

Abstract
Objective:
Polygenic risk scores (PRSs) assess the individual genetic propensity to a condition by combining sparse information scattered across genetic loci, often displaying small effect sizes. Most PRSs are constructed in European-ancestry populations, limiting their use in other ethnicities. Here we constructed and validated a PRS for late-onset Alzheimer’s Disease (LOAD) in Caribbean Hispanics (CH).
Methods:
We used a CH discovery (n = 4,312) and independent validation sample (n = 1,850) to construct an ancestry-specific PRS (“CH-PRS”) and evaluated its performance alone and with other predictors using the area under curve (AUC) and logistic regression (strength of association with LOAD and statistical significance). We tested if CH-PRS predicted conversion to LOAD in a subsample with longitudinal data (n = 1,239). We also tested the CH-PRS in an independent replication CH cohort (n = 200) and brain autopsy cohort (n = 33). Finally, we tested the effect of ancestry on PRS by using European and African American discovery cohorts to construct alternative PRSs (“EUR-PRS”, “AA-PRS”).
Results:
The full model (LOAD ~ CH-PRS + sex + age + APOE-ϵ4), achieved an AUC = 74% (ORCH-PRS = 1.51 95% CI = 1.36–1.68), raising to >75% in APOE-ϵ4 non-carriers. CH-PRS alone achieved an AUC = 72% in the autopsy cohort, raising to AUC = 83% in full model. Higher CH-PRS was significantly associated with clinical LOAD in the replication CH cohort (OR = 1.61, 95%CI = 1.19–2.17) and significantly predicted conversion to LOAD (HR = 1.93, CI = 1.70–2.20) in the longitudinal subsample. EUR-PRS and AA-PRS reached lower prediction accuracy (AUC = 58% and 53%, respectively).
Interpretation:
Enriching diversity in genetic studies is critical to provide an effective PRS in profiling LOAD risk across populations.
Introduction
Genome-wide association studies (GWAS) have identified hundreds of disease-associated loci in several complex diseases such as late-onset Alzheimer’s Disease (LOAD), mostly single-nucleotide polymorphisms (SNPs) characterized by small effect sizes. The polygenic risk score (PRS)1 has the potential to identify individual’s disease risk by combining sparse information distributed across this large number of SNPs. PRS can highlight at-risk individuals and allow for better diagnostic and application of early intervention strategies.
Several efforts to develop PRSs have been conducted for LOAD2,3 or LOAD-endophenotypes,4 achieving good sensitivity and specificity in detecting disease status, and in improving the diagnostic algorithm in association with well-established variables such as sex, age and APOE. PRS leverages summary statistics derived from large GWAS efforts (here referred as “training” or “discovery” dataset)5 to construct an informative predictive variable in an independent dataset (“validation” dataset) phenotyped for the same outcome or related endophenotypes. One major limitation is that PRS performances are hindered when discovery and validation datasets do not share the same ancestral background. Most existing GWASs datasets have focused on population of European descent,6 which limits the incorporation of PRS in other race/ethnic groups, limiting the generalizability across under-represented populations. Indeed, earlier studies have used European ancestry GWAS to construct PRS in other ethnic groups due to a lack of large matching genetic studies7 with lukewarm results.
In this study we developed a PRS using a large Caribbean Hispanic (CH) dataset phenotyped for LOAD. This population is an invaluable asset to investigate LOAD because of their unique genetic background and the high disease incidence and prevalence (two-fold that of non-Hispanic Whites). To our knowledge, no rigorous investigation for PRS within the Caribbean Hispanic population has been conducted so far.
Methods
Data analyzed were obtained from three studies recruiting individuals of CH ancestry: (1) the Washington Heights and Inwood Columbia aging project (WHICAP study); (2) Estudio Familiar de Influencia Genetica en Alzheimer (EFIGA) family study; (3) 10/66 Puerto Rico study (10/66 PR). WHICAP8 is a Northern Manhattan based community-based study of randomly selected elderly individuals of three ethnic groups: non-Hispanic whites, Caribbean Hispanics and African American. EFIGA9 is a family-based study recruiting families with at least two living relatives with dementia history as well as sporadic LOAD cases and healthy controls. 10/66 PR is part of the large 10/66 consortium (it refers to the 66% people with dementia that reside in the developing nations with less than one-tenth of the population-based research conducted in those settings). The 10/66 study is led by King’s College London, recruits people over the age of 65 and is conducted in several countries10,11 focused mainly on lower-income economies.
Informed consent was obtained from all participants. For the WHICAP and EFIGA, the study protocol was approved by the Institutional Review Board (IRB) of Columbia university Medical Center (CUMC). The study protocol for the 10/66 PR population-based study and the consent procedures were approved by the King’s College London research ethics committee and University of Puerto Rico, Medical Sciences Campus Institutional Review Board (IRB). The study was conducted according to the principles expressed in the Declaration of Helsinki.
