ekesten B, Komáromy AM, Ofri R, Petersen-Jones SM, Narfström K, 2013. Guidelines for clinical electroretinography in the dog: 2012 update. Documenta Ophthalmologica 127, 79–87. 10.1007/s10633-013-9388-8 [DOI] [PubMed] [Google Scholar]
- Frugier T, Mitchell NL, Tammen I, Houweling PJ, Arthur DG, Kay GW, van Diggelen OP, Jolly RD, Palmer DN, 2008. A new large animal model of CLN5 neuronal ceroid lipofuscinosis in Borderdale sheep is caused by a nucleotide substitution at a consensus splice site (c.571+1G>A) leading to excision of exon 3. Neurobiology of Disease 29, 306–315. 10.1016/j.nbd.2007.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge L, Li HY, Hai Y, Min L, Xing L, Min J, Shu HX, Mei OY, Hua L, 2018. Novel Mutations in CLN5 of Chinese Patients With Neuronal Ceroid Lipofuscinosis. Journal of Child Neurology 33, 837–850. 10.1177/0883073818789024 [DOI] [PubMed] [Google Scholar]
- Gelatt KN, Gilger BC, Kern TJ (Eds.), 2013. Veterinary Ophthalmology, 5th Ed. ed. John Wiley & Sons, Inc., Ames, IA. [Google Scholar]
- Gilliam D, Kolicheski A, Johnson GS, Mhlanga-Mutangadura T, Taylor JF, Schnabel RD, Katz ML, 2015. Golden Retriever dogs with neuronal ceroid lipofuscinosis have a two-base-pair deletion and frameshift in CLN5. Molecular Genetics and Metabolism 115, 101–109. 10.1016/j.ymgme.2015.04.001 [DOI] [PubMed] [Google Scholar]
- Gouras P, Brown KR, Mattison JA, Neuringer M, Nagasaki T, Ivert L, 2018. The Ultrastructure, Spatial Distribution, and Osmium Tetroxide Binding of Lipofuscin and Melanosomes in Aging Monkey Retinal Epithelium. Current Eye Research 43, 1019–1023. 10.1080/02713683.2018.1464194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg V, Lauronen L, Autti T, Santavuori P, Savukoski M, Uvebrant P, Hofman I, Peltonen L, Järvelä I, 2000. Phenotype-genotype correlation in eight patients with Finnish variant late infantile NCL (CLN5). Neurology 55, 579–581. 10.1212/WNL.55.4.579 [DOI] [PubMed] [Google Scholar]
- Houweling PJ, Cavanagh JAL, Palmer DN, Frugier T, Mitchell NL, Windsor PA, Raadsma HW, Tammen I, 2006. Neuronal ceroid lipofuscinosis in Devon cattle is caused by a single base duplication (c.662dupG) in the bovine CLN5 gene. Biochimica et Biophysica Acta - Molecular Basis of Disease 1762, 890–897. 10.1016/j.bbadis.2006.07.008 [DOI] [PubMed] [Google Scholar]
- Hughes SM, Hope KM, Xu JB, Mitchell NL, Palmer DN, 2014. Inhibition of storage pathology in prenatal CLN5-deficient sheep neural cultures by lentiviral gene therapy. Neurobiology of Disease 62, 543–550. 10.1016/j.nbd.2013.11.011 [DOI] [PubMed] [Google Scholar]
- Isosomppi J, Vesa J, Jalanko A, Peltonen L, 2002. Lysosomal localization of the neuronal ceroid lipofuscinosis CLN5 protein. Human Molecular Genetics 11, 885–891. 10.1093/hmg/11.8.885 [DOI] [PubMed] [Google Scholar]
- Jules F, Sauvageau E, Dumaresq-Doiron K, Mazzaferri J, Haug-Kröper M, Fluhrer R, Costantino S, Lefrancois S, 2017. CLN5 is cleaved by members of the SPP/SPPL family to produce a mature soluble protein. Experimental Cell Research 357, 40–50. 10.1016/j.yexcr.2017.04.024 [DOI] [PubMed] [Google Scholar]
- Katz ML, Coates JR, Cooper JJ, O’Brien DP, Jeong M, Narfström K, 2008. Retinal Pathology in a Canine Model of Late Infantile Neuronal Ceroid Lipofuscinosis. Investigative Opthalmology & Visual Science 49, 2686. 10.1167/iovs.08-1712 [DOI] [PubMed] [Google Scholar]