Diagnosis of Probable/Possible AD
For EFIGA and WHICAP, LOAD diagnosis was carried out according to the National Institute of Neurological and Communication Disorders and Stroke–Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA).12 For 10/66 PR, diagnosis of dementia was assigned according to 10/66 protocol.13 Sex and age (age at onset for incident cases, age at baseline for prevalent cases, age at last evaluation for cognitive healthy controls) were used as main covariates in all statistical models.
Genotyped QC and Imputation
Genotyped data were cleaned with standard QC measures using PLINK (v1.9).14 In brief, SNPs were excluded if MAF≤1%, Hardy–Weinberg equilibrium p value<1-E06 and imputation “INFO” quality <40%; individuals were removed if genotype missingness ≥2%. Data were imputed employing the Haplotype Reference Consortium reference panel15 as described earlier.16 Supervised global ancestry analysis was conducted using ADMIXTURE tool17 using data from the Human Genome Diversity Project (HGDP)18 which comprised Europeans (n = 70), Africans (n = 104) and Native Americans (n = 64) as surrogates of respective ancestry. Individuals that failed to meet criterion of three-way admixture were culled from analysis (ie, showing ≤1% of each European, African and Native American ancestry). Principal component (PC) analysis was conducted using the king software to account for variance in population stratification. We selected PCs to be included in sub-sequential models based on stepwise regression. We excluded from analyses those individuals that deviated ±6 standard deviations from the mean.
Training and Validation Samples
Starting from the entire CH cohort (n = 8,819) that include both sporadic cases and controls as well as large family pedigrees, we selected an unrelated sub-sample (n = 6,162) using the software king (v 2.2.4).19 Subsequently, individuals were randomly assigned to an independent training and validation datasets (70%:30% ratio). The training sub-sample (n = 4,312) was used to perform a traditional genome-wide association study (GWAS) using a generalized mixed model with LOAD as an outcome and first three PCs, sex, age and genetic relationship matrix (GRM) as fixed and random effects, respectively. GRM was constructed using the GEMMA software (v0.98).20 GWAS analyses were conducted employing the GMMAT (v1.1)21 R package. Genomic inflation was calculated using median χ2 statistics. Schematic representation of analysis pipeline is shown in Fig 1.
FIGURE 1:
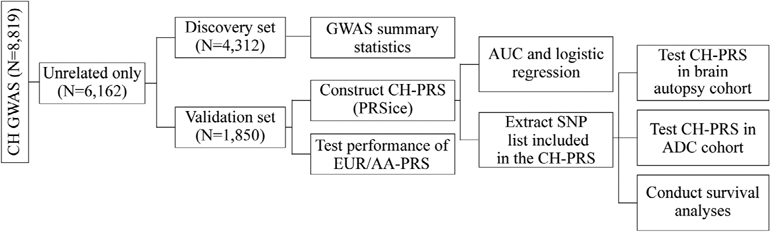
Schematic representation of the analyses pipeline.
PRS Construction and Evaluation of Performance
We used the PRSice software (v 2.3.2)22 to construct the CH-PRS. Analyses were conducted with standard parameters, where SNPs were LD-clumped (–clump-kb 250 –clump-r2 0.1 –clump p1) and filtered out if minor allele frequency (MAF) < 1%. Analyses were conducted on autosomal chromosomes. We included the first three PCS and removed the APOE region (1 MB around index SNPs) from CH-PRS calculation and treated APOE as an independent covariate when modeling LOAD prediction performance. Details about PRSice bioinformatics code can be found on GitHub (https://github.com/sariya/CUMC_taub/tree/master/PRS_CH).
We estimated the area under the curve (AUC)23 to evaluate PRS’s accuracy in the validation dataset to identify predictive ability, with values ranging from 50 to 100%, where 50% is random classification and 100% is perfect classification. We also used regression models to study significance and strength of association between PRS and LOAD or other endophenotypes.
We tested three main statistical models:
LOAD ~ CH-PRS (“model A”)
LOAD ~ CH-PRS + APOE locus (e4 and/or e2 alleles) (“model B”)
LOAD ~ CH-PRS + APOE locus + sex + age (“full model”)
We also computed the maximal possible predictive power (AUCmax) according to disease prevalence and heritability of LOAD. AUCmax is the maximum AUC that could be achieved for a disease when the test classifier is a “perfect” predictor of genetic risk and was computed using the approach proposed by Wray and colleagues,24 and implemented in their online calculator. We tested three possible heritability scenarios according to published data25 in tandem with the disease prevalence as reported in.26
As secondary analyses, we tested PRS performances by cohort strata: (1) APOE-Ɛ4 status (carrier vs non-carrier); (2) sex (men vs women); and in the entire CH cohort (ie, including discovery + validation + related individuals);
AUCs obtained across models were compared using the roc.test function in pROC (R library) (bootstrap method).
Due to three-way admixed nature of the CH population (European, African, and Native American ancestry), we sought to assess the role of ethnicity and its effect on PRS performance. To do so, we built two alternative PRSs using European (EUR)27 and African American (AA)28 GWAS as discovery datasets (from here on labeled as “EUR-PRS” and “AA-PRS”). EUR-PRS and AA-PRS were constructed with identical LD clumping and SNP filters. In addition, the first three PCs were used to generate the PRS. APOE region (1 MB around index SNPs) was excluded. We then compared their performances with that achieved by our original CH-PRS again using the roc.test R function.