- Katz ML, Coates JR, Sibigtroth CM, Taylor JD, Carpentier M, Young WM, Wininger FA, Kennedy D, Vuillemenot BR, O’Neill CA, 2014. Enzyme replacement therapy attenuates disease progression in a canine model of late-infantile neuronal ceroid lipofuscinosis (CLN2 Disease). Journal of Neuroscience Research 92, 1591–1598. 10.1002/jnr.23423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz ML, Drea CM, Eldred GE, Hess HH, Robison WG, 1986. Influence of early photoreceptor degeneration on lipofuscin in the retinal pigment epithelium. Experimental Eye Research 43, 561–573. 10.1016/S0014-4835(86)80023-9 [DOI] [PubMed] [Google Scholar]
- Katz ML, Redmond MT, 2001. Effect of Rpe65 knockout on accumulation of lipofuscin fluorophores in the retinal pigment epithelium. Investigative Ophthalmology and Visual Science 42, 3023–3030. [PubMed] [Google Scholar]
- Katz ML, Robison WG, 1984. Age-related changes in the retinal pigment epithelium of pigmented rats. Experimental Eye Research 38, 137–151. 10.1016/0014-4835(84)90098-8 [DOI] [PubMed] [Google Scholar]
- Katz ML, Shanker MJ, 1989. Development of lipofuscin-like fluorescence in the retinal pigment epithelium in response to protease inhibitor treatment. Mechanisms of Ageing and Development 49, 23–40. 10.1016/0047-6374(89)90065-1 [DOI] [PubMed] [Google Scholar]
- Katz ML, Tecedor L, Chen Y, Williamson BG, Lysenko E, Wininger F. a, Young WM, Johnson GC, Whiting REH, Coates JR, Davidson BL, 2015. AAV gene transfer delays disease onset in a TPP1-deficient canine model of the late infantile form of Batten disease. Science Translational Medicine 7, 313ra180–313ra180. 10.1126/scitranslmed.aac6191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz ML, Wendt KD, Sanders DN, 2005. RPE65 gene mutation prevents development of autofluorescence in retinal pigment epithelial phagosomes. Mechanisms of Ageing and Development 126, 513–521. 10.1016/j.mad.2004.11.004 [DOI] [PubMed] [Google Scholar]
- Kimotsuki T, Yasuda M, Tamahara S, Matsuki N, Ono K, 2005. Topographic analysis of flash visual evoked potentials in dogs. Journal of Veterinary Medical Science 67, 869–875. 10.1292/ivms.67.869 [DOI] [PubMed] [Google Scholar]
- Kimotsuki T, Yasuda M, Tamahara S, Tomihari M, Matsuki N, Ono K, 2006. Age-associated changes of flash visual evoked potentials in dogs. Journal of Veterinary Medical Science. 68(1):79–82. [DOI] [PubMed] [Google Scholar]
- Kolicheski A, Johnson GS, O’Brien DP, Mhlanga-Mutangadura T, Gilliam D, Guo J, Anderson-Sieg TD, Schnabel RD, Taylor JF, Lebowitz A, Swanson B, Hicks D, Niman ZE, Wininger FA, Carpentier MC, Katz ML, 2016. Australian Cattle Dogs with Neuronal Ceroid Lipofuscinosis are Homozygous for a CLN5 Nonsense Mutation Previously Identified in Border Collies. Journal of Veterinary Internal Medicine 30, 1149–1158. 10.1111/jvim.13971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin H, Ribeiro MG, Lavoie C, 2013. Topology and Membrane Anchoring of the Lysosomal Storage Disease-Related Protein CLN5. Human Mutation 34, 1688–1697. 10.1002/humu.22443 [DOI] [PubMed] [Google Scholar]