PRS Replication
We used an in-house script to re-construct PRS using the SNP list prioritized by PRScise and test its association with LOAD in two different scenarios:
1) the ADC dataset (n = 200), a small CH cohort, part of the Alzheimer’s disease centers (ADC-CH). The NIA ADC cohorts include patients and controls ascertained and evaluated by the clinical and neuropathology cores of the 39 past and present NIA-funded Alzheimer’s Disease Centers (ADC). Data collection is coordinated by the National Alzheimer’s Coordinating Center (NACC). NACC coordinates collection of phenotype data from the ADCs, cleans all data, coordinates implementation of definitions of AD cases and controls, and coordinates collection of samples. Biological specimens are collected, stored, and distributed by the National Cell Repository for Alzheimer’s Disease (NCRAD).29 Data comprised 102 controls (51%), 135 women (67.5%); the mean (±standard deviation) age of cases and controls was 72 ± 7 and 73 ± 7, respectively.
2) a small CH cohort (n = 33) with brain autopsy data.30 Braak staging31,32 and neuropathological diagnosis of AD33 were used as main outcomes. Detailed description of the CH autopsy cohort can be found elsewhere.30 Briefly, the autopsy brains were ascertained from the brain bank of the Alzheimer’s Disease Research Center (ADRC). Neuropathological evaluation is also reported elsewhere.34,35 Neurofibrillary tangles and neuritic plaques were assessed using hematoxylin–eosin and the modified Bielschowsky. Participants that met the criterion of Braak stage ≥4 neurofibrillary pathological, and also the CERAD neuropathological criteria36 (ABC score for intermediate/high) were deemed as cases. We used the clm and glm R functions to test association between PRS and Braak staging and AD pathological diagnosis, respectively. Data comprised 16 controls (48.5%), 21 women (63.6%); the mean (±SD) age of cases and controls was 79 ± 10 and 67 ± 12, respectively.
Longitudinal Analyses
We used a Cox proportional hazards analysis37 implemented in the survival R package to test the CH-PRS performance in predicting conversion from cognitive healthy to LOAD status over time. We restricted our analysis to the EFIGA study only and selected individuals with at least one year of follow up, retaining incident cases and healthy controls only. CH-PRS was entered as a main predictor with age at baseline, sex and APOE-ϵ4 status as covariates. CH-PRS was modeled as continuous variable and also binned in quartiles for graphical representation. We also tested a mixed effects Cox regression model using the coxme R package to account for relatedness between individuals. Finally, we conducted sensitivity analyses restricted to those EFIGA individuals that were part of the validation sample only; because of the small sample size and low number of conversion events, CH-PRS was binned in tertiles.
Results
Random split of unrelated samples resulted in 4,312 individuals assigned to discovery dataset and 1,850 to the validation dataset. We found no significant differences in sex, APOE or age distribution between discovery and validation dataset (χ2 p = 0.48; χ2 p = 0.99; t-test p = 0.32, respectively) (Table 1 shows sample demographics).
Table 1.
Demographics for Caribbean Hispanics (CH) in Validation and Discovery Cohort. “SD” Represents Standard Deviations
| Dataset | EFIGA | WHICAP | 10/66 Puerto Rico | |
|---|---|---|---|---|
| Validation | n | 967 | 478 | 405 |
| Cases | 491 | 233 | 35 | |
| Controls | 476 | 245 | 370 | |
| Age (mean ± SD) | 74.6 ± 8.7 | 80.0 ± 6.7 | 75.1 ± 6.9 | |
| Sex, n (%) | ||||
| Female | 662 (68.4) | 326 (68.2) | 273 (67.4) | |
| APOE | ||||
| Ɛ4− | 609 | 364 | 303 | |
| Ɛ4+ | 356 | 114 | 98 | |
| missing | 2 | 0 | 4 | |
| Discovery | n | 2272 | 1054 | 986 |
| Cases | 1144 | 552 | 72 | |
| Controls | 1128 | 502 | 914 | |
| Age (mean ± SD) | 74.0 ± 8.8 | 79.9 ± 6.7 | 75.9 ± 7.1 | |
| Sex, n (%) | ||||
| Female | 1532(67.4) | 711 (67.4) | 655 (67.4) | |
| APOE | ||||
| Ɛ4− | 1422 | 786 | 762 | |
| Ɛ4+ | 843 | 266 | 210 | |
| missing | 7 | 2 | 14 |
Discovery Dataset GWAS
In the discovery dataset, we did not identify genome-wide significant signals besides APOE locus (rs429358, p = 4.51E-15). No significant genomic inflation was observed (λ = 1.01). Fig 2 shows Manhattan plot (APOE region was removed as previously discussed in Methods).
FIGURE 2:

Manhattan plot for genome-wide association analysis using the CH discovery sample. On the X-axis are represented chromosomes; on the Y-axis −log (p-value).