- Lebrun A-H, Storch S, Rüschendorf F, Schmiedt M-L, Kyttälä A, Mole SE, Kitzmüller C, Saar K, Mewasingh LD, Boda V, Kohlschütter A, Ullrich K, Braulke T, Schulz A, 2009. Retention of lysosomal protein CLN5 in the endoplasmic reticulum causes neuronal ceroid lipofuscinosis in Asian Sibship. Human Mutation 30, E651–E661. 10.1002/humu.21010 [DOI] [PubMed] [Google Scholar]
- Leinonen H, Keksa-Goldsteine V, Ragauskas S, Kohlmann P, Singh Y, Savchenko E, Puranen J, Malm T, Kalesnykas G, Koistinaho J, Tanila H, Kanninen KM, 2017. Retinal Degeneration in A Mouse Model of CLN5 Disease Is Associated with Compromised Autophagy. Scientific Reports 7, 1–12. 10.1038/s41598-017-01716-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamo A, Jules F, Dumaresq-Doiron K, Costantino S, Lefrancois S, 2012. The Role of Ceroid Lipofuscinosis Neuronal Protein 5 (CLN5) in Endosomal Sorting. Molecular and Cellular Biology 32, 1855–1866. 10.1128/mcb.06726-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmor MF, Holder GE, Seeliger MW, Yamamoto S, 2004. Standard for clinical electroretinography (2004 update). Documenta Ophthalmologica 108, 107–114. 10.1023/B:DOOP.0000036793.44912.45 [DOI] [PubMed] [Google Scholar]
- Melville SA, Wilson CL, Chiang CS, Studdert VP, Lingaas F, Wilton AN, 2005. A mutation in canine CLN5 causes neuronal ceroid lipofuscinosis in Border collie dogs☆. Genomics 86, 287–294. 10.1016/j.ygeno.2005.06.005 [DOI] [PubMed] [Google Scholar]
- Mitchell NL, Russell KN, Wellby MP, Wicky HE, Schoderboeck L, Barrell GK, Melzer TR, Gray SJ, Hughes SM, Palmer DN, 2018. Longitudinal In Vivo Monitoring of the CNS Demonstrates the Efficacy of Gene Therapy in a Sheep Model of CLN5 Batten Disease. Molecular Therapy 26, 2366–2378. 10.1016/j.ymthe.2018.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moharir A, Peck SH, Budden T, Lee SY, 2013. The Role of N-Glycosylation in Folding, Trafficking, and Functionality of Lysosomal Protein CLN5. PLoS ONE 8, e74299. 10.1371/journal.pone.0074299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan BR, Coates JR, Johnson GC, Shelton GD, Katz ML, 2014. Characterization of thoracic motor and sensory neurons and spinal nerve roots in canine degenerative myelopathy, a potential disease model of amyotrophic lateral sclerosis. Journal of Neuroscience Research 92, 531–541. 10.1002/jnr.23332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray SJ, Russell KN, Melzer TR, Gray SJ, Heap SJ, Palmer DN, Mitchell NL, 2021. Intravitreal gene therapy protects against retinal dysfunction and degeneration in sheep with CLN5 Batten disease. Experimental Eye Research 108600. 10.1016/i.exer.2021.108600 [DOI] [PubMed] [Google Scholar]
- Narfström K, Katz ML, Bragadottir R, Seeliger M, Boulanger A, Redmond TM, Caro L, La i C-M., Rakozcy E, 2003. Functional and structural recovery of the retina after gene therapy in the RPE65 null mutation dog. Invest. Ophthalmol. Vis. Sci 44:1663–1672. [DOI] [PubMed] [Google Scholar]
- Narfström K, Vaegan, Katz ML, Bragadottir R, Rakoczy EP, Seeliger M, 2005. Assessment of Structure and Function over a 3-Year Period after Gene Transfer in RPE65−/− Dogs. Doc. Ophthalmol 111:39–48. [DOI] [PubMed] [Google Scholar]
- NCL Mutation and Patient Database [WWW Document], n.d. URL https://www.ucl.ac.uk/ncl-disease/ncl-resource-gateway-batten-disease/mutation-and-patient-database/mutation-and-patient-datasheets-4 (accessed 5.2.21).