CH-PRS Performance
PRSice generated a PRS using 146,608 SNPs with an optimal p-value threshold = 0.3. AUC in validation set was found to be 62.21% (model A) while logistic regression found a significant association between CH-PRS and LOAD (p < 0.001, OR = 1.53, CI = 1.38–1.69). In model B, we achieved an AUC = 63.43% (p < 0.001, OR = 1.52, CI = 1.38–1.68). The full model achieved an AUC of 74.02% (p < 0.001, OR = 1.51, CI = 1.38–1.68). Details can be found in Table 2. As a reference, we show in the last column the published AUCs computed in non-Hispanic Whites (NHW; the International Genomics of Alzheimer’s Project [IGAP] study27) When ROCs were compared among different statistical models (eg, those including age, sex without and with PRS as predictors), we observed that including the CH-PRS significantly improved the AUC (p = 3.82E-04). Fig 3 shows comparison of ROC before and after PRS addition to the covariates.
TABLE 2.
Polygenic Risk Score (PRS) in the CH validation Sample; AUC Represents Area Under Curve Shown in Percentage (%); for Logistic Regression Models We Report Confidence Interval (CI) and Odds Ratio (OR). Last Column Shows AUCs Reported in IGAP PRS Paper [2]
| CH validation |
model | AUC (%) | OR | 95% CI | P-value | IGAP AUC (%) |
|---|---|---|---|---|---|---|
| Entire sample | LOAD ~ APOE-Ɛ4 | 55.0 | 1.60 | 1.31–1.96 | 3.31E-06 | 68.3 |
| LOAD ~ PRS | 62.2 | 1.53 | 1.38–1.69 | <1E-16 | Not reported | |
| LOAD ~ PRS+ APOE-Ɛ4 + APOE-Ɛ2 | 63.4 | 1.53 | 1.38–1.68 | <1E-16 | 74.0 | |
| LOAD ~ PRS + APOE-Ɛ4 + APOE-Ɛ2 + sex + age | 74.0 | 1.51 | 1.36–1.68 | 1.75E-14 | 78.2 | |
| APOE-Ɛ4− | LOAD ~ PRS + sex + age | 75.3 | 1.61 | 1.41–1.84 | 1.25E-12 | Not reported |
| APOE-Ɛ4+ | LOAD ~ PRS + sex + age | 69.8 | 1.34 | 1.12–1.61 | 1.16E-03 | |
| Men | LOAD ~ PRS + APOE-Ɛ4 + age | 72.4 | 1.58 | 1.32–1.92 | 7.62E-07 | |
| Women | LOAD ~ PRS + APOE-Ɛ4 + age | 74.8 | 1.47 | 1.29–1.67 | 3.95E-09 |
FIGURE 3:

Receiver operating characteristic (ROC) curve analyses for distinguishing LOAD group from cognitive healthy control group in the CH validation dataset.
AUCmax estimates ranged from 75 to 83%, according to published prevalence and heritability estimates. All scenarios are presented in Table 3.
TABLE 3.
AUCmax (%) Stratified by Age Group Along With Heritability Estimates According to Wray and Colleagues Online Calculator
| Age group | Disease prevalence (%) | AUCmax Heritability = 38.9% |
Heritability = 34.8% | heritability = 28% | |
|---|---|---|---|---|---|
| 64–74 | 10 | 83 | 82 | 79 | |
| 75–84 | 20 | 81 | 79 | 76 | |
| >85 | 30 | 80 | 78 | 75 |
CH-PRS Performance in Autopsy Cohort
In the autopsy cohort, we found that higher CH-PRS scores were significantly associated with higher odds of a pathological AD diagnosis (Model A: AUC = 72.06%; OR = 2.34, CI = 1–5.51, p = 0.05) as well as with higher Braak staging (p = 0.06; OR = 1.83 CI = 0.97–3.45). When the full model was tested, we achieved an AUC = 83.09% (OR = 2.34, CI = 0.95–6.91, p = 0.08). Fig 4 shows CH-PRS scores in cases vs. controls.
FIGURE 4:

Box plot for CH-PRS in the autopsy sample. On the X-axis are represented cases and controls, Y-axis represents CH-PRS scores.
Secondary Analyses
Stratified analyses in APOE-Ɛ4 carriers (n = 568) and APOE-Ɛ4 non-carriers (n = 1,276) showed that PRS performed better at trend (bootstrap p = 0.08) in the latter (full model: AUC = 75.33%; OR = 1.61, CI = 1.41–1.84, p < 0.001) as compared to the former (full model: AUC = 69.78%; OR = 1.34, CI = 1.12–1.61, p = 1.16E-03). No significant differences were observed in sex-strata (bootstrap p = 0.7).
CH-PRS AUC in the full cohort (discovery set + validation set + related individuals initially excluded; n = 8,819) reached 78.9% in Model A, and 85.5% in the full model. APOE-stratified analyses mirrored findings as above, where PRS performed better Ɛ4 non-carriers (n = 5,688; Model A: AUC = 79.64%; OR = 1.28; CI = 1.26–1.29 p < 0.001) than carriers (n = 3,065; Model A: AUC = 78.30%; OR = 1.27; CI = 1.25–1.29, p < 0.001).
Survival Analyses
Analyses were conducted in 1,239 individuals (913 censored and 326 conversion events). We found CH-PRS significantly associated with conversion from healthy cognition to LOAD. In model adjusted for sex, age at baseline (Surv(time, LOAD) ~ sex + age at baseline + CH-PRS) we observed a 94% increase in the expected hazard for CH-PRS (p < 0.001, HR = 1.94, CI = 1.71–2.21). Results were confirmed adding APOE-ϵ4 and APOE-ϵ2 to the model (p < 0.001, HR = 1.93, CI = 1.70–2.20). When CH-PRS was binned in quartiles, we observed increasing HRs for higher quartiles, keeping the first one as reference (fourth quartile HR = 4.34, CI = 2.98–6.32, p < 0.001) (Fig 5). Results obtained with the mixed effect Cox regression test overlapped to those presented above.
FIGURE 5:

Survival plot for CH-PRS (binned in quartile).
Sensitivity analyses included individuals that were part of the validation sample only (n = 177, 40 conversion events). Because of the small sample size, instead of quartiles, CH-PRS was binned in tertiles, with the higher one significantly predicting conversion to LOAD (Surv (time, LOAD) ~ sex + age at baseline + CH-PRS, HR = 2.25, CI = 1.02–4.94, p = 0.043).
PRS in ADC-CH
PRS constructed in CH-ADC was significantly associated with LOAD (OR = 1.60. CI = 1.19–2.17, p = 1.97E-03) with an AUC = 63.59%. We observed the best AUC in the full model (67.25%; OR = 1.56, CI = 1.12–2.18, p = 8.32E-03).
PRS Using European and African American Discovery Cohorts
Using IGAP GWAS as a discovery dataset, we constructed a new EUR-PRS in the CH validation sample: the optimal model included 97,875 SNPs (p-value threshold = 0.5) and obtained an AUC = 58.47% (CI = 55.84–61.10) (OR = 1.34, CI = 1.22–1.48, p = 9.39E-10). Using the AA GWAS, we obtained a new AA-PRS that included 15 SNPs (p-value threshold = 1E-05) and reached an AUC = 53.23% (CI = 50.62–55.85) (OR = 1.10, CI = 1.00–1.21, p = 4.03E-02).
When these two PRS were compared to the CH-PRS, we observed that CH-PRS was superior to both the EUR-PRS (p = 0.04) and the AA-PRS (p < 0.001), although the first instance does not survive after multiple testing correction.
Nevertheless, when we used the CH-PRS and EUR-PRS in Cox regression models, with or without restricting the analyses to the validation sample only, we observed that neither the EUR-PRS nor the AA-PRS significantly predicted conversion from cognitive healthy to LOAD (eg, Surv(time, LOAD) ~ sex + age at baseline + EUR-PRS, HR = 0.75, CI = 0.32–1.73, p = 0.5).
Discussion
To our knowledge, this is the first PRS developed in Caribbean Hispanics for LOAD. We showed a good prediction accuracy when combined with other well-established risk factors such as age, sex and APOE, comparable to those reported in larger studies of European ancestry. Adding the PRS to these risk factors significantly improved accuracy and we were able to demonstrate the strong association between higher PRS scores and neuropathological features in an independent CH autopsy cohort. We also replicated such association in an independent case-control CH cohort and showed that PRS significantly predicts conversion from healthy status to LOAD.
Most PRS have been developed and validated in NHW populations2 with reasonable prediction values. These PRS also showed optimal performance in predicting conversion from a healthy status to AD,38 or association with LOAD-endophenotypes.39 Our empirical AUC in the model combing the PRS and APOE reached a 64% prediction accuracy, lower than what was observed in NHW (AUC = 71.7%) or the estimated AUCmax (75–83%, depending on age strata). Two aspects need to be considered to explain this discrepancy:
First, in NHW the APOE genotype alone reaches an AUC = 68.3%: in other words, a large part of diagnosis accuracy derives from the APOE locus. This represents the largest difference between NHW and our study, where the APOE-ϵ4 allele reaches a lukewarm AUC = 55%.
Second, in NHW, the addition of the PRS to the APOE locus little improves the overall prediction accuracy (AUC = 68.3% vs AUC = 74.5%, respectively). The addition of the 20 genome-wide associated loci to APOE further reduced this gap (AUC = 72% vs AUC = 74.5%, respectively). Consequently, much of the model performance can be attributed to APOE and the genome-wide top-loci only. On the contrary, in our study we observed a much larger improvement for the APOE + PRS model compared to the one with APOE alone (AUC = 64% vs AUC = 55%).