- Perentos N, Martins AQ, Cumming RJM, Mitchell NL, Palmer DN, Sawiak SJ, Morton AJ, 2016. An EEG Investigation of Sleep Homeostasis in Healthy and CLN5 Batten Disease Affected Sheep. Journal of Neuroscience 36, 8238–8249. 10.1523/JNEUROSCI.4295-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perentos N, Martins AQ, Watson TC, Bartsch U, Mitchell NL, Palmer DN, Jones MW, Jennifer Morton A, 2015. Translational neurophysiology in sheep: Measuring sleep and neurological dysfunction in CLN5 Batten disease affected sheep. Brain 138, 862–874. 10.1093/brain/awv026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda-Trujillo N, Cornejo W, Carrizosa J, Wheeler RB, Múnera S, Valencia A, Agudelo-Arango J, Cogollo A, Anderson G, Bedoya G, Mole SE, Ruíz-Linares A, 2005. A CLN5 mutation causing an atypical neuronal ceroid lipofuscinosis of juvenile onset. Neurology 64, 740–742. 10.1212/01.WNL.0000151974.44980.F1 [DOI] [PubMed] [Google Scholar]
- Russell KN, Mitchell NL, Anderson NG, Bunt CR, Wellby MP, Melzer TR, Barrell GK, Palmer DN, 2018. Computed tomography provides enhanced techniques for longitudinal monitoring of progressive intracranial volume loss associated with regional neurodegeneration in ovine neuronal ceroid lipofuscinoses. Brain and Behavior 8, 1–8. 10.1002/brb3.1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santavuori P, Rapola J, Nuutila A, Raininko R, Lappi M, Launes J, Herva R, Sainio K, 1991. The Spectrum of Jansky-Bielschowsky Disease. Neuropediatrics 22, 92–96. 10.1055/S-2008-1071423 [DOI] [PubMed] [Google Scholar]
- Santavuori P, Rapola J, Sainio K, Raitta Ch., 1982. A Variant of Jansky-Bielschowsky Disease. Neuropediatrics 13, 135–141. 10.1055/s-2008-1059612 [DOI] [PubMed] [Google Scholar]
- Santavuori P, Vanhanen SL, Autti T, 2001. Clinical and neuroradiological diagnostic aspects of neuronal ceroid lipofuscinoses disorders. European Journal of Paediatric Neurology 5, 157–161. 10.1053/ejpn.2000.0454 [DOI] [PubMed] [Google Scholar]
- Savukoski M, Klockars T, Holmberg V, Santavuori P, Lander ES, Peltonen L, 1998. CLN5, a novel gene encoding a putative transmembrane protein mutated in Finnish variant late infantile neuronal ceroid lipofuscinosis. Nature Genetics 19, 286–288. 10.1038/975 [DOI] [PubMed] [Google Scholar]
- Schmiedt ML, Bessa C, Heine C, Ribeiro MG, Jalanko A, Kyttälä A, 2010. The neuronal ceroid lipofuscinosis protein CLN5: New insights into cellular maturation, transport, and consequences of mutations. Human Mutation 31, 356–365. 10.1002/humu.21195 [DOI] [PubMed] [Google Scholar]
- Schulz A, Ajayi T, Specchio N, de Los Reyes E, Gissen P, Ballon D, Dyke JP, Cahan H, Slasor P, Jacoby D, Kohlschütter A, 2018. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. New England Journal of Medicine 378, 1898–1907. 10.1056/nejmoa1712649 [DOI] [PubMed] [Google Scholar]
- Simonati A, Williams RE, Nardocci N, Laine M, Battini R, Schulz A, Garavaglia B, Moro F, Pezzini F, Santorelli FM, 2017. Phenotype and natural history of variant late infantile ceroid-lipofuscinosis 5. Developmental Medicine & Child Neurology 59, 815–821. 10.1111/dmcn.13473 [DOI] [PubMed] [Google Scholar]
- Sisk DB, Levesque DC, Wood PA, Styer EL, 1990. Clinical and pathologic features of ceroid lipofuscinosis in two Australian cattle dogs. Journal of the American Veterinary Medical Association 197, 361–4. [PubMed] [Google Scholar]
- Skerritt G (Ed.), 2018. King’s Applied Anatomy of the Central Nervous System of Domestic Mammals, 2nd Ed. ed. John Wiley & Sons, Inc., West Sussex, UK. [Google Scholar]
- Strain GM, Jackson RM, Tedford BL, 1990. Visual Evoked Potentials in the Clinically Normal Dog. Journal of Veterinary Internal Medicine 4, 222–225. 10.1111/j.1939-1676.1990.tb00901.x [DOI] [PubMed] [Google Scholar]
- Studdert V, Mitten R, 1991. Clinical features of ceroid lipofuscinosis in border collie dogs. Australian Veterinary Journal 68, 137–140. 10.1111/j.1751-0813.1991.tb03156.x [DOI] [PubMed] [Google Scholar]