The lower AUC in Hispanics for APOE is not surprising because of the weaker effect of APOE in admixed populations compared with NHW. For example, African Americans are 1.4 times more likely to carry the Ɛ4 allele than NHW, yet the APOE effect size is lower and comparable to that of other loci (eg, ABCA7).28 Indeed, other genetic loci not yet identified could explain the discrepancy between the theoretical prediction accuracy (AUCmax) and the observed AUC for our CH-PRS. The contribution of rare variants is not captured by traditional PRSs, which are constructed over common variants. Rare variants could play a larger role in CH than in NHW. The frequency of underlying genetic risk variants varies substantially between populations and admixed groups (such as CH and AA) are enriched with rare variants.40 Higher degree of genetic variability for admixed populations is well-established: recent studies have shown that the number of variants per genome is higher among individuals of African ancestry (~5 million variants) compared with individuals of East Asian, European, or South Asian ancestry (~4 million variants). Consequently, populations carrying a significant proportion of African genome (such as CH) will have a greater variability in the number of variants. CH also carry a significant proportion of Amerindian genome. Populations of indigenous ancestry in the Americas show exacerbated genetic divergence due to extreme isolation and serial founder effects, leading to an increased fraction of population-specific variation.41
PRS performance in APOE-ϵ4 non carriers showed even higher AUC in the full model (~75%), which is very close to prediction accuracy obtained in NHW. Thus, while the APOE locus has poor prediction accuracy by itself, it still contributes to heterogeneity of the genetic risk profile in CH. Furthermore, prediction accuracy could be hampered by the heterogeneity in LOAD definition and differential contribution of concomitant conditions across ethnic groups. For example, it is well established that cardiovascular and cerebrovascular conditions are strongly associated with LOAD and improve disease prediction (Brickman and Tosto, 2019). Indeed, these conditions have higher incidence and prevalence in Hispanic populations compared to NHW.
PRS performance appears even more compelling in the brain autopsy cohort with an associated AUC = 72%, which increases to 83% in the full model. The observed AUC is therefore close to the estimated AUCMAX (75–83%) and comparable to the higher accuracy observed in NHW autopsy cohorts: a recent investigation conducted in a large pathological case-control series (part of the IGAP study) reported a predictive AUC = 84% for the PRS.42 This AUC is even higher than the theoretical maximal genetic variance (AUCmax ~ 82%), indicating that in NHW the PRS: (1) captures almost entirely the genetic component of LOAD; and (2) genetic prediction is better in autopsy-confirmed case-control series than clinical ones. Similar findings were reported in other NHW studies,38 where PRS and APOE along with gender and age at death produced a final AUC = 82.5%. Again, in NHW the APOE genotypes alone showed a predictability of 81.8%. Results observed in autopsy samples suggest that the accuracy of clinical diagnosis could explain the lower AUC performances in clinical samples.
Alternative PRS constructed in our CH validation sample using NHW or AA discovery GWASs confirmed the importance of wider representation in genetic studies and ethnic-specific pipelines implementation. Despite our CH-PRS being generated using a smaller discovery GWAS (~ 4,000), its performance was comparable if not superior to the one generated using IGAP, a dataset approximately 15 times bigger. Notably, survival analyses showed that only the CH-PRS was significantly associated with conversion to LOAD, whereas the EUR-PRS did not show any significant association. Although CH carry a significant proportion of European and African descent genome (56% and 35%, respectively) the unique admixture of CH limits the use of other ethnic groups to construct an efficient PRS. Furthermore, better performance of the EUR-PRS compared to the AA-PRS (AUC = 58% vs 53%) could have two explanations: (1) CH have higher European ancestry than African ancestry, therefore the NHW GWAS better captures the risk profile and the allele frequencies of our target CH cohort (although AA show as well a significant proportion of European ancestry [~20%]); (2) the NHW GWAS is better powered than the AA GWAS (~74,000 vs ~6,000) which could explain the better performance of the EUR-PRS. Consequently, higher representation of Hispanics and AA is needed to obtain consistent comparisons across ethnicities.
Strengths and Limitations
The replication of our findings indicated that CH-PRS was significantly associated with higher likelihood of LOAD diagnosis in the ADC-CH cohort. This is highly relevant as the ADC study is independent from our CH samples in terms of recruitment sites, demographic distribution, ascertainment methods etc. Furthermore, the effect sizes observed for the newly-developed CH-PRS are highly comparable (OR = 1.61 vs OR = 1.51 in ADC-CH vs our study, respectively). Validation in an independent brain autopsy cohort and in a sub-sample with longitudinal data are additional strengths.
We used the AUC throughout our investigation as it is an established measure for determining the efficacy of tests in correctly classifying diseased and non-diseased individuals and to compare our findings to those of previous studies. Nevertheless, AUCs have been shown to be misleading and are problematic as comparative measures across studies.43
This study has limitations. First, the sample sizes of both our CH validation and replication datasets (CH-ADC and the brain autopsy datasets) are relatively small, which may underestimate the observed AUCs. In addition, we did not have a third large independent CH sample to carry out heritability estimates, thus the decision to use published data to compute the AUCmax. Independent cohorts for replication in non-European descent populations are often difficult to obtain because of the limited number of such cohorts with available genetic data. In particular, autopsy cases are extremely rare, as Hispanics tend to not participate in either organ donation in general, or brain donation more specifically, to the same extent as non-Hispanic Whites.44
Second, GWAS summary statistics that are publicly available for AA comprised a significantly smaller sample size compared to the NHW GWAS, hence, it remains to be investigated if the poorer AA-PRS performance should be attributed to its smaller sample size or it is truly due to a lower AFR admixture component in the CH population.
Third, although the PRS in the full cohort reached optimal AUC (>85%), this model includes individuals that are part of both the discovery and validation sub-samples, as well as other related individual initially excluded from analyses. This results in an overfitting model that could overestimate the true PRS’s sensitivity and specificity. The EFIGA sub-sample used for survival analyses is also part of both the discovery and validation samples: despite the already discussed risk of overfitting, it has to be noted that in this case the outcome is different (ie, time to conversion vs. LOAD in our main analysis). Nevertheless, when analyses were restricted to validation sample only, we confirmed the validity of our results.
Lastly, our CH-PRS focused on common variants which, with the exception of APOE, are usually proxies for the causal (rare) variants. Future studies should evaluate the contribution of rare variants in PRS performances, possibly deploying them based on their deleterious nature. Recent strategies have focused on constructing PRS using rare variants, which could be particularly useful in admixed populations.45 Alternative approaches have relied on population prediction at the gene level rather than the single polymorphism since the effect of genes on traits is likely to be more highly conserved across ethnicities. This approach converts SNP’s effect sizes in predicted transcript abundance46 resulting in a polygenic transcriptome risk scores.
Acknowledgments
The National Institutes of Health, National Institute on Aging (NIH-NIA) supported this work through the following grants: R56AG069118 and R56AG066889.
For WHICAP: Data collection and sharing for this project was supported by the Washington Heights-Inwood Columbia Aging Project (WHICAP, R01AG037212, RF1AG054023, RF1AG066107) funded by the National Institute on Aging (NIA) and by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant Number UL1TR001873. This manuscript has been reviewed by WHICAP investigators for scientific content and consistency of data interpretation with previous WHICAP Study publications. We acknowledge the WHICAP study participants and the WHICAP research and support staff for their contributions to this study.
For EFIGA: Data collection for this project was supported by the Genetic Studies of Alzheimer’s disease in Caribbean Hispanics (EFIGA) funded by the National Institute on Aging (NIA) and by the National Institutes of Health (NIH) (5R37AG015473, RF1AG015473, R56AG051876, R01AG067501, R56AG063908, RF1AG015473. We acknowledge the EFIGA study participants and the EFIGA research and support staff for their contributions to this study.
For ADI 10/66 PR Alzheimer’s Disease International Epidemiological Study: Data collection for this project was supported by a recurrent PR Legislature grant, Pfizer Co. Grant # GA9001NE, and for PR Apo-E labs: Human Genetics Core Award from Columbia University Irving Institute for Clinical and Translational Research. For ADC study. Alzheimer’s Disease Genetics Consortium (ADGC), U01AG032984, RC2AG036528; National Alzheimer’s Coordinating Center (NACC), U01AG016976. Samples from the National Cell Repository for Alzheimer’s Disease (NCRAD), which receives government support under a cooperative agreement grant (U24 AG21886) awarded by the National Institute on Aging (NIA), were used in this study. We thank contributors who collected samples used in this study, as well as patients and their families, whose help and participation made this work possible; Data for this study were prepared, archived, and distributed by the National Institute on Aging Alzheimer’s Disease Data Storage Site (NIAGADS) at the University of Pennsylvania (U24-AG041689-01).
Footnotes
Potential Conflict of interests
None declared.
References
- 1.Lewis CM, Vassos E. Polygenic risk scores: from research tools to clinical instruments. Genome Med 2020;12:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Escott-Price V, Sims R, Bannister C, et al. Common polygenic variation enhances risk prediction for Alzheimer’s disease. Brain 2015;138:3673–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Escott-Price V, Shoai M, Pither R, et al. Polygenic score prediction captures nearly all common genetic risk for Alzheimer’s disease. Neurobiol Aging 2017;49:214. e7–e11. [DOI] [PubMed] [Google Scholar]
- 4.Walhovd KB, Fjell AM, Sorensen O, et al. Genetic risk for Alzheimer disease predicts hippocampal volume through the human lifespan. Neurol Genet 2020;6:e506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi SW, Mak TS, O’Reilly PF. Tutorial: a guide to performing polygenic risk score analyses. Nat Protoc 2020;15:2759–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mills MC, Rahal C. A scientometric review of genome-wide association studies. Commun Biol 2019;2:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tosto G, Bird TD, Tsuang D, et al. Polygenic risk scores in familial Alzheimer disease. Neurology 2017;88:1180–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brickman AM, Schupf N, Manly JJ, et al. Brain morphology in older African Americans, Caribbean Hispanics, and whites from northern Manhattan. Arch Neurol 2008;65:1053–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Romas SN, Santana V, Williamson J, et al. Familial Alzheimer disease among Caribbean Hispanics: a reexamination of its association with APOE. Arch Neurol 2002;59:87–91. [DOI] [PubMed] [Google Scholar]