- Tracy CJ, Whiting REH, Pearce JW, Williamson BG, Vansteenkiste DP, Gillespie LE, Castaner LJ, Bryan JN, Coates JR, Jensen CA, Katz ML, 2016. Intravitreal implantation of TPP1-transduced stem cells delays retinal degeneration in canine CLN2 neuronal ceroid lipofuscinosis. Experimental Eye Research 152, 77–87. 10.1016/j.exer.2016.09.003 [DOI] [PubMed] [Google Scholar]
- Tyynelä J, Suopanki J, Santavuori P, Baumann M, Haltia M, 1997. Variant Late Infantile Neuronal Ceroid-lipofuscinosis. Journal of Neuropathology and Experimental Neurology 56, 369–375. 10.1097/00005072-199704000-00005 [DOI] [PubMed] [Google Scholar]
- Villani NA, Bullock G, Michaels JR, Yamato O, O’Brien DP, Mhlanga-Mutangadura T, Johnson GS, Katz ML, 2019. A mixed breed dog with neuronal ceroid lipofuscinosis is homozygous for a CLN5 nonsense mutation previously identified in Border Collies and Australian Cattle Dogs. Molecular Genetics and Metabolism 127, 107–115. 10.1016/j.ymgme.2019.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Eisenhart-Rothe P, Grubman A, Greferath U, Fothergill LJ, Jobling AI, Phipps JA, White AR, Fletcher EL, Vessey KA, 2018. Failure of Autophagy–Lysosomal Pathways in Rod Photoreceptors Causes the Early Retinal Degeneration Phenotype Observed in Cln6nclf Mice. Investigative Ophthalmology & Visual Science 59, 5082–5097. 10.1167/iovs.18-24757 [DOI] [PubMed] [Google Scholar]
- Wavre-Shapton ST, Calvi AA, Turmaine M, Seabra MC, Cutler DF, Futter CE, Mitchison HM, 2015. Photoreceptor phagosome processing defects and disturbed autophagy in retinal pigment epithelium of Cln3Δex1-6 mice modelling juvenile neuronal ceroid lipofuscinosis (Batten disease). Human Molecular Genetics 24, 7060–7074. 10.1093/hmg/ddv406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting REH, Jensen CA, Pearce JW, Castaner LJ, Gillespie LE, Bristow DE, Katz ML, 2016. Intracerebroventricular gene therapy that delays neurological disease progression is associated with selective preservation of retinal ganglion cells in a canine model of CLN2 disease. Experimental Eye Research 146:276–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting REH, Narfström K, Yao G, Pearce JW, Coates JR, Castaner LJ, Jensen CA, Dougherty BN, Vuillemenot BR, Kennedy D, O’Neill CA, Katz ML, 2014. Enzyme Replacement Therapy Delays Pupillary Light Reflex Deficits in a Canine Model of Late Infantile Neuronal Ceroid Lipofuscinosis. Experimental Eye Research 125:164–172. [DOI] [PubMed] [Google Scholar]
- Whiting REH, Pearce JW, Castaner LJ, Jensen CA, Katz RJ, Gilliam DH, Katz ML, 2015. Multifocal retinopathy in Dachshunds with CLN2 neuronal ceroid lipofuscinosis. Experimental Eye Research 134, 123–132. 10.1016/j.exer.2015.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting REH, Pearce JW, Vansteenkiste DP, Bibi K, Lim S, Robinson Kick G, Castaner LJ, Sinclair J, Chandra S, Nguyen A, O’Neill CA, Katz ML, 2020a. Intravitreal enzyme replacement preserves retinal structure and function in canine CLN2 neuronal ceroid lipofuscinosis. Experimental Eye Research 197, 108130. 10.1016/j.exer.2020.108130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting REH, Robinson Kick G, Ota-Kuroki J, Lim S, Castaner LJ, Jensen CA, Kowal J, Nguyen A, Corado C, O’Neill CA, Katz ML, 2020b. Intravitreal enzyme replacement inhibits progression of retinal degeneration in canine CLN2 neuronal ceroid lipofuscinosis. Experimental Eye Research 197, 108135. 10.1016/i.exer.2020.108135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting REH, Yao G, Narfström K, Pearce JW, Coates JR, Dodam JR, Castaner LJ, Katz ML, 2013. Quantitative Assessment of the Canine Pupillary Light Reflex. Invest. Ophthalmol. Vis. Sci 54:5432–5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin W, Mullen TE, Kiely R, Min J, Feng X, Cao Y, O’Malley L, Shen Y, Chu-Shore C, Mole SE, Goebel HH, Sims K, 2010. CLN5 mutations are frequent in juvenile and late-onset non-Finnish patients with NCL. Neurology 74, 565–571. 10.1212/WNL.0b013e3181cff70d [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.