- 10.Prina AM, Acosta D, Acosta I, et al. Cohort profile: the 10/66 study. Int J Epidemiol 2017;46:406–i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prina AM, Wu YT, Kralj C, et al. Dependence- and disability-free life expectancy across eight low- and middle-income countries: a 10/66 study. J Aging Health 2020;32:401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prince M, Ferri CP, Acosta D, et al. The protocols for the 10/66 dementia research group population-based research programme. BMC Public Health 2007;7:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang CC, Chow CC, Tellier LC, et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCarthy S, Das S, Kretzschmar W, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 2016;48:1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sariya S, Lee JH, Mayeux R, et al. Rare variants imputation in admixed populations: comparison across reference panels and bioinformatics tools. Front Genet 2019;10:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alexander DH, Lange K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform 2011;12:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenberg NA, Mahajan S, Ramachandran S, et al. Clines, clusters, and the effect of study design on the inference of human population structure. PLoS Genet 2005;1:e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manichaikul A, Mychaleckyj JC, Rich SS, et al. Robust relationship inference in genome-wide association studies. Bioinformatics 2010;26:2867–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet 2012;44:821–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen H, Wang C, Conomos MP, et al. Control for population structure and relatedness for binary traits in genetic association studies via logistic mixed models. Am J Hum Genet 2016;98:653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi SW, O’Reilly PF. PRSice-2: polygenic risk score software for biobank-scale data. Gigascience 2019;1:8(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hajian-Tilaki K. Receiver operating characteristic (ROC) curve analysis for medical diagnostic test evaluation. Caspian J Intern Med 2013;4:627–635. [PMC free article] [PubMed] [Google Scholar]
- 24.Wray NR, Yang J, Goddard ME, Visscher PM. The genetic interpretation of area under the ROC curve in genomic profiling. PLoS Genet 2010;6:e1000864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med 2016;18:421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vega IE, Cabrera LY, Wygant CM, et al. Alzheimer’s disease in the Latino community: intersection of genetics and social determinants of health. J Alzheimers Dis 2017;58:979–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kunkle BW, Grenier-Boley B, Sims R, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet 2019;51:414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reitz C, Jun G, Naj A, et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E 4,and the risk of late-onset Alzheimer disease in African Americans. JAMA 2013;309:1483–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 2011;43:436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Felsky D, Sariya S, Santa-Maria I, et al. The Caribbean-Hispanic Alzheimer’s brain transcriptome reveals ancestry-specific disease mechanisms. bioRxiv. 2020:2020.05.28.122234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 32.Braak H, Alafuzoff I, Arzberger T, et al. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006;112:389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener 2019;14:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vonsattel JP, Aizawa H, Ge P, et al. An improved approach to prepare human brains for research. J Neuropathol Exp Neurol 1995;54:42–56. [DOI] [PubMed] [Google Scholar]
- 35.Vonsattel JP, Del Amaya MP, Keller CE. Twenty-first century brain banking. Processing brains for research: the Columbia University methods. Acta Neuropathol 2008;115:509–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fillenbaum GG, van Belle G, Morris JC, et al. Consortium to establish a registry for Alzheimer’s disease (CERAD): the first twenty years. Alzheimers Dement 2008;4:96–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cox DR. Regression models and life-tables. J R Stat Soc B Methodol 1972;34:187–220. [Google Scholar]
- 38.Chaudhury S, Brookes KJ, Patel T, et al. Correction: Alzheimer’s disease polygenic risk score as a predictor of conversion from mild-cognitive impairment. Transl Psychiatry 2019;9:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan D, Hu B, Darst BF, et al. Biobank-wide association scan identifies risk factors for late-onset Alzheimer’s disease and endophenotypes. bioRxiv. 2018:468306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Igartua C, Myers RA, Mathias RA, et al. Ethnic-specific associations of rare and low-frequency DNA sequence variants with asthma. Nat Commun 2015;6:5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Avila-Arcos MC, McManus KF, Sandoval K, et al. Population history and gene divergence in native Mexicans inferred from 76 human exomes. Mol Biol Evol 2020;37:994–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Escott-Price V, Myers AJ, Huentelman M, Hardy J. Polygenic risk score analysis of pathologically confirmed Alzheimer disease. Ann Neurol 2017;82:311–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lobo J, Jiménez-Valverde A, Real R. AUC: a misleading measure of the performance of predictive distribution models. Glob Ecol Biogeogr 2008;17:145–151. [Google Scholar]
- 44.Frates J, Garcia BG. Hispanic perceptions of organ donation. Prog Transplant 2002;12:169–175. [DOI] [PubMed] [Google Scholar]
- 45.Lali R, Chong M, Omidi A, et al. Calibrated rare variant genetic risk scores for complex disease prediction using large exome sequence repositories. bioRxiv. 2020:2020.02.03.931519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liang Y, Pividori M, Manichaikul A, et al. Polygenic transcriptome risk scores improve portability of polygenic risk scores across ancestries. bioRxiv. 2020:2020.11.12.373647. [DOI] [PMC free article] [PubMed] [Google Scholar